
Abstract
Presenilin-1 (PS1) gene encodes the catalytic component of γ-secretase, which proteolytically processes several type I transmembrane proteins. We here present evidence that the cytosolic peptide efnB2/CTF2 produced by the PS1/γ-secretase cleavage of efnB2 ligand promotes EphB4 receptor-dependent angiogenesis in vitro. EfnB2/CTF2 increases endothelial cell sprouting and tube formation, stimulates the formation of angiogenic complexes that include VE-cadherin, Raf-1 and Rok-α, and increases MLC2 phosphorylation. These functions are mediated by the PDZ-binding domain of efnB2. Acute downregulation of PS1 or inhibition of γ-secretase inhibits the angiogenic functions of EphB4 while absence of PS1 decreases the VE-cadherin angiogenic complexes of mouse brain. Our data reveal a mechanism by which PS1/γ-secretase regulates efnB2/EphB4 mediated angiogenesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The generation of new capillaries from pre-existing vessels, a process known as angiogenesis, is an essential part of growth, development, and wound healing for both embryonic and adult organisms. Insufficient blood vessel branching would impair the transport of oxygen and nutrients to the brain.
Presenilin1 (PS1) is a transmembrane protein that serves as the proteolytic component of the γ-secretase complex that cleaves several type I transmembrane proteins close to the transmembrane/cytoplasm interface [1]. There is evidence that PS1 is essential to vascular integrity [2,3,4]; however, the mechanisms via which this regulation is achieved are unknown.
The EphB4 receptor/ephrinB2 (efnB2) ligand signaling mediates angiogenesis and angiogenic remodeling. Deletion of efnB2 leads to vascular abnormalities and embryonic lethality in mice [5] and removal of the cytoplasmic domain of efnB2, the only known ligand of the EphB4 receptor, shows the same abnormal phenotype as the EphB4 KO mice [6] indicating that the cytoplasmic domain of efnB2 is a key component of this process.
We found that EphB4 stimulates angiogenic complex formation between vascular endothelial cadherin (VE-cadherin), Raf-1 and Rok-α kinases and that EphB4 increases phosphorylation of myosin light chain 2 (MLC2). Both functions of EphB4 depend on PS1/γ-secretase. The VE-cadherin complexes are known to mediate angiogenic function of other factors such as FGF-2 and cAMP-inducible Rap1 GEF (guanine nucleotide exchange factor) EPAC. Recruitment of Rok-α to VE-cadherin results in the phosphorylation and activation of MLC2 and the actomyosin contractile apparatus leading to maturation of adherens junctions, a step essential for cell cohesion during sprouting angiogenesis [7, 8].
We found that PS1/γ-secretase promotes the EphB4/efnB2-mediated angiogenesis by cleaving efnB2 ligand in endothelial cells to produce the cytoplasmic peptide efnB2/CTF2, which has angiogenic functions. This peptide increases sprouting and tube formation, stimulates VE-cadherin angiogenic complex formation, and promotes the phosphorylation of MLC2 at VE-cadherin cell–cell contact sites in vitro, raising the possibility that it has similar angiogenic activity in vivo. Consistent with these findings, absence of PS1 in PS1 knockout (KO) mice inhibits the VE-cadherin angiogenic complexes of the brain. Our findings identify a molecular mechanism via which the efnB2/EphB4 system promotes angiogenesis and show that this mechanism is regulated by PS1/γ-secretase.
Results
EphB4-Fc stimulates γ-secretase cleavage of efnB2 and the production of peptide efnB2/CTF2 that regulates endothelial cell sprouting and tube formation
We showed in mouse fibroblasts and HEK293 cells that the EphB2 receptor induces cleavage of efnB2 ligands by metalloproteinases to generate a transmembrane peptide efnB2/CTF1, which is further processed by PS1/γ-secretase to generate cytosolic efnB2/CTF2 [9]. We tested whether EphB4 receptor stimulates the PS1/γ-secretase processing of efnB2 in primary endothelial BAMEC cells. Figure 1 shows that EphB4-Fc stimulates the accumulation of the fragment efnB2/CTF1 at ~ 21 kDa (bottom panel, arrow) known to be produced by the EphB-stimulated MP cleavage of efnB2. This fragment is then cleaved by PS1/γ-secretase to produce peptide efnB2/CTF2 at ~ 18 kDa (bottom panel, arrow). As expected, efnB2/CTF2 accumulation is inhibited by γ-secretase inhibitors (lanes. D).

EphB4-Fc stimulates efnB2 cleavage by γ-secretase, producing peptide efnB2/CTF2 that regulates sprouting and tube formation. a. Upper: BAMECs transduced with efnB2 triple myc-tagged (myc3) for detection purposes in pMX retroviral vector were stimulated with 2 μg/ml pre-clustered Fc or EphB4-Fc for the indicated time points in the presence or absence of the γ-secretase inhibitor DAPT (D; 1 μM). All samples were treated with the proteasomal inhibitor lactacystin (10 μM). The extracts were IPed with a rabbit polyclonal antibody against c-myc to isolate exogenous myc-tagged efnB2. Cell extracts were immunoblotted with a rabbit polyclonal antibody against the C-terminus of efnB. Arrows indicate full-length efnB2-myc3 (FL) as well as the fragments produced by metalloproteinase and PS1/γ-secretase cleavage of efnB2-myc3 (CTF1 and CTF2, respectively). Lower: quantification of full-length efnB2 and efnB2/CTF2. N = 3. b BAMECs were transduced with pMX alone or vector expressing the cytoplasmic domain of efnB2 triple myc-tagged (CTF2-myc3). Cells were grown on collagen-coated Cytodex® microcarrier beads, transferred to fibrinogen solution, and solidified with thrombin to form fibrin gels. Upper panel: phase-contrast image of BAMEC cells sprouting from a microcarrier bead. Lower panel: Hoechst nuclear staining shows the number of endothelial cells that make up individual sprouts. The percentage of sprouts exceeding the diameter of the bead (~ 175 μm) relative to control vector (pMX) was determined. Wilcoxon matched-pairs signed-rank (n = 6, *p < 0.05, error bars = SEM). c BAMECs transduced with pMX alone or vector expressing CTF2-myc3 were seeded on a 24-well plate pre-coated with Matrigel™ Basement Membrane Matrix and left at 37°C/5% CO2 for 8–12 h to spontaneously form tubes in the presence or absence of L685,458 (1 μM). The plate was observed under a light microscope and the number of tubes formed was determined as a percentage relative to CTF2-myc3. Paired t test (n = 3, ***p < 0.001, error bars = SEM)
To test whether efnB2/CTF2 peptide has angiogenic activity, we examined whether it stimulates endothelial cell sprouting in vitro. To this end, BAMEC cells were transduced with efnB2/CTF2-myc3 and sprouting was measured with the microcarrier bead-based sprouting assay [10]. Figure 1b shows that BAMEC overexpressing efnB2/CTF2 sprout significantly more than cells overexpressing vector (pMX) alone. EfnB2/CTF2 non-tagged with myc tag significantly induces sprouting of cells on beads (Additional file 1: Fig. S1) ruling out the possibility that this effect is mediated by the myc tag of efnB2/CTF2-myc3.
We then tested whether the efnB2/CTF2 peptide promotes tube formation of endothelial cells. This is the next biological process that endothelial cells undergo to ultimately form a complete microvessel [11]. BAMECs overexpressing efnB2/CTF2-myc3 or pMX were seeded on Matrigel as previously described [12, 13]. As shown in Fig. 1c, cells expressing efnB2/CTF2-myc3 form significantly more tubes than the cells expressing vector alone and this induction is not inhibited by γ-secretase inhibitor L685,458, indicating that cleavage of other γ-secretase substrates does not significantly contribute to this angiogenic function of efnB2/CTF2. In support of the above, γ-secretase inhibitor does not inhibit the efnB2/CTF2-induced sprouting (Fig. S1).
Sequences of efnB2/CTF2 that mediate its angiogenic function
The angiogenic function of efnB2/CTF2 may be mediated by its C-terminal PDZ-binding motif. It has been shown that this motif of efnB2 mediates neovascularization in adult mice as mice expressing a mutant form of efnB2 which lacks the PDZ-binding domain have decreased angiogenesis in vivo [14, 15]. In addition, tyrosine residues of the cytoplasmic domain of efnB ligands are phosphorylated by Src-family kinases and mediate sprouting angiogenesis [16]. To this end, we created a mutant efnB2/CTF2 in which the PDZ-binding motif of efnB2 (YYKV) was deleted (ΔPDZ) and a mutant efnB2/CTF2 in which the five conserved tyrosine residues were mutated to phenylalanines (Tyr:Phe). A diagram of the mutants is illustrated in Fig. 2a and expression of these constructs in BAMEC cells is shown in Fig. 2b. In contrast to WT efnB2/CTF2, BAMECs expressing 5Tyr:Phe-myc3 or ΔPDZ-myc3 mutants were unable to sprout more or form more tubes in a Matrigel™ tube formation assay compared to cells transduced with vector alone (Fig. 2c–e) These results show that the in vitro angiogenic activity of efnB2/CTF2 peptide is mediated by its PDZ-binding domain and its tyrosine residues.

Mutant efnB2/CTF2 peptides do not promote sprouting and tube formation. a WT and mutant efnB2/CTF2-myc3 peptides. b BAMECs expressing efnB2/CTF2-myc3 constructs were pre-treated with the proteasomal inhibitor lactacystin (10 μM) and cell extracts were probed on western blot with a rabbit polyclonal antibody against the C-terminus of efnB. The immunoblot shows the expression of wild-type efnB2/CTF2 and mutants 5Tyr:Phe and ΔPDZ all tagged with myc3 (CTF2-myc3, 5Tyr:Phe-myc3, ΔPDZ-myc3) at ~ 18 kDa while mutant ΔPDZ-myc3 interestingly migrates at ~ 20 kDa. NT non-transduced. c Cells were grown on collagen-coated Cytodex® microcarrier beads, transferred to fibrinogen solution, and solidified with thrombin to form fibrin gels. Upper panel: phase-contrast image of BAMEC cells sprouting from a microcarrier bead. Lower panel: Hoechst nuclear staining shows the number of endothelial cells that make up individual sprouts. d The number of sprouts exceeding the diameter of the bead (~ 175 μm) was determined and analyzed as a percentage relative to control vector (pMX, grey bars). Paired t test (n = 5, **p < 0.01, error bars = SEM). ns non-significant. e Cells were seeded in random wells on a 24-well plate pre-coated with Matrigel™ Basement Membrane Matrix and placed in a stage top incubation chamber controlled at 37°C/5% CO2. The number of tubes formed per construct was determined and analyzed as a percentage relative to CTF2-myc3. Paired t test (n = 3, **p < 0.01, error bars = SEM). ns non-significant
Interestingly, mutant ΔPDZ-myc3 migrated at ~ 20 kDa (lane 5), at a higher apparent molecular weight than the WT efnB2/CTF2-myc3. This could be due to conformational changes in this peptide or even dimerization, which in the case of other proteins has been shown to be SDS resistant [17, 18].
EphB4- and efnB2/CTF2-induced sprouting depends on Rap1 activity
Angiogenic activity of FGF-2, EPAC and VEGF depends on small GTPase Rap1 activity [7, 19]. We examined whether EphB4-Fc-induced sprouting also depends on Rap1 activity. We used a chemical inhibitor of Rap1 activity, GGTi-298 that functions as a selective inhibitor of Rap1 prenylation [20]. GGTi-298 significantly decreases EphB4-Fc-induced BAMEC sprouting on beads Fig. 3a. Similarly to EphB4-Fc, GGTi-298 significantly decreases efnB2/CTF2-myc3-induced sprouting, showing that peptide-induced sprouting depends on Rap1 activity as well (Fig. 3b).

Rap1 activity is required for EphB4-Fc- and efnB2/CTF2-induced sprouting. a BAMECs were grown on collagen-coated Cytodex® microcarrier beads and transferred to fibrinogen solution. Pre-clustered Fc or EphB4-Fc (4 μg/ml) was added to the fibrinogen solution as well as DMSO or GGTi-298 (0.5 μM) then solidified with thrombin to form fibrin gels. (Left) Phase-contrast images show BAMEC cells sprouting from a microcarrier bead. (Right) The percentage of sprouts exceeding the diameter of the bead relative to control (Fc) was determined. Paired t test (n = 3, *p < 0.05, error bars = SEM). b Cells were transduced with pMX or pMX expressing efnB2/CTF2-myc3. Cells were grown on collagen-coated Cytodex® microcarrier beads and allowed to sprout in the presence of DMSO (vehicle) or GGTi-298 (0.5 μM). (Left) Phase-contrast image of BAMEC cells sprouting from a microcarrier bead. (Right) The percentage of sprouts exceeding the diameter of the bead relative to control (pMX) was determined. Unpaired t test (*p < 0.05, error bars = SEM) and paired t test (*p < 0.05, error bars = SEM) n = 4
To test whether EphB4-Fc or efnB2/CTF2-myc3 affect Rap1 activity, we treated BAMEC cells with Fc or EphB4-Fc and measured Rap1 activity in cell extracts. Rap1 activity was also measured in BAMEC extracts overexpressing efnB2/CTF2-myc3 and was compared to vector-expressing cells. We found that neither EphB4-Fc nor efnB2/CTF2-myc3 affect Rap1 activity (Fig. S3). To test whether inhibition of Rap1 activity affects the γ-secretase processing of efnB2 and the EphB4-induced accumulation of efnB2/CTF2, we treated cells with Fc or EphB4-Fc in the presence or absence of GGTi-298 and we probed for efnB2/CTF2 as in Fig. 1. We found that GGTi-298 does not affect the EphB4-induced accumulation of efnB2/CTF2 (not shown).
EphB4 stimulates Raf-1/VE-cadherin and Rok-α/VE-cadherin complexes in a γ-secretase- and Rap1-dependent manner
Since endothelial cell sprouting requires formation of Raf-1/VE-cadherin and Rok-α/VE-cadherin complexes [7], we asked whether EphB4-Fc stimulates these complexes. BAMEC cells were treated with Fc or EphB4-Fc and cell extracts were immunoprecipitated (IPed) with anti-Raf-1 or anti-Rok-α antibodies. VE-cadherin was detected in the IPs. As shown in Fig. 4a, d, EphB4-Fc significantly increases the complexes between VE-cadherin and Raf-1 and between VE-cadherin and Rok-α. In addition, since the EphB4-Fc-induced sprouting of BAMECs depends on γ-secretase activity [9], we asked whether the EphB4-induced complexes depend on γ-secretase using γ-secretase inhibitors L685,458 and Compound E. As shown in Fig. 4b, e both inhibitors block the EphB4-Fc-induced complexes between VE-cadherin and Raf-1 and between VE-cadherin and Rok-α suggesting that γ-secretase activity promotes the EphB4-induced complexes. The result was confirmed with a third γ-secretase inhibitor DAPT (not shown). Rap1 activity is necessary for the induction of these complexes by EphB4-Fc as the specific Rap1 inhibitor GGTi-298 strongly inhibits this induction (Fig. 4c, f).

EphB4-Fc increases the Raf-1/VE-cadherin and Rok-α/VE-cadherin complexes in a γ-secretase- and Rap1-dependent manner. a Left: BAMECs were treated with 2 μg/ml EphB4-Fc or Fc for the indicated times. Cells were lysed in 1%(v/v) TX-100 in the presence of protease inhibitors and extracts were IPed with anti-Raf-1 antibodies. IPs were probed on WB with anti-VE-cadherin (upper panel) or anti-Raf-1 (lower panel) antibodies. EphB4-Fc increases the Raf-1/VE-cadherin complexes (lanes 3, 6). Right: Bar graph shows quantification of results. Paired t test, n = 4, **p < 0.01, error bars = SEM. b Left: cells were treated with 2 μg/ml EphB4-Fc or Fc for 1 h in the presence or absence of γ-secretase inhibitors L685,458 (1 μM) or Compound E (1 μM) added 3 h prior to EphB4-Fc. Cells were lysed and IPed as in a. EphB4-Fc-induced increase in Raf-1/VE-cadherin complexes (lane 2) was inhibited by both γ-secretase inhibitors (lanes 4,6). Right: bar graph shows quantification of results. Paired t test, n = 4, **p < 0.01, error bars = SEM. c Left: cells were treated with 2 μg/ml Fc or EphB4-Fc in the presence or absence of 0.5 μM Rap1 inhibitor GGTi-298 as indicated in the figure. Cell extracts were IPed as in a. Rap1 inhibitor strongly inhibits EphB4-Fc-induced complexes between Raf-1 and VE-cadherin. Right: bar graph shows quantification of results. Paired t test, n = 4, ***p < 0.001, error bars = SEM. d Left: cells were treated with 2 μg/ml EphB4-Fc or Fc for the indicated times. Cells were lysed in TX-100 and extracts were IPed with anti-Rok-α antibodies. IPs were probed on WB with anti-VE-cadherin (upper panel) or anti-Rok-α (middle panel) antibodies. EphB4-Fc increases the VE-cadherin/Rok-α complexes (lanes 3 and 6) compared to Fc (lanes 2 and 5). Right: bar graph shows quantification of results. Paired t test, n = 4, *p < 0.05, **p < 0.01, error bars = SEM. e Left: cells were treated with γ-secretase inhibitors L685,458 (1 μM) or Compound E (1 μM) as above or DMSO for 3 h and then treated with 2 μg/ml EphB4-Fc or Fc for 1 h. Cell extracts were lysed and IPed as in d. EphB4-Fc-induced Rok-α/VE-cadherin complexes (lane 2) were inhibited by both inhibitors (lanes 4,6). Right: bar graph shows quantification of results. Paired t test, n = 4, *p < 0.05, error bars = SEM. f Left: cells were treated as in c and cell extracts were IPed as in d. Rap1 inhibitor strongly inhibits EphB4-Fc-induced complexes between Rok-α and VE-cadherin. Right: bar graph shows quantification of results. Paired t test, n = 4, **p < 0.01, error bars = SEM
The PDZ-binding domain of efnB2/CTF2 interacts with VE-cadherin and stimulates its angiogenic complexes with Raf-1 and Rok-α
As peptide efnB2/CTF2 promotes sprouting of endothelial cells (Figs. 1, 2), we asked whether it also affects the complexes between VE-cadherin, Raf-1 and Rok-α. We overexpressed efnB2/CTF2 in BAMEC cells and analyzed the VE-cadherin/Raf-1 and the VE-cadherin/Rok-α complexes in cell extracts as in Fig. 4. Transient overexpression of efnB2/CTF2 significantly increases the complexes between Raf-1 and VE-cadherin and between Rok-α and VE-cadherin compared to vector alone (Fig. 5a). A similar effect of efnB2/CTF2 on these complexes is observed in cells stably expressing this peptide (not shown). The effect is specific since the product of the cleavage of N-cadherin by γ-secretase (N-cad/CTF2) [21] did not affect the complexes (Fig. 5b). These data suggest that EphB4-Fc promotes the VE-cadherin/Raf-1 and VE-cadherin/Rok-α complexes by increasing the γ-secretase cleavage of efnB2 producing cytoplasmic efnB2/CTF2 peptide which stimulates these complexes.

WT but not mutant efnB2/CTF2 forms complexes with VE-cadherin, Raf1 and Rok-α and increases angiogenic complexes. a Left: BAMECs were infected with HSV particles expressing efnB2/CTF2-myc3 or vector at 1MOI. 24 h later cells were lysed, extracts were IPed with anti-Raf-1 antibodies and IPs were probed on WB for VE-cadherin (upper panel) or Raf-1 (middle panel). Extracts were also IPed with anti-myc antibody and efnB2/CTF2-myc3 was detected on WB with anti-ephrinB antibody (lower panel, lane 3). efnB2/CTF2-myc3 increases the Raf-1/VE-cadherin complexes (upper panel, lane 3). Right: quantification of results. Paired t test, n = 4, **p < 0.01, error bars = SEM. b Left: BAMECs were infected as in a. Cell extracts were IPed with anti-Rok-α antibodies and IPs were probed on WB for VE-cadherin (upper panel) or Rok-α (middle panel). efnB2/CTF2-myc3 increases the Rok-α/VE-cadherin complexes (upper panel, lane 3) while N-cadherin/CTF2 (N-cad/CTF2) had no effect on these complexes (lane 4). Right: quantification of results. Paired t test, n = 4, *p < 0.05, error bars = SEM. c Left: cells were transduced with pMX vectors expressing WT efnB2/CTF2-myc3 or mutants 5Tyr:Phe-myc3 or ΔPDZ-myc3. Cell extracts were IPed with anti-Raf-1 antibodies and obtained IPs were probed on WBs for VE-cadherin (upper panel) or Raf-1 (lower panel). WT efnB2/CTF2 increases the Raf-1/VE-cadherin complexes compared to vector pMX (lanes 4 and 5). Mutant efnB2/CTF2 peptides showed no increase (lanes 2, 3). Quantification of results. Paired t test, n = 4, **p < 0.01, error bars = SEM. Right: extracts from the above cells were IPed with anti-VE-cadherin antibodies and IPs were probed for Raf-1 (upper panel) or VE-cadherin (lower panel) on WB. WT but not mutant efnB2/CTF2 increases the amount of Raf-1/VE-cadherin complexes (lanes 1–4). Quantification of results. Paired t test, n = 4, **p < 0.01, error bars = SEM. d Left: cells were transduced with WT efnB2/CTF2-myc3 or mutant ΔPDZ-myc3. Cell extracts were IPed with anti-Rok-α and IPs were probed on WBs for VE-cadherin (upper panel) or Rok-α (lower panel). WT but not mutant efnB2/CTF2 increases the Rok-α/VE-cadherin complexes. Right: quantification of results. Paired t test, n = 4, *p < 0.05, **p < 0.01, error bars = SEM. e Left: cells were transduced with WT efnB2/CTF2-myc3 (CTF2-myc3) or mutants 5Tyr:Phe-myc3 or ΔPDZ-myc3. Cells were extracted and IPed as described in Figs. 4 and 5. All three proteins, VE-cadherin, Raf-1 and Rok-α co-IPed with WT efnB2/CTF2 and to a much lesser extent with mutant peptides. Right: quantification of results. Paired t test, n = 4, *p < 0.05, **p < 0.01, error bars = SEM
To identify sequences of efnB2/CTF2 that mediate its effect on VE-cadherin complexes, we tested whether mutants that inhibit its angiogenic function (Fig. 2) affect these complexes. As shown in Fig. 5c, d, although WT efnB2/CTF2 increases the VE-cadherin/Raf-1 and VE-cadherin/Rok-α complexes, mutant peptides fail to do so. These data indicate that PDZ-binding domain and tyrosine residues of efnB2/CTF2 mediate its effect on VE-cadherin complexes.
To examine whether efnB2/CTF2 associates with VE-cadherin, Raf-1 and Rok-α, we overexpressed efnB2/CTF2-myc3 or vector alone in BAMECs and we performed co-IP experiments. We found that efnB2/CTF2-myc3 forms complexes with all members of the VE-cadherin angiogenic complex (Fig. 5e) while both ΔPDZ and 5Tyr:Phe mutants form complexes to a lesser extent compared to WT peptide (Fig. 5e). EfnB2/CTF2 stimulates sprouting in a Rap1-dependent manner (Fig. 3b); however, the peptide does not affect Rap1 activity (Fig. S3). We tested whether efnB2/CTF2 forms complexes with Rap1. We did not detect any such complexes (not shown). The above indicates that efnB2/CTF2 exerts its angiogenic effects by associating with members of VE-cadherin angiogenic complex, possibly acting as a scaffolding protein.
EphB4-Fc-induced sprouting and angiogenic complex formation depends on PS1
EphB4-Fc-induced sprouting and tube formation depend on γ-secretase activity (9, see also Figure S2). To determine whether PS1 promotes EphB4-Fc-induced sprouting and tube formation, PS1 was downregulated in BAMEC cells using siRNA and in vitro angiogenesis assays were performed as described in Fig. 1. Downregulation of PS1 is maintained for 7 days, which accounts for the duration of both tube formation and sprouting assays (Fig. 6a). EphB4-Fc-induced sprouting is abolished in the presence of PS1 siRNA (Fig. 6b). Interestingly, downregulation of PS1 by siRNA reduces also basal (unstimulated) sprouting (Fc + DMSO compared to Fc + PS1 siRNA). This could be due to the inhibition of accumulation of constitutive low amounts of efnB2/CTF2 (Fig. 1a) in the absence of PS1. Furthermore, these data suggest that in addition to efnB2/EphB4 system, absence of PS1 may affect other angiogenic pathways as well. EphB4-induced sprouting depends on Rap1 (Fig. 3a). To test whether downregulation of PS1 affects Rap1 expression, we examined the effect of PS1 siRNA on Rap1 expression. We found that downregulation of PS1 did not affect Rap1 expression (Figure S4).

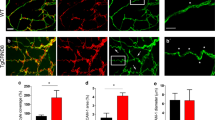
EphB4-Fc-induced sprouting and angiogenic complex formation depends on PS1. PS1 promotes angiogenic complexes in vivo. a Cells were nucleofected with various concentrations of PS1 siRNA or buffer alone. After the indicated days, cells were lysed in 1% SDS lysis buffer and immunoblotted with 33B10 mouse monoclonal antibodies against PS1/CTF and actin. Cortical neuronal extracts from PS1 WT and KO embryonic mice were used as a positive control for PS1. b Cells were nucleofected with 7.5 μM of bovine-specific PS1 siRNA or non-targeting siRNA. Cells were then used for the microcarrier bead assay in the presence of pre-clustered Fc or EphB4-Fc as described. The percentage of sprouts exceeding the diameter of the bead relative to control (Fc + non-targeting siRNA) was determined. Paired t test (n = 4, *p < 0.05, error bars = SEM). c Left: cells were nucleofected with PS1 siRNA or control non-targeting sequences as described in b and treated with 2 μg/ml Fc or EphB4-Fc as indicated. Cell extracts were IPed with anti-Raf1 antibody and IPs were probed on WB for VE-cadherin (upper panel) or Raf-1 (middle panel). Input panel shows expression of VE-cadherin. Right: quantification of results. Paired t test, n = 3, *p < 0.05, error bars = SEM. d Left: cell extracts from (c) were IPed with anti-Rok-α antbody and IPs were probed on WB for VE-cadherin (upper panel) or Rok-α (middle panel). Input panel shows expression of VE-cadherin. Right: quantification of results. Paired t test, n = 3, *p < 0.05, error bars = SEM. e Left: brain extracts from WT or PS1 KO mouse embryos were IPed with anti-VE-cadherin antibodies and IPs were probed on WB for Rok-α (first panel) or VE-cadherin (second panel). Input shows Rok-α and VE-cadherin expression. Right: quantification of results. Paired t test, n = 3, *p < 0.05, error bars = SEM
We next tested whether the increase of VE-cadherin angiogenic complexes by EphB4-Fc depends on PS1. We found that downregulation of PS1 inhibits the EphB4-Fc-induced complex of Raf-1 with VE-cadherin (Fig. 6c, lanes 3, 4) and of Rok-α with VE-cadherin (Fig. 6d, lanes 3, 4). Therefore, PS1 is necessary for the induction of these complexes by EphB4-Fc. Consistent with the above findings in vitro, we observed inhibition of Rok-α/VE-cadherin complexes in vivo in brain extracts of mice lacking PS1 expression (PS1 KO), supporting the role of PS1 in the VE-cadherin angiogenic complexes (Fig. 6e).
EphB4-Fc induces phosphorylation of myosin light chain 2 in a γ-secretase-dependent manner
Angiogenic factor-induced recruitment of Raf-1/Rok-α complex to VE-cadherin results in phosphorylation of myosin light chain 2 (MLC2) [7]. Activated MLC2 has to be tightly controlled for adherens junctions to be plastic and stable enough to allow sprouting [8, 22]. To test whether EphB4-Fc induces phosphorylation of MLC2, BAMEC cells were treated with EphB4-Fc or Fc and phosphorylated MLC2 was detected on WB using specific antibodies. We found that EphB4-Fc increases p-MLC2 (Fig. 7a, b). This increase was inhibited by two different γ-secretase inhibitors (Fig. 7a, b) showing that this function of EphB4 is mediated by γ-secretase activity. Then, we tested whether phosphorylated MLC2 is present in the VE-cadherin-containing junctions. Immunostaining intensity of p-MLC2 localized at VE-cadherin- containing cell–cell contact sites was quantified as described (see Materials and Methods and Fig. S5). We found that EphB4-Fc significantly increases levels of p-MLC2 at VE-cadherin-containing cell–cell contact sites in a γ-secretase-dependent manner (Fig. 7c). MLC2 intensity at cell–cell contact sites was not affected by EphB4-Fc or γ-secretase inhibitors showing that increase in p-MLC2 was not due to increase of total MLC2 at cell–cell contact sites (not shown). Combined our data shows that EphB4-Fc stimulates phosphorylation of p-MLC2 which in close proximity with junctional VE-cadherin is known to promote junction growth and stabilization allowing angiogenic sprouting. Cadherins associate with actomyosin; however, there is no conclusive evidence for direct binding between the two [23]. We tested whether EphB4-Fc affects the association of VE-cadherin with MLC2. Using co-IP experiments, we saw that EphB4-Fc does not affect VE-cadherin/MLC2 complexes (not shown).

EphB4-Fc increases phosphorylation of p-MLC2 at VE-cadherin-containing cell–cell contact sites in a γ-secretase-dependent manner. a BAMECs were treated with 2 μg/ml Fc or EphB4-Fc for 10 min in the presence or absence of 1 μM L685,458 or 100 nM RO 4929097. Cells were extracted in 1% SDS buffer and p-MLC2 and MLC2 were detected in cell extracts with WB using anti-p-MLC (Thr18/Ser19) and anti-MLC2 antibodies, respectively. b Densitometric quantification of the WB bands showed statistically significant increase in p-MLC2 after EphB4-Fc treatment, an increase that was inhibited by both γ-secretase inhibitors. Paired t test, n = 3, ***p < 0.001, error bars = SEM. c Cells were grown on glass coverslips and treated with Fc, EphB4-Fc and γ-secretase inhibitors as above. VE-cadherin and p-MLC2 were detected by immunostaining with specific antibodies. Intensity of p-MLC2 colocalizing with VE-cadherin at cell–cell contact sites was measured in images obtained with a Zeiss confocal laser scanning microscope (see Fig. S5). EphB4-Fc significantly increases p-MLC2 at VE-cadherin cell–cell contact sites and γ-secretase inhibitors inhibit that function of EphB4-Fc. Paired t test, n = 3, *p < 0.05, **p < 0.01, error bars = SEM. WT but not mutant efnB2/CTF2 increases phosphorylation of p-MLC2 at VE-cadherin-containing cell–cell contact sites. d BAMECs were transduced with efnB2/CTF2-myc3 or N-cad/CTF2 or vector alone. Cells were extracted in 1% SDS buffer and p-MLC2 and MLC2 were detected in cell extracts with WB using anti-p-MLC (Thr18/Ser19) and anti-MLC2 antibodies, respectively. e Densitometric quantification of the WB bands showed statistically significant increase in p-MLC2 in efnB2/CTF2-myc3 expressing cells but not in N-cad/CTF2 expressing cells. Paired t test, n = 3, **p < 0.01, error bars = SEM. f Cells transduced with WT or mutant efnB2/CTF2 were grown on glass coverslips. VE-cadherin and p-MLC2 were detected by immunostaining with specific antibodies. Intensity of p-MLC2 colocalizing with VE-cadherin at cell–cell contact sites was measured as described (Fig. S5). efnB2/CTF2-myc3 but not mutant CTF2 overexpression significantly increases p-MLC2 at VE-cadherin cell–cell contact sites. Paired t test, n = 3, *p < 0.05, error bars = SEM
WT but not mutant efnB2/CTF2 induces myosin light chain 2 phosphorylation
We next tested whether efnB2/CTF2 affects phosphorylation of MLC2. CTF2-myc3 was overexpressed in BAMECs and p-MLC2 was detected in cell extracts as described in Fig. 7a. We found that efnB2/CTF2-myc3 significantly increases p-MLC2 and this is specific since the product of γ-secretase cleavage of N-cadherin (N-cad/CTF2) did not affect this phosphorylation (Fig. 7d, e). Furthermore, using immunocytochemistry as described in Fig. 7c, we found that overexpression of WT but not mutant efnB2/CTF2 significantly increases p-MLC2 at VE-cadherin cell–cell contact sites (Fig. 7f).
Discussion
We show that the EphB4 receptor increases specific angiogenic complexes between VE-cadherin and kinases Raf-1 and Rok-α. This is a novel function of EphB4 which we found to be inhibited by both acute downregulation of PS1 and γ-secretase inhibitors, indicating that PS1/γ-secretase system regulates the EphB4-induced angiogenesis. Consistent with the above in vitro findings, we observed that absence of PS1 in KO mice inhibits the VE-cadherin angiogenic complexes in the brain. The VE-cadherin/Rok-α complexes in brain extracts (Fig. 6E) may exist in cell types other than endothelial; however, since VE-cadherin is mainly expressed in endothelial cells, it is more likely that the complexes that we detect are from brain endothelial cells. BAMEC microvessel endothelial cells are prepared from capillaries, supporting our hypothesis that the PS1/efnB2/EphB4 system affects angiogenesis by targeting capillaries.
We also found that stimulation of sprouting and angiogenic complex formation by EphB4-Fc requires Rap1 activity. Since binding of EphB1 receptor to efnB1 ligand promotes Rap1 activation and platelet adhesion [24], we tested whether EphB4-Fc affects Rap1 activity. Using an in vitro activity assay, we found that EphB4-Fc has no effect on Rap1 activity. In addition, we found that Rap1 inhibitor does not affect the EphB4-Fc-induced processing of efnB2 by γ-secretase and the accumulation of proteolytic fragment efnB2/CTF2. The above show that both Rap1 and γ-secretase activities are independently necessary for the in vitro angiogenic function of the EphB4/efnB2 system.
Our finding that EphB4-Fc increases phosphorylation of MLC2 suggests that EphB4-Fc-induced assembly of VE-cadherin angiogenic complexes results in phosphorylation of MLC2, a step known to result in activation of the actinomysin contractile apparatus. We also found that the EphB4-Fc-induced phosphorylation of MLC2 is regulated by PS1/γ-secretase. This phosphorylation has been shown to allow VE-cadherin to be connected to the apparatus and to be positioned at the cell surface to cluster together and form tight adherens junctions with neighboring cells [7].
Since EphB4-Fc-induced sprouting, tube formation, angiogenic complex formation and MLC2 phosphorylation depend on PS1/γ-secretase, we examined whether the product of PS1/γ-secretase processing of efnB2, cytoplasmic peptide efnB2/CTF2, affects the above angiogenic functions of endothelial cells. This peptide is produced after treatment of cells with EphB4-Fc which increases both degradation of full-length ephrinB2 and accumulation of proteolytic fragment efnB2/CTF2 (Fig. 1a).
We found that efnB2/CTF2 significantly promotes endothelial cell sprouting and tube formation. Interestingly γ-secretase inhibitors did not inhibit the efnB2/CTF2-induced sprouting or tube formation indicating that cleavage of other substrates by γ-secretase does not affect angiogenic activity of efnB2/CTF2. We also show that efnB2/CTF2 increases formation of VE-cadherin/Raf-1/Rok-α complexes. This indicates that this peptide exerts its angiogenic function by promoting the assembly of these angiogenic complexes to VE-cadherin adherens junctions. EfnB2/CTF2-induced sprouting requires active Rap1 and it also increases phosphorylation of junctional MLC2, further supporting our theory that this peptide promotes assembly and maturation of adherens junctions to stimulate angiogenesis.
To identify the residues that mediate efnB2/CTF2 angiogenic function, we examined whether the PDZ-binding domain and tyrosine residues of this peptide are necessary for these functions. This was based on previous observations that efnB-induced sprouting is mediated by phosphorylation of efnB by Src kinase on tyrosine residues [16] and that the PDZ-binding domain of efnB2 is critical for its angiogenic activity [14, 15]. BAMEC cells expressing efnB2/CTF2 mutants either lacking the PDZ-binding domain or having all tyrosine residues mutated to phenylalanine were unable to sprout or form tubes.
Mutation of the five conserved tyrosine residues on efnB2/CTF2 inhibits the ability of this peptide to promote sprouting and tube formation, indicating that one, if not all, of these tyrosine residues are required for these functions. However, two of the five tyrosine residues are located within the PDZ-binding motif of efnB2 (YYKV); therefore, it is possible that this mutant is unable to promote sprouting and tube formation due to alteration of its PDZ-binding domain. Site-directed mutagenesis of individual tyrosine residues will help identify the specific residues responsible for the peptide’s angiogenic function.
Mutant efnB2/CTF2 failed to increase VE-cadherin/Raf-1/Rok-α angiogenic complexes indicating that the inability of these mutants to promote sprouting is due to their inability to stimulate the VE-cadherin angiogenic complexes. To test whether efnB2/CTF2 acts as a scaffold that brings together components of the VE-cadherin complex, we tested its association with all three members of this complex. We found that the peptide co-IPs with all three proteins (VE-cadherin, Raf-1 and Rok-α) while ΔPDZ and 5Tyr:Phe mutants associate to a much lesser extent. This indicates that efnB2/CTF2 interacts with VE-cadherin, Raf-1 and Rok-α promoting their association and this function is mediated by its PDZ-binding domain. It will be interesting to identify additional protein components of this interaction to understand the role of efnB2/CTF2 in the assembly of this angiogenic complex. Identifying the minimal sequence of efnB2/CTF2 that mediates its angiogenic function can be used to design peptides that modulate these complexes and eventually the angiogenic function of endothelial cells.
EfnB2/CTF2 promotes Src phosphorylation and activation [25]. This peptide was recently found to increase Src and FAK phosphorylation and activation in microglia promoting cell migration [26]. EphB4-induced sprouting depends on Src activity [16] so it is possible that efnB2/CTF2 regulates angiogenesis by targeting various cellular pathways including Src activation and assembly of VE-cadherin complexes. It will be interesting to explore the interplay between pathways affected by efnB2/CTF2 to fine tune these cellular functions.
Other ligand-receptor systems such as VEGF/VEGFR and DLL4/Notch1 that regulate angiogenesis are also processed by PS1/γ-secretase [27]. Interestingly, VEGFR function in angiogenesis has been shown to be regulated by efnB2 [28, 29]; therefore, the effect of PS1/γ-secretase on EphB4/efnB2 system may be affecting VEGFR angiogenic functions. Notch1 is also processed by γ-secretase-producing cytoplasmic fragment Notch1 ICD (intracellular domain); however, Notch1 ICD, when overexpressed in ECs, decreases sprouting on beads [30] and it has been established that activation of Notch signaling inhibits angiogenesis [31,32,33].
There has been a large amount of evidence suggesting that EphB4 plays a critical role in angiogenesis [6, 16, 34]; however, the mechanisms of this function of EphB4 have yet to be understood. Here, we identify a mechanism via which EphB4 exerts its proangiogenic function and, in addition, we show that this function of EphB4 is regulated by PS1/γ-secretase, which cleaves efnB2 ligand to produce cytoplasmic peptide efnB2/CTF2. This peptide potently stimulates sprouting and tube formation of endothelial cells, angiogenic complex formation and phosphorylation of MLC2. A schematic representation of this model is shown in Fig. 8. Mutations in PS1 have been implicated in familial Alzheimer’s disease (FAD); it will, therefore, be interesting to examine the effects of PS1 FAD mutants in endothelial cell function as it may shed light in the mechanisms leading to vascular impairment observed in AD brains.

PS1/γ-secretase promotes EphB4-induced sprouting via processing of efnB2. EphB4 binding to efnB2 induces cleavage of the latter by PS1/γ-secretase, producing cytoplasmic peptide efnB2/CTF2. This peptide acts as a scaffolding protein, promoting the assembly of angiogenic complexes including VE-cadherin, Raf-1 and Rok-α in a Rap1-dependent manner and increasing phosphorylation of MLC2 at VE-cadherin-containing cell–cell contact sites. Peptide efnB2/CTF2 potently increases endothelial cell sprouting and tube formation and all above functions are mediated by its PDZ-binding domain. All functions are inhibited by downregulation of PS1 or presence of γ-secretase inhibitors
Materials and methods
Chemicals and antibodies
Cytodex® microcarrier (MC) beads, fibrinogen (type I-S from bovine plasma), aprotinin, thrombin (from bovine plasma), Hoechst 33258 and hexadimethrine bromide solution were from Sigma-Aldrich Inc. Recombinant mouse EphB4-Fc chimera and recombinant human IgG1 Fc were from R&D Systems. Rabbit anti-human Fc and peroxidase-conjugated AffiniPure goat anti-mouse IgG light chain-specific were from Jackson Immunoresearch Laboratories Inc. L685,458 and lactacystin were from VWR Scientific. Compound E (γ-secretase inhibitor XXI) and rabbit anti-Rap1 polyclonal antibody were from EMD Millipore Corp. DAPT and RO4929097 were from Selleck Chemicals LLC. GGTi-298 was from Fisher Scientific. Rabbit polyclonal antibodies against c-myc (A-14) and efnB (C-18) were from Santa Cruz. Rabbit monoclonal antibodies against VE-cadherin (D87F2) and Rok-α (D1B1), rabbit polyclonal antibodies against Raf-1 (c-Raf antibody), myosin light chain 2 and phospho-myosin light chain 2 (Thr18/Ser19) and mouse monoclonal antibody against myc-tag (9B11) were from Cell Signaling Technology. Mouse monoclonal antibody 33B10 against PS1/CTF has been previously described [9]. Alexa Fluor 488 goat anti-rabbit IgG and Alexa Fluor 568 goat anti-mouse IgG were from Thermo Fisher Scientific.
Cell culture, transfection, retroviral transduction and HSV infection
The pMX retroviral-based expression system pMXs-IG [35] was used for stable expression of Mus musculus efnB2 and WT or mutant efnB2/CTF2 all of which were conjugated with a triple myc tag (myc3) at the C-terminus. The same vector was used for stable expression of untagged efnB2/CTF2 and N-cad/CTF2. Bovine adrenal microvessel endothelial cells (BAMEC; VEC Technologies) were cultured according to company instructions. Transduction and sorting of cells was performed as previously described [25]. For HSV infection, Herpes Simplex Virus (HSV) particles expressing efnB2/CTF2-myc3 or N-cad/CTF2 were obtained from MIT Viral Gene Transfer Core. BAMEC cells were infected with particles expressing the above constructs or control vector at 1 multiplicity of infection (MOI 1) for 48 h before experiments were performed.
Western Blot (WB) and immunoprecipitation (IP)
Western blot (WB), immunoprecipitation (IP), densitometric scanning and quantification of the X-ray films were performed as described [9, 25]. WT and PS1 KO mouse embryos were collected at E14.5. Brains were homogenized and extracted as described [9] and extracts were IPed as above.
Microcarrier bead-based sprouting assay
The assay has been previously described [9]. Briefly, BAMEC cells were cultured with pre-swollen microcarrier beads at 37°C/5% CO2 for 2–4 days to allow cells to sprout from the beads. Dishes were fixed with 4% PFA in PBS pH 7.4 and cell nuclei were stained with 20 μg/ml Hoechst 33258 in DPBS for 1 h at room temperature. 50 beads were chosen at random and the total number of sprouts that exceeded the diameter of the bead (~ 175 μm) was manually counted. Representative phase-contrast and Hoechst-stained florescent images were taken on an inverted Olympus IX-50 microscope.
Tube formation assay
BD Matrigel™ LDEV-free matrix was prepared on a 24-well plate according to manufacturer’s instructions (BD Biosciences). Briefly, BAMECs suspended in MCDB-131 complete medium were added to the Matrigel at a density of 12 × 104 cells per 300 μl medium. Each experimental condition was assigned three wells for plating at random. The plate was placed in a LiveCell incubation chamber (37°C, 5% CO2, and 95% humidity) and InVivo software was used to mark five random positions within each well. Phase-contrast pictures were taken from each marked position every 10 min for up to 9 h using the Olympus IX-70 inverted microscope with the Evolution QEi CCD monochrome digital camera attachment. A time point was arbitrarily chosen for the purposes of counting tubes. The number of tubes from each marked positions for each well was manually determined using the ImageJ Cell Counter plugin.
Immunofluorescence and quantification of colocalization
For detection of VE-cadherin and p-MLC2, BAMEC cells grown on glass coverslips were fixed with − 20 °C cold methanol for 10 min, blocked with SuperBlock® Blocking Buffer in TBS (Thermo Fisher Scientific) for 1 h and stained with antibodies against VE-cadherin, p-MLC2 or MLC2 overnight. Fluorescently labeled secondary antibodies (Alexa Fluor 488 and Alexa Fluor 568) were added for 1 h, cells were washed with TBS, mounted with Vectashield (Vector Laboratories Inc), and photographed on a Leica SP5 DMI confocal laser scanning microscope. Cell Profiler was used to quantify colocalization between VE-cadherin and p-MLC2 at cell–cell contact sites (see Fig. S5).
Downregulation of PS1 with siRNA
To downregulate PS1 in BAMEC cells, three specific siRNA sequences targeting bovine PS1 were designed (GE Healthcare Dharmacon Inc). A non-targeting siRNA sequence was used as control (D-001210-02-05). Sequences were nucleofected into BAMEC cells following the manufacturer’s protocol (Lonza Walkersville). To test the efficiency of siRNA tansfection siGLO Green transfection indicator was used (Thermo Fisher Scientific). Different amounts of siRNA were nucleofected for different time points using the DharmaFECT 4 Transfection Reagent. The siRNA that showed significant downregulation of PS1 expression was used for experiments.
Abbreviations
- EfnB2:
-
EphrinB2
- CTF2:
-
Carboxy terminal fragment 2
- CTF1:
-
Carboxy terminal fragment 1
- PS1:
-
Presenilin1
- WT:
-
Wild type
- KO:
-
Knockout
- VE-cadherin:
-
Vascular Endothelial cadherin
- N-cad:
-
Neuronal cadherin
- Rap1:
-
Ras-proximate-1 or Ras-related protein 1
- Rok-α:
-
Rho-associated, coiled-coil-containing protein kinase 2
- BAMEC:
-
Bovine adrenal microvessel endothelial cells
- MLC2:
-
Myosin light chain 2
- FAD:
-
Familial Alzheimer’s disease
- AD:
-
Alzheimer’s disease
References
Barthet G, Georgakopoulos A, Robakis NK (2012) Cellular mechanisms of gamma-secretase substrate selection, processing and toxicity. Prog Neurobiol 98(2):166–175. https://doi.org/10.1016/j.pneurobio.2012.05.006
Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S (1997) Skeletal and CNS defects in Presenilin-1-deficient mice. Cell 89(4):629–639
Nakajima M, Yuasa S, Ueno M, Takakura N, Koseki H, Shirasawa T (2003) Abnormal blood vessel development in mice lacking presenilin-1. Mech Dev 120(6):657–667
Gama Sosa MA, Gasperi RD, Rocher AB, Wang AC, Janssen WG, Flores T, Perez GM, Schmeidler J, Dickstein DL, Hof PR, Elder GA (2010) Age-related vascular pathology in transgenic mice expressing presenilin 1-associated familial Alzheimer’s disease mutations. Am J Pathol 176(1):353–368. https://doi.org/10.2353/ajpath.2010.090482
Gerety SS, Wang HU, Chen ZF, Anderson DJ (1999) Symmetrical mutant phenotypes of the receptor EphB4 and its specific transmembrane ligand ephrin-B2 in cardiovascular development. Mol Cell 4(3):403–414
Adams RH, Diella F, Hennig S, Helmbacher F, Deutsch U, Klein R (2001) The cytoplasmic domain of the ligand ephrinB2 is required for vascular morphogenesis but not cranial neural crest migration. Cell 104(1):57–69
Wimmer R, Cseh B, Maier B, Scherrer K, Baccarini M (2012) Angiogenic sprouting requires the fine tuning of endothelial cell cohesion by the Raf-1/Rok-alpha complex. Dev Cell 22(1):158–171. https://doi.org/10.1016/j.devcel.2011.11.012
Abraham S, Yeo M, Montero-Balaguer M, Paterson H, Dejana E, Marshall CJ, Mavria G (2009) VE-Cadherin-mediated cell-cell interaction suppresses sprouting via signaling to MLC2 phosphorylation. Curr Biol 19(8):668–674. https://doi.org/10.1016/j.cub.2009.02.057
Georgakopoulos A, Litterst C, Ghersi E, Baki L, Xu C, Serban G, Robakis NK (2006) Metalloproteinase/presenilin1 processing of ephrinB regulates EphB-induced Src phosphorylation and signaling. EMBO J 25(6):1242–1252. https://doi.org/10.1038/sj.emboj.7601031
Nehls V, Drenckhahn D (1995) A novel, microcarrier-based in vitro assay for rapid and reliable quantification of three-dimensional cell migration and angiogenesis. Microvasc Res 50(3):311–322. https://doi.org/10.1006/mvre.1995.1061
Eilken HM, Adams RH (2010) Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr Opin Cell Biol 22(5):617–625. https://doi.org/10.1016/j.ceb.2010.08.010
Madri JA, Pratt BM, Tucker AM (1988) Phenotypic modulation of endothelial cells by transforming growth factor-beta depends upon the composition and organization of the extracellular matrix. J Cell Biol 106(4):1375–1384
Grant DS, Kibbey MC, Kinsella JL, Cid MC, Kleinman HK (1994) The role of basement membrane in angiogenesis and tumor growth. Pathol Res Pract 190(9–10):854–863
Makinen T, Adams RH, Bailey J, Lu Q, Ziemiecki A, Alitalo K, Klein R, Wilkinson GA (2005) PDZ interaction site in ephrinB2 is required for the remodeling of lymphatic vasculature. Genes Dev 19(3):397–410. https://doi.org/10.1101/gad.330105
Kida Y, Ieronimakis N, Schrimpf C, Reyes M, Duffield JS (2013) EphrinB2 reverse signaling protects against capillary rarefaction and fibrosis after kidney injury. J Am Soc Nephrol 24(4):559–572. https://doi.org/10.1681/ASN.2012080871
Palmer A, Zimmer M, Erdmann KS, Eulenburg V, Porthin A, Heumann R, Deutsch U, Klein R (2002) EphrinB phosphorylation and reverse signaling: regulation by Src kinases and PTP-BL phosphatase. Mol Cell 9(4):725–737
Grigorian AL, Bustamante JJ, Hernandez P, Martinez AO, Haro LS (2005) Extraordinarily stable disulfide-linked homodimer of human growth hormone. Protein Sci 14(4):902–913. https://doi.org/10.1110/ps.041048805
Isbert S, Wagner K, Eggert S, Schweitzer A, Multhaup G, Weggen S, Kins S, Pietrzik CU (2012) APP dimer formation is initiated in the endoplasmic reticulum and differs between APP isoforms. Cell Mol Life Sci 69(8):1353–1375. https://doi.org/10.1007/s00018-011-0882-4
Chrzanowska-Wodnicka M (2010) Regulation of angiogenesis by a small GTPase Rap1. Vascul Pharmacol 53(1–2):1–10. https://doi.org/10.1016/j.vph.2010.03.003
Qian Y, Vogt A, Vasudevan A, Sebti SM, Hamilton AD (1998) Selective inhibition of type-I geranylgeranyltransferase in vitro and in whole cells by CAAL peptidomimetics. Bioorg Med Chem 6(3):293–299
Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, Robakis NK (2003) A CBP binding transcriptional repressor produced by the PS1/epsilon-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell 114(5):635–645
Liu Z, Tan JL, Cohen DM, Yang MT, Sniadecki NJ, Ruiz SA, Nelson CM, Chen CS (2010) Mechanical tugging force regulates the size of cell-cell junctions. Proc Natl Acad Sci USA 107(22):9944–9949. https://doi.org/10.1073/pnas.0914547107
Weis WI, Nelson WJ (2006) Re-solving the cadherin-catenin-actin conundrum. J Biol Chem 281(47):35593–35597. https://doi.org/10.1074/jbc.R600027200
Prevost N, Woulfe DS, Tognolini M, Tanaka T, Jian W, Fortna RR, Jiang H, Brass LF (2004) Signaling by ephrinB1 and Eph kinases in platelets promotes Rap1 activation, platelet adhesion, and aggregation via effector pathways that do not require phosphorylation of ephrinB1. Blood 103(4):1348–1355. https://doi.org/10.1182/blood-2003-06-1781
Georgakopoulos A, Xu J, Xu C, Mauger G, Barthet G, Robakis NK (2011) Presenilin1/gamma-secretase promotes the EphB2-induced phosphorylation of ephrinB2 by regulating phosphoprotein associated with glycosphingolipid-enriched microdomains/Csk binding protein. FASEB J 25(10):3594–3604. https://doi.org/10.1096/fj.11-187856
Kemmerling N, Wunderlich P, Theil S, Linnartz-Gerlach B, Hersch N, Hoffmann B, Heneka MT, de Strooper B, Neumann H, Walter J (2017) Intramembranous processing by gamma-secretase regulates reverse signaling of ephrin-B2 in migration of microglia. Glia 65(7):1103–1118. https://doi.org/10.1002/glia.23147
Boulton ME, Cai J, Grant MB (2008) gamma-Secretase: a multifaceted regulator of angiogenesis. J Cell Mol Med 12(3):781–795. https://doi.org/10.1111/j.1582-4934.2008.00274.x
Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Luthi U, Barberis A, Benjamin LE, Makinen T, Nobes CD, Adams RH (2010) Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465(7297):483–486. https://doi.org/10.1038/nature09002
Sawamiphak S, Seidel S, Essmann CL, Wilkinson GA, Pitulescu ME, Acker T, Acker-Palmer A (2010) Ephrin-B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature 465(7297):487–491. https://doi.org/10.1038/nature08995
Leong KG, Hu X, Li L, Noseda M, Larrivee B, Hull C, Hood L, Wong F, Karsan A (2002) Activated Notch4 inhibits angiogenesis: role of beta 1-integrin activation. Mol Cell Biol 22(8):2830–2841
Zheng W, Tammela T, Yamamoto M, Anisimov A, Holopainen T, Kaijalainen S, Karpanen T, Lehti K, Yla-Herttuala S, Alitalo K (2011) Notch restricts lymphatic vessel sprouting induced by vascular endothelial growth factor. Blood 118(4):1154–1162. https://doi.org/10.1182/blood-2010-11-317800
Sainson RC, Aoto J, Nakatsu MN, Holderfield M, Conn E, Koller E, Hughes CC (2005) Cell-autonomous notch signaling regulates endothelial cell branching and proliferation during vascular tubulogenesis. FASEB J 19(8):1027–1029. https://doi.org/10.1096/fj.04-3172fje
Larrivee B, Prahst C, Gordon E, del Toro R, Mathivet T, Duarte A, Simons M, Eichmann A (2012) ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev Cell 22(3):489–500. https://doi.org/10.1016/j.devcel.2012.02.005
Adams RH, Wilkinson GA, Weiss C, Diella F, Gale NW, Deutsch U, Risau W, Klein R (1999) Roles of ephrinB ligands and EphB receptors in cardiovascular development: demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes Dev 13(3):295–306
Kitamura T, Koshino Y, Shibata F, Oki T, Nakajima H, Nosaka T, Kumagai H (2003) Retrovirus-mediated gene transfer and expression cloning: powerful tools in functional genomics. Exp Hematol 31(11):1007–1014
Acknowledgements
This work was supported by NIH Grants 2R01-NS047229-11, P50AG05138, AG-17926, AG-008200 and by Alzheimer’s Association Grant IIRG-11-205149.
Author information
Authors and Affiliations
Contributions
AG conceived the project, designed the research, supervised the project and wrote the manuscript with input from all authors. NW, GV, YY performed experiments and contributed to writing of the manuscript. NR supervised the work and contributed to research design and writing of the manuscript.
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Warren, N., Voloudakis, G., Yoon, Y. et al. The product of the γ-secretase processing of ephrinB2 regulates VE-cadherin complexes and angiogenesis. Cell. Mol. Life Sci. 75, 2813–2826 (2018). https://doi.org/10.1007/s00018-018-2762-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-018-2762-7