
Abstract
Clinical trial results presented in 2019 suggest that antibody-based removal of cerebral amyloid β (Aβ) plaques may possibly clear tau tangles and modestly slow cognitive decline in symptomatic Alzheimer’s disease (AD). Although regulatory approval of this approach is still pending, preparing the healthcare system for the advent of disease-modifying therapies against AD is imperative. In particular, it will be necessary to identify the most suitable biomarkers to facilitate appropriate treatment of AD. Here, we give an update on recent developments in fluid and imaging biomarkers for AD-related pathologies and discuss potential approaches that could be adopted to screen for and clarify the underlying pathology in people seeking medical advice because of cognitive symptoms. We succinctly review recent data regarding biomarkers for Aβ and tau pathology, neurodegeneration, synaptic dysfunction, and inflammation, highlight the need for further research into common copathologies, and suggest how different biomarkers could be used (most likely in combination) to facilitate the development and clinical implementation of novel drug candidates against AD.
Similar content being viewed by others

Introduction
Alzheimer’s disease (AD) is a slowly progressive neurodegenerative disease that currently lacks effective treatment. The first discernible pathology is the accumulation of 42 amino acid-long amyloid β (Aβ) protein in extracellular plaques in the brain, occurring decades before clinical onset [1]. Biomarker studies suggest that Aβ accumulation is followed by synaptic dysfunction and increased phosphorylation and secretion of tau, a microtubule-binding axonal protein that is highly expressed in cortical neurons [2]. This dysfunctional tau metabolism places neurons at increased risk of degeneration with the development of intraneuronal neurofibrillary tangles that are composed of hyperphosphorylated and truncated tau proteins. Neurodegeneration eventually translates into the AD clinical syndrome, with cognitive symptoms that worsen as the disease progresses [3]. This is the most common account of the pathophysiological cascade in AD (the so-called amyloid cascade hypothesis [4]), which has been largely validated in familial forms of the disease that are caused by mutations in genes encoding Aβ turnover-related proteins. Sporadic AD (the age-related and more common form of the disease), appears to be characterized by a more complex interplay between Aβ aggregation, vascular changes, microglial and astrocytic activation, other copathologies (e.g., neuronal α-synuclein and TDP-43 inclusions), genetic susceptibility (with the ε4 allele of the apolipoprotein E [APOE] gene being the strongest risk factor) and factors that impact brain health in development and aging potentially influencing resilience to AD pathology [5,6,7].
Among these pathological processes, Aβ remains the most prominent target in clinical trials of disease-modifying drug candidates against AD [8], both at the symptomatic and the preclinical stage. Exactly what starts the process of concentration-dependent initiation of prion-like Aβ misfolding due to increased production and/or defective Aβ clearance, is presently unclear, but a wealth of experimental and observational data suggest that Aβ accumulation is not an innocent by-stander but toxic to synapses and neurons [9]. Primary approaches to alter the amyloid-initiated cascade include immunotherapy, β-secretase 1 (BACE1) inhibitors, and vaccines. Potential mediators of Aβ toxicity, including the APOE protein and microglial and glial activation [10,11,12], have also been targeted in preclinical and early-phase attempts. Strong evidence points toward a critical role for neurofibrillary tangles in the development of cognitive impairment, spurring on the development and testing of antitau therapies, with several clinical trials ongoing [8, 13].
Although a new drug for AD has not been approved for clinical use in over 15 years, a confluence of recent factors suggest that this may soon change. These include increases in funding for AD research, improved understanding of the biology of AD, and promising results from a recent amyloid immunotherapy trial coupled with the likelihood of regulatory approval. The number of individuals waiting for a disease-modifying therapy is large and growing. Within the US alone, there are 5.8 million individuals living with AD dementia, 9.1 million people in EU member states (https://ec.europa.eu/health/sites/health/files/state/docs/2018_healthatglance_rep_en.pdf), and an estimated 50 million people with dementia worldwide (https://www.who.int/news-room/fact-sheets/detail/dementia), the majority of these cases due to AD. On top of these, a currently unknown number of individuals harbor presymptomatic pathology, presenting an additional population of individuals who may benefit from treatment. When one or more new disease-modifying therapies becomes available clinically, healthcare systems will need to be prepared to provide these new treatments.
A critical component of preparing for the approval of new treatments, is the selection, validation, and deployment of tools for disease screening and treatment monitoring. Here, we provide an overview of the biomarkers that reflect the core components of AD, including biomarkers for Aβ and tau pathology, as well as biomarkers for other components of the pathological cascade, including neurodegeneration, synaptic dysfunction and inflammation (Fig. 1). We also discuss the pathophysiological processes these biomarkers reflect, highlight the need for further research into common copathologies, and suggest how biomarkers could be used (particularly in combination) in clinical practice, considering potential challenges in clinical practice, and how to facilitate the development, clinical implementation and monitoring of novel drug candidates against AD.

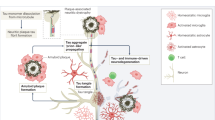
The figure illustrates neural cells and Alzheimer’s disease (AD) pathology, with AD-related biomarkers indicated in text boxes. Cerebrospinal fluid (CSF) and plasma Aβ42/Aβ40 ratio, as well as amyloid positron emission tomography (PET) are direct markers of Aβ pathology. In response to Aβ pathology, neurons phosphorylate and secrete tau at increased rate, resulting in increased total and phosphorylated tau (T-tau and P-tau, respectively) concentrations in CSF and in increased P-tau concentration in plasma. CSF and plasma tau may thus be considered neuronal response markers to Aβ. The most direct biomarker for tangle pathology is tau PET. CSF and plasma P-tau concentrations also increase in tau PET-positive individuals but the increase happens well before tau pathology is detectable on PET, at least using current tracers. Axons are rich in tau and neurofilament light (NfL) that leak into the CSF and blood during neuroaxonal degeneration. The best-established imaging biomarker for neurodegeneration is volumetric magnetic resonance imaging (MRI) of the brain. Leading synaptic biomarkers are neurogranin (Ng) in CSF, as well as SV2A- and fluorodeoxyglucose (FDG) PET. The best-established astrocytic biomarker is CSF YKL-40 and there are also promising data on CSF and plasma glial fibrillary acidic protein (GFAP) as an astrocytic activation/degeneration marker. The best-established biomarkers for microglia are CSF soluble triggering receptor expressed on myeloid cells 2 (sTREM2) and translocator protein (TSPO) PET.
Biomarkers for Aβ pathology
Extracellular deposition of Aβ, generated through the cleavage of amyloid precursor protein (APP) by BACE1 and γ-secretase, into plaques is the key pathological feature of AD, and has been proposed as the main pathogenic event in the disease [14]. While the amyloid cascade hypothesis was proposed three decades ago [4, 15], development of tools to measure Aβ pathology in vivo and prior to autopsy, via diagnostic biomarkers in cerebrospinal fluid (CSF) and through amyloid positron emission tomography (PET) imaging [16], has improved the knowledge on the molecular processes underlying AD and facilitated the development of targeted treatments.
AD CSF is characterized by reduced—by ~50% of normal levels—concentration of the 42 amino acid-long and aggregation-prone form of Aβ (Aβ42) [17]. Aβ42 is a secreted degradation product of APP that normally is mobilized from the brain interstitial fluid into the CSF and blood, likely via the glymphatic system [18]. In AD, it aggregates in the brain parenchyma, resulting in reduced CSF levels. The diagnostic accuracy for Aβ pathology can be increased by dividing the concentration of aggregation-prone Aβ42 by soluble Aβ40 (both products of the same APP-processing pathway but Aβ40, in contrast to Aβ42, remains soluble in AD). The CSF Aβ42/Aβ40 ratio, which accounts for interindividual differences in amyloidogenic APP-processing (high vs. low Aβ producers), is close to 100% concordant with amyloid PET [19], and discordant cases which are typically CSF-positive and PET-negative, often turn PET-positive within a few years [19,20,21].
With regard to imaging, three amyloid PET tracers have been approved by both the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for Aβ plaque imaging in cognitively impaired patients being clinically evaluated for AD, namely [18F]florbetapir (Amyvid), [18F]flutemetamol (Vizamyl), and [18F]florbetaben (Neuraceq) [16]. [11C]PIB is widely used in the research context, but given the short half-life of carbon-11 (~20 min), it requires an onsite cyclotron for production. An example amyloid PET image is shown in Fig. 2a. Since amyloid PET has been in use for the past 15 years, it has passed a number of hurdles. Amyloid PET has been validated against neuropathology [16], has undergone extensive standardization regarding how to quantify Aβ pathology and how to define cut-points for abnormality [22], and has appropriate use criteria [23]. Amyloid PET is the most widely used biomarker in current clinical trials, and is likely to be a first choice for clinical use, particularly in the US and Europe, when an antiamyloid therapy is approved. An important consideration in the use of amyloid PET is the availability of PET scanners and cyclotron-equipped production facilities, which vary by country. The availability of PET scanner time and the production of radiotracers for screening and treatment monitoring of amyloid therapies will impact initial patient wait times for treatment until the availability of facilities can be scaled up. The RAND Corporation has modeled this for several countries, including the US [24], Canada [25], some EU member states [26], and Japan [27], and has suggested that use of CSF biomarkers in lieu of or in addition to amyloid PET would reduce wait times for amyloid therapy.

a Warm/red color in the standardized uptake value ratio (SUVR) image denotes regions of amyloid deposition, as imaged with [11C] Pittsburgh Compound-B (PiB) positron emission tomography (PET) in an individual with Alzheimer’s disease (AD) dementia. b Warm/red color in the SUVR image denotes areas of [18F]AV1451-binding, indicating neurofibrillary tangle pathology in an individual with AD dementia. c Cooler colors (green/blue) are indicative of hypometabolism as shown on [18F] fluorodeoxyglucose (FDG) PET in an individual with mild cognitive impairment due to AD. d The compound [C11]UCB-J binds to synaptic vesicle glycoprotein 2A (SV2A). Red color in the SUVR image denotes regions of greater synaptic density in a healthy cognitively unimpaired (CU) late middle-aged adult.
The regulatory status of CSF biomarkers for clinical evaluation of AD is less clear, but both the FDA and the EMA have encouraged the further study of CSF biomarkers in the context of AD, and the Coalition Against Major Diseases CSF Biomarker Team is working toward seeking formal qualification from the FDA on the use of CSF biomarkers for clinical trial enrichment at the pre-dementia stage of the disease [28]. Reference methods and materials for CSF Aβ42 assay standardization, as well as high-precision clinical chemistry tests on fully automated instruments, are in place [29], which bodes well for full implementation of these biomarkers in clinical laboratory practice with uniform reference limits around the globe; in many European countries CSF biomarkers are already used in clinical laboratory practice in accordance with country-specific regulations. Work on the reference measurement procedures for CSF Aβ40 is ongoing under the auspices of the International Federation of Clinical Chemistry and Laboratory Medicine (IFCC) CSF Proteins working group and should be concluded during 2020.
Given that CSF sampling may be considered invasive, and amyloid PET expensive with limited availability (and non-negligible radiation exposure), a blood biomarker for cerebral Aβ pathology would be an important step toward precision medicine in the clinical work-up of patients with cognitive complaints. In contrast to earlier reports [17], recent findings suggest that plasma Aβ42 in ratio with Aβ40 (measured by immunoprecipitation mass spectrometry or ultrasensitive enzyme-linked immunosorbent assays) reflects cerebral Aβ pathology with relatively high accuracy against both amyloid PET and CSF Aβ42/Aβ40 ratio [30,31,32,33], although evaluation against neuropathology is still needed. A recent validation study utilizing a fully automated immunoassay (Elecsys) to measure plasma Aβ42 and Aβ40 further underscores the promising capabilities of plasma Aβ in clinical laboratory practice [34].
The Aβ42/Aβ40 ratio is reduced by only 14–20% in plasma [30,31,32,33], compared with 50% in CSF [17], with a greater overlap between Aβ-positive and –negative individuals, and the correlation with CSF is weak, which could be explained by production of Aβ peptides in platelets and other noncerebral tissues, but the concordant research findings using high-precision analytical tools still represent an important research advancement toward clinical implementation (following much needed standardization work), perhaps using staged testing (e.g., a blood Aβ test favoring sensitivity over specificity, followed by a more specific CSF- or imaging-based test in clinical practice).
Biomarkers for tau pathology
The aggregation of hyperphosphorylated forms of the axonal protein tau in the neuronal soma, forming neurofibrillary tangles, is a key pathological feature of AD, although tau inclusions in neurons or glial cells are also found in other neurodegenerative dementias [35]. The cornerstone markers total tau (T-tau) and phosphorylated tau (P-tau) have been proposed, together with CSF Aβ42/Aβ40 ratio, as biomarkers for biologically defining AD [36], and are considered diagnostic in the research criteria for AD [37]. Both CSF T-tau and P-tau concentrations reflect AD-related pathophysiology across the neurodegenerative dementias [38, 39]. The most likely explanation for this is that the increased CSF levels of tau are due to increased phosphorylation and secretion of tau from neurons, as a neuronal response to Aβ exposure [40, 41]. CSF T-tau and P-tau may thus be regarded predictive markers of AD-type neurodegeneration and tangle formation but not direct markers of these processes (and not markers of non-AD tauopathies, for which better biomarkers still need to be developed). Fully automated assays for clinical use are available [42, 43], and standardization work is ongoing in collaborative efforts between IFCC and the Global Biomarker Standardization Consortium.
Whilst ultrasensitive plasma T-tau assays can detect neuronal injury in acute brain disorders, such as stroke and traumatic brain injury [44, 45], they work relatively poorly in AD settings [46], and the correlation with CSF is weak [47]. A potential explanation for this is that the assay set up is vulnerable to proteolytic degradation of tau in the blood (the half-life of tau measured using currently available T-tau assays is 10 h [48], compared with around 20 days in CSF [41]).
Fortunately, we have recently seen a number of real breakthroughs in the plasma tau biomarker field. Mielke et al. originally demonstrated a correlation between P-tau181, and amyloid and tau PET, which indicates that plasma P-tau181 is a good predictor of brain AD pathology [49]. These findings were replicated in a recent study by Palmqvist et al., demonstrating that plasma P-tau181 associates with amyloid PET positivity and correlates strongly with CSF P-tau181 [2]. Interestingly, the change in plasma P-tau181 became significant before amyloid PET, but after CSF and plasma Aβ42, i.e., already at sub-PET threshold Aβ pathology [2]. Thus, plasma P-tau181 might be useful both diagnostically to detect early Aβ-related tau dys-metabolism, as well as for disease staging (albeit without anatomical precision). Recent large validation studies show very similar results [50, 51], corroborating plasma P-tau as a robust blood biomarker for AD pathology that should be relatively easy to standardize and implement in clinical laboratory practice.
A drawback of fluid biomarkers is the inability to determine brain region-specific changes, which may limit staging of disease severity (e.g., Braak staging [52]) and limiting their use as progression markers. While lagging behind amyloid PET, the recent development of PET ligands to visualize, map and quantify tau pathology has provided new information on the temporal and spatial characteristics of tau accumulation in the living brain. First generation tracers include compounds in the THK series ([18F]THK5317, [18F]THK5351), [11C]PBB3, and [18F]AV1451 [53]. Of these, THK tracers are unlikely to be used clinically given that they also bind to monoamine oxidase-B, complicating the localization of neurofibrillary tangle pathology [54]. [11C]PBB3 has high affinity for AD tau pathology, but decomposes to a polar radiometabolite with brain penetrance, impacting accurate quantification of PBB3-binding [55]. [18F]AV1451, the most widely used compound, corresponds with post-mortem tau pathology [56], and has been used in several clinical trials; off-target binding is known a limitation [57]. An example [18F]AV1451 image is shown in Fig. 2b. The second generation tracer, [18F]MK-6240, is gaining wider use and unlike [18F]AV1451, shows limited off-target-binding near brain regions where tau tangle pathology develops [58]. Other tau tracers under evaluation include [18F]RO-948 PET [59], [18F]PI-2620 [60], and [18F]GTP1 [61].
In terms of accessibility and standardized use, tau imaging is still in its infancy compared with amyloid PET, but it could conceivably become a valuable clinical tool for evaluating the efficacy of amyloid, tau, or combination therapy. Prior to implementation in the clinical setting, additional developments are needed, including standardization of processing methods, cut-points for positivity (or standardized staging), and additional evidence supporting clinical use. The results accrued to date via PET imaging corroborate the Braak model of distinct spatial and temporal stages of tau pathology progression in the AD brain [52], and tau PET has already proven useful in clinical trials to detect pharmacodynamic effects of disease-modifying drug candidates against both Aβ (to monitor downstream tau changes) and tau pathology (target engagement) [62].
Biomarkers for neurodegeneration
Volumetric magnetic resonance imaging (MRI) and CSF tau have been used commonly as biomarkers for AD-type neurodegeneration [36, 63]. Typical MRI-based measures are derived from T1-weighted imaging and include whole brain atrophy [64], gray matter atrophy, and regional atrophy (e.g., medial temporal areas, hippocampus, and hippocampal subfields [65]), as well as surface-based cortical thickness [66]. Advances in neuroimaging acquisition and modeling of water diffusion have also facilitated the characterization of cortical microstructure, which may provide a sensitive measure of neurodegeneration that precedes overt cell loss [67]. Clinically, volumetric imaging is already used to monitor progression of neurodegeneration; multi-shell diffusion-weighted imaging is available as part of commercial scanning packages, but would need specialized processing for interpretation of neurodegeneration in the context of AD.
CSF T-tau was proposed as a strong candidate biomarker of neurodegeneration, but given that it reflects to a greater extent the increased tau secretion from neurons affected by Aβ pathology, rather than neuronal cell loss, it is a predictive but not a direct biomarker for neurodegeneration in AD [46]. Recently, neurofilament light (NfL) has emerged as a general biomarker for neuroaxonal degeneration and injury, irrespective of cause [68]. The biomarker can be measured in both CSF and plasma (or serum), and virtually all CSF findings have been replicated in blood with sensitive assays [69]. The highest NfL levels are seen in frontotemporal, vascular and HIV-associated dementias [70]. However, the findings in familial AD are also very clear; mutation carriers show a sudden change in their blood NfL levels around a decade before expected clinical onset, which probably marks the onset of neurodegeneration, and the higher the increase, the more rapid clinical disease progression [71, 72]. In sporadic AD, there is a clear association of Aβ and tau positivity, as well as longitudinal neurodegeneration as determined by MRI, with increased plasma NfL concentration, but with a larger overlap across groups than in familial AD [73], most likely due to the multitude of neurodegenerative changes that may result in NfL increase in people older than 70 years of age.
Biomarkers for synaptic dysfunction
Synaptic dysfunction appears to be an early event in AD, and synaptic loss is traditionally regarded the best correlate of cognitive decline in the disease [74]. Fluorodeoxyglucose (FDG) PET has long been used clinically for differential diagnosis of AD. Patients with AD dementia show a characteristic pattern of hypometabolism in precuneus, posterior cingulate, parietal cortices, lateral temporal cortex, frontal cortices, and medial temporal lobe [75]. An example [18F]FDG PET image with medial temporal lobe hypometabolism is shown in Fig. 2c Hypometabolism likely reflects a combination of synaptic dysfunction, neuronal cell loss, and metabolic dysfunction [36, 76], in addition to potentially being influenced by astroglial glutamate transport [77]. Despite the complexity of the signal, FDG PET remains an attractive biomarker given its wide clinical use for differential diagnosis of AD, its likely sensitivity to synaptic dysfunction, and its capability of detecting abnormalities in the preclinical asymptomatic stage of AD.
Another emerging method to detect synaptic loss in neurodegenerative dementias is PET tracers targeting synaptic vesicle protein 2A (SV2A), which is expressed ubiquitously in synapses [78]. [11C]UCB-J is a high-affinity ligand for SV2A, and imaging with the tracer demonstrates ~40% lower SV2A-binding in the hippocampus among individuals with AD [78]. Fluorine-18-labeled SV2A PET compounds would facilitate broader use, and are currently in development and testing [79, 80]. An example [11C]UCB-J PET scan acquired in a healthy control is shown in Fig. 2d. The extent to which SV2A ligands provide information above and beyond that provided by FDG PET remains to be determined.
In the biofluids, the most promising biomarker candidate for synaptic dysfunction in AD is the dendritic protein neurogranin (Ng). CSF Ng concentration is increased in AD and correlates with T-tau and P-tau concentrations, as well as cognitive deterioration over time, in an Aβ-dependent manner [81]. CSF Ng concentration is normal or slightly reduced in non-AD neurodegenerative dementias [82, 83], suggesting that it is not a general biomarker for synaptic loss but rather reflects an AD-specific and probably Aβ-driven change in its metabolism and secretion (similar to tau). CSF Ng concentration appears to relate to cognitive function more closely than the other CSF biomarkers [84]. Ng can be measured in plasma but without correlation with CSF levels, probably due to extra-cerebral production of the protein [85]. There are currently no available blood-based biomarkers for synaptic dysfunction or loss.
Biomarkers for glial activation and neuroinflammation
Neuroinflammation, as well as activation of microglial cells and astrocytes, are key features of neurodegenerative dementias, with most research having been performed in AD. During the last decade, it has been debated whether neuroinflammation and astrogliosis are important drivers of neurodegeneration, or downstream effects of the accumulation of Aβ and tau. Variants in the triggering receptor expressed on myeloid cells 2 (TREM2) gene, encoding a receptor of the innate immune system, which is highly expressed in microglia, have been found to increase the risk of late-onset AD by two- to fourfold, similar to what has been observed in patients with one copy of APOE ε4 [86]. This suggests that the innate immune system may be an active player in the AD process, potentially as a mediator of Aβ toxicity.
A number of candidate markers have been examined in relation to inflammation/astroglial activation in neurodegenerative dementias, of which YKL-40, a glycoprotein expressed in both astrocytes and microglia, and the soluble form of TREM2 (sTREM2) have proven to be the most promising [87]. Several cross-sectional, as well as longitudinal, studies in the past years have shown that CSF YKL-40 and sTREM2 levels are modestly increased in patients with AD and correlate with CSF tau levels in Aβ-positive individuals [87]. Data from the Dominantly Inherited Alzheimer’s Network (DIAN) study of familial AD mutation carriers suggest that CSF sTREM2 concentration increases before symptomatic disease onset, and just after CSF Aβ42 and T-tau have turned positive [88]. sTREM2 appears to peak in the mild cognitive impairment stage of the clinical presentation, with a slight decline in the clinical AD dementia stage [88], which may complicate the development of clinical cut-points. Currently, there are no established blood tests for neuroinflammation or astrocytic or microglial activation, but recent data on plasma glial fibrillary acidic protein (GFAP, an intermediate filament protein that is selectively expressed by astrocytes in the central nervous system) showed increased concentrations in AD patients compared with cognitively normal controls [89]. The currently available CSF GFAP data are less clear [17].
Activated, but not resting, microglia express the translocator protein (TSPO) on the outer membranes of their mitochondria [90]. [11C]PK11195, [11C]PBR28, and [18F]FEPPA are in vivo markers of the TSPO expressed by activated microglia. Recently, it was reported that 80% of Aβ-positive MCI patients showed active inflammation and increased TSPO ligand retention in the brain [91]. It has also been reported in established AD that levels of temporoparietal cortical microglial activation correlate inversely with cognitive function [92], suggesting a disease-promoting effect of the detected activation. Despite the relatively established role for TSPO PET imaging in detecting activated microglia in vivo, several challenges remain, particularly for clinical deployment. A major consideration is that there is genetic variation in the TSPO gene, making individuals more or less prone to bind the ligand. Thus, participants need to be genotyped for the single nucleotide polymorphism (rs6971) in exon 4 of the TSPO gene, to either exclude low binders or stratify participants into high, medium, and low affinity binders [93]. Another challenge is that acute vs. chronic inflammation cannot be differentiated, and it is presently not possible to differentiate more or less anti- and pro-inflammatory phenotypes of microglia with TSPO-targeting radioligands. This challenge is also relevant for the fluid-based biomarkers, and developing better biomarkers for different states of microglial activation is an active research field.
Several other indicators of neuroinflammation have been considered in CSF and blood in AD, including markers of pro- and anti-inflammatory cytokines and chemokines, e.g., soluble tumor necrosis factor receptor 1 and 2, α1-antichymotrypsin, soluble CD40 ligand, IL-10 and transforming growth factor-β1 [94]. These markers could be used for monitoring treatments, but are nonspecific to AD and may show variable expression depending on the general health of the patient and when they are measured in relation to disease stage.
Biomarkers for common copathologies: α-synuclein and TDP-43
Misfolding of α-synuclein plays a major role in the development of common neurodegenerative diseases, such as Parkinson’s disease (PD) and dementia with Lewy bodies (DLB). α-Synuclein is the main constituent of Lewy bodies. TAR DNA-binding protein 43 (TDP-43) is another inclusion-forming protein that is frequently seen in some forms of frontotemporal dementia and in amyotrophic lateral sclerosis. Furthermore, both these pathologies are often seen together with classical AD pathology; neuropathological studies show that around 50% of AD patients have Lewy bodies [95], whilst the corresponding figure for TDP-43 is 30–40% [96]. There are currently no established imaging biomarkers for α-synuclein or TDP-43 inclusions, and it has been difficult to develop fluid biomarker tests that are pathology-specific (both proteins can be measured in biofluids, but there is no correlation with pathology nor reproducible group differences, except for a slight decrease in CSF α-synuclein concentration in PD [97]). Nevertheless, the fact that α-synuclein oligomers may spread in a prion-like manner has sparked the idea that seeding aggregation assays, such as real-time quaking-induced conversion (RT-QuIC) or protein-misfolding cyclic amplification, could be used to qualitatively detect pathological forms of α-synuclein in CSF [98]. Studies analyzing CSF with RT-QuIC of α-synuclein have been able to distinguish synucleinopathies from nonsynucleinopathies with excellent diagnostic accuracy, detecting DLB and multiple system atrophy with 100 and 80% sensitivity, respectively [99]. Other studies from independent research groups have shown very similar results [100, 101], which is encouraging. Unsurprisingly, these assays do not discriminate between different synucleinopathies. As development of biomarkers for these copathologies progresses, the extent to which they provide information on clinical disease progression in AD will become clearer.
An integrated model
Biomarkers for AD have significantly advanced the field in several ways, and hold tremendous promise for diagnosis, staging pathology, and measuring treatment response. However, identifying the best biomarkers, either alone or in combination, requires that the biomarker modalities discussed above be examined largely within the same individuals, and preferably in longitudinal cohort studies. Recently, the results of a large head-to-head comparison of biomarkers for Aβ and tau pathology, neuroinflammation, synaptic dysfunction, and neurodegeneration in the Swedish BioFINDER cohort were published [2]. In Fig. 3, we present a model of the temporal staging of AD-related biomarker modalities along the phases of the AD continuum and incorporating the BioFINDER results. As predicted by previously published hypothetical models [102] and the amyloid cascade hypothesis [4], the earliest changes in the BioFINDER study were found in Aβ42, followed closely by P‐tau and T‐tau, as Aβ response markers. Change points in amyloid positivity were similar whether CSF or plasma were utilized, but in both cases, fluid amyloid abnormalities preceded change in amyloid PET. Neurodegeneration, indexed by hippocampal volume and CSF NfL, and synaptic dysfunction, indexed by CSF Ng, occurred after amyloid PET positivity. YKL‐40 increased after amyloid, P-tau, and neurodegeneration. (It is likely that GFAP would follow a similar pattern, although we still lack longitudinal data for this marker.) Of special note, CSF and plasma P-tau had similar dynamic ranges [2], highlighting the potential for plasma P-tau to be used as a leading blood-based biomarker of AD. The CSF sTREM2 changes depicted in Fig. 3 are modeled on the basis of data generated in the DIAN study, in which changes in CSF sTREM2 occurred after alterations were observed in markers of brain amyloidosis and in close association with onset of neurodegeneration [103]. sTREM2 appears to peak in early disease stages and declines later in the AD dementia stage [88].

The first biomarkers to change are cerebrospinal fluid (CSF) and plasma Aβ42/Aβ40 ratio. This is shortly followed by CSF and plasma tau increases, as a neuronal response to the amyloid changes. Shortly thereafter amyloid PET turns positive. Then microglia react and secrete soluble triggering receptor expressed on myeloid cells 2 (sTREM2), which reaches its maximum in the MCI stage of the disease, whereafter it declines in the dementia phase (there is not enough data on translocator protein PET, as a biomarker for microglial activation, to put it into the model). CSF neurogranin (Ng) is an early marker of synaptic dysfunction and increases in close association with amyloid PET positivity. When tau PET turns positive, a range of neurodegeneration and synaptic dysfunction biomarkers (CSF and serum/plama neurofilament light [NfL], hippocampal volume, and SV2A- and fluorodeoxyglucose [FDG] PET) change more or less in parallel. Most studies suggest that astrocytic biomarkers (YKL-40 and glial fibrillary acidic protein [GFAP]) change relatively late in the disease process.
Considerations regarding the clinical interpretation of biomarker results
As detailed above, AD-related pathologies appear many years before clinical onset of the disease. While positive Aβ and tau biomarkers suggest that the patient has plaque and tangle pathology, the challenge will be for clinicians to determine if these pathologies are likely explanations for patient symptoms or not. A depressed patient with some cognitive problems may well have preclinical AD pathology that might develop into AD in 10 years, whilst his or her current symptoms are mainly caused by the depression. The role of comorbid AD in other neurodegenerative diseases represents another but related challenge. For example, a patient with a typical frontotemporal dementia syndrome with positive AD biomarkers is likely to have comorbid AD pathology but it is possible that this AD copathology is silent and does not contribute to the clinical syndrome at all. Age is another consideration, in particular with the oldest old. Among older adults who are cognitively unimpaired, knowing amyloid status may only partially inform lifetime risk of dementia. For example, a cognitively unimpaired 60 to 65-year-old woman who is positive for amyloid has a lifetime risk of developing AD dementia that is around 30%, while for an amyloid-positive 85-year-old woman, the lifetime risk is lower: around 14% [104]. Likewise, the relationship between pathological brain lesions and clinical status appears to attenuate at advanced ages. Post-mortem evaluation of almost 300 older subjects without neurological impairment showed that approximately half of the subjects displayed Aβ deposition whereas some degree of tau pathology could be seen in almost all brains [105]. Accordingly, the specificities of CSF tau and Aβ biomarkers for subjects without AD and the areas under the receiver-operating characteristics curves for distinguishing AD from non-AD patients decrease with age [106]. Similarly, volumetric MRI changes typical of AD are commonly seen in cognitively healthy subjects older than 80 years of age [107, 108].
Biomarkers for AD in a new era of disease-modifying therapies
Recent clinical trial results suggest that antibody-based removal of cerebral Aβ plaques may possibly clear tau tangles and modestly slow cognitive decline in symptomatic AD [109]. Although it is yet unclear whether this will result in regulatory approval, the results have reinvigorated the AD field, and it is imperative that the healthcare system readies itself for disease-modifying therapies against AD. Biomarkers for AD will be essential to this process, as will improve interpretation of biomarker results, considering potential caveats as discussed above.
Synthesizing the recent biomarker breakthroughs above, it is relatively easy to envision blood-based testing for Aβ and tau pathology using plasma Aβ42/Aβ40 ratio and plasma P-tau as screening tools. Whilst the difference in plasma Aβ42/Aβ40 ratio between Aβ-positive and –negative individuals is rather modest (a 14–20% reduction [30,31,32,33]), the increase in plasma P-tau concentration is around threefold, with very high diagnostic accuracies for AD (85–95%) [50, 51], suggesting that plasma P-tau could serve as a screening blood test in primary care. Positive patients could then be referred to a specialized memory clinic to be more closely examined, undergo amyloid PET imaging where available, and commence treatment with an anti-Aβ antibody therapy. Plasma P-tau (representing a neuronal reaction to Aβ) and NfL levels (representing neurodegeneration) could be monitored throughout therapy (e.g., at each antibody infusion or every 3rd month), followed by yearly amyloid PET scans. Repeat MRIs will be needed initially to monitor amyloid-related imaging abnormalities (ARIA), but in the future, it is plausible that increases in plasma NfL concentration could substitute for MRI to detect clinically relevant ARIA. The patient would then be treated until amyloid PET is negative and plasma P-tau concentration has normalized. Post-treatment, the patient could be followed with annual plasma P-tau measurements to gauge need for additional therapy. As additional therapies are developed, for example, microglia modulators or treatments improving synaptic function, biomarkers related to these processes are expected to facilitate tracking of therapy efficacy. Future clinical trials should incorporate both neuroimaging and fluid biomarker approaches, to assess biological response at the same time as they provide information needed to develop the more readily accessible biomarkers. Given the potential barriers which may impede access to AD therapy, and the need to expand treatment options beyond specialized centers, fluid biomarkers provide an attractive option for screening and monitoring treatment efficacy. While the proposed recommendations need empirical data for support, they represent a testable scenario regarding how upcoming clinical trials could be designed and AD treatments could be delivered in the clinic with the support of biomarkers.
References
DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener. 2019;14:32.
Palmqvist S, Insel PS, Stomrud E, Janelidze S, Zetterberg H, Brix B, et al. Cerebrospinal fluid and plasma biomarker trajectories with increasing amyloid deposition in Alzheimer’s disease. EMBO Mol Med. 2019;11:e11170.
Jack CR Jr., Holtzman DM. Biomarker modeling of Alzheimer’s disease. Neuron. 2013;80:1347–58.
Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–5.
De Strooper B, Karran E. The cellular phase of Alzheimer’s disease. Cell. 2016;164:603–15.
Sala Frigerio C, Wolfs L, Fattorelli N, Thrupp N, Voytyuk I, Schmidt I, et al. The major risk factors for Alzheimer’s disease: age, sex, and genes modulate the microglia response to abeta plaques. Cell Rep. 2019;27:1293–306. e6.
Kivipelto M, Mangialasche F, Ngandu T. Lifestyle interventions to prevent cognitive impairment, dementia and Alzheimer disease. Nat Rev Neurol. 2018;14:653–66.
Loera-Valencia R, Cedazo-Minguez A, Kenigsberg PA, Page G, Duarte AI, Giusti P, et al. Current and emerging avenues for Alzheimer’s disease drug targets. J Intern Med. 2019;286:398–437.
Walsh DM, Teplow DB. Alzheimer’s disease and the amyloid beta-protein. Prog Mol Biol Transl Sci. 2012;107:101–24.
Huang LK, Chao SP, Hu CJ. Clinical trials of new drugs for Alzheimer disease. J Biomed Sci. 2020;27:18.
Suidan GL, Ramaswamy G. Targeting apolipoprotein E for Alzheimer’s disease: an industry perspective. Int J Mol Sci. 2019;20:2161. https://doi.org/10.3390/ijms20092161.
Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement. 2019;5:272–93.
Bittar A, Bhatt N, Kayed R. Advances and considerations in AD tau-targeted immunotherapy. Neurobiol Dis. 2020;134:104707.
Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8:595–608.
Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharm Sci. 1991;12:383–8.
Cohen AD, Landau SM, Snitz BE, Klunk WE, Blennow K, Zetterberg H. Fluid and PET biomarkers for amyloid pathology in Alzheimer’s disease. Mol Cell Neurosci. 2019;97:3–17.
Olsson B, Lautner R, Andreasson U, Ohrfelt A, Portelius E, Bjerke M, et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol. 2016;15:673–84.
Rasmussen MK, Mestre H, Nedergaard M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018;17:1016–24.
Hansson O, Lehmann S, Otto M, Zetterberg H, Lewczuk P. Advantages and disadvantages of the use of the CSF amyloid beta (Abeta) 42/40 ratio in the diagnosis of Alzheimer’s Disease. Alzheimers Res Ther. 2019;11:34.
Lewczuk P, Matzen A, Blennow K, Parnetti L, Molinuevo JL, Eusebi P, et al. Cerebrospinal fluid abeta42/40 corresponds better than abeta42 to amyloid PET in Alzheimer’s disease. J Alzheimers Dis. 2017;55:813–22.
Mattsson N, Palmqvist S, Stomrud E, Vogel J, Hansson O. Staging beta-amyloid pathology with amyloid positron emission tomography. JAMA Neurol. 2019;76. https://doi.org/10.1001/jamaneurol.2019.2214 [Online ahead of print].
Klunk WE, Koeppe RA, Price JC, Benzinger TL, Devous MD Sr., Jagust WJ, et al. The centiloid project: standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement. 2015;11:1–15. e1–4.
Johnson KA, Minoshima S, Bohnen NI, Donohoe KJ, Foster NL, Herscovitch P, et al. Update on appropriate use criteria for amyloid PET imaging: dementia experts, mild cognitive impairment, and education. Amyloid imaging task force of the Alzheimer’s association and society for nuclear medicine and molecular imaging. Alzheimers Dement. 2013;9:e106–9.
Liu JL, Hlavka JP, Hillestad R, Mattke S. Assessing the preparedness of the U.S. health care system infrastructure for an Alzheimer’s treatment. Santa Monica, CA: RAND Corporation; 2017.
Liu JL, Hlavka JP, Coulter DT, Baxi SM, Mattke S, Gidengil CA. Assessing the preparedness of the Canadian health care system infrastructure for an Alzheimer’s treatment. Santa Monica, CA: RAND Corporation; 2019.
Hlavka JP, Mattke S, Liu JL. Assessing the preparedness of the health care system infrastructure in six European countries for an Alzheimer’s treatment. Santa Monica, CA: RAND Corporation; 2018.
Mattke S, Hlávka JP, Yoong J, Wang M, Goto R. Assessing the preparedness of the Japanese health care system infrastructure for an Alzheimer’s treatment. Santa Monica, CA: RAND Corporation; 2019.
Arneric SP, Batrla-Utermann R, Beckett L, Bittner T, Blennow K, Carter L, et al. Cerebrospinal fluid biomarkers for Alzheimer’s disease: a view of the regulatory science qualification landscape from the coalition against major diseases CSF biomarker team. J Alzheimers Dis. 2017;55:19–35.
Kuhlmann J, Andreasson U, Pannee J, Bjerke M, Portelius E, Leinenbach A, et al. CSF abeta1-42—an excellent but complicated Alzheimer’s biomarker—a route to standardisation. Clin Chim Acta. 2017;467:27–33.
Janelidze S, Stomrud E, Palmqvist S, Zetterberg H, van Westen D, Jeromin A, et al. Plasma beta-amyloid in Alzheimer’s disease and vascular disease. Sci Rep. 2016;6:26801.
Nakamura A, Kaneko N, Villemagne VL, Kato T, Doecke J, Dore V, et al. High performance plasma amyloid-beta biomarkers for Alzheimer’s disease. Nature. 2018;554:249–54.
Ovod V, Ramsey KN, Mawuenyega KG, Bollinger JG, Hicks T, Schneider T, et al. Amyloid beta concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimers Dement. 2017;13:841–9.
Schindler SE, Bollinger JG, Ovod V, Mawuenyega KG, Li Y, Gordon BA, et al. High-precision plasma beta-amyloid 42/40 predicts current and future brain amyloidosis. Neurology. 2019;93:e1647–59.
Palmqvist S, Janelidze S, Stomrud E, Zetterberg H, Karl J, Zink K, et al. Performance of fully automated plasma assays as screening tests for Alzheimer disease-related beta-amyloid status. JAMA Neurol. 2019;76:1060–9.
Irwin DJ. Tauopathies as clinicopathological entities. Parkinsonism Relat Disord 2016;22 Suppl 1:S29–33.
Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14:535–62.
Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K, et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol. 2014;13:614–29.
Itoh N, Arai H, Urakami K, Ishiguro K, Ohno H, Hampel H, et al. Large-scale, multicenter study of cerebrospinal fluid tau protein phosphorylated at serine 199 for the antemortem diagnosis of Alzheimer’s disease. Ann Neurol. 2001;50:150–6.
Skillback T, Farahmand BY, Rosen C, Mattsson N, Nagga K, Kilander L, et al. Cerebrospinal fluid tau and amyloid-beta(1-42) in patients with dementia. Brain. 2015;138:2716–31.
Maia LF, Kaeser SA, Reichwald J, Hruscha M, Martus P, Staufenbiel M, et al. Changes in amyloid-beta and tau in the cerebrospinal fluid of transgenic mice overexpressing amyloid precursor protein. Sci Transl Med. 2013;5:194re2.
Sato C, Barthelemy NR, Mawuenyega KG, Patterson BW, Gordon BA, Jockel-Balsarotti J, et al. Tau kinetics in neurons and the human central nervous system. Neuron. 2018;98:861–4.
Leitao MJ, Silva-Spinola A, Santana I, Olmedo V, Nadal A, Le Bastard N, et al. Clinical validation of the lumipulse G cerebrospinal fluid assays for routine diagnosis of Alzheimer’s disease. Alzheimers Res Ther. 2019;11:91.
Blennow K, Shaw LM, Stomrud E, Mattsson N, Toledo JB, Buck K, et al. Predicting clinical decline and conversion to Alzheimer’s disease or dementia using novel Elecsys Abeta(1-42), pTau and tTau CSF immunoassays. Sci Rep. 2019;9:19024.
De Vos A, Bjerke M, Brouns R, De Roeck N, Jacobs D, Van den Abbeele L, et al. Neurogranin and tau in cerebrospinal fluid and plasma of patients with acute ischemic stroke. BMC Neurol. 2017;17:170.
Bogoslovsky T, Wilson D, Chen Y, Hanlon D, Gill J, Jeromin A, et al. Increases of plasma levels of glial fibrillary acidic protein, tau, and amyloid beta up to 90 days after traumatic brain injury. J Neurotrauma. 2017;34:66–73.
Zetterberg H. Review: tau in biofluids—relation to pathology, imaging and clinical features. Neuropathol Appl Neurobiol. 2017;43:194–9.
Pereira JB, Westman E, Hansson O, Alzheimer’s Disease Neuroimaging I. Association between cerebrospinal fluid and plasma neurodegeneration biomarkers with brain atrophy in Alzheimer’s disease. Neurobiol Aging. 2017;58:14–29.
Randall J, Mortberg E, Provuncher GK, Fournier DR, Duffy DC, Rubertsson S, et al. Tau proteins in serum predict neurological outcome after hypoxic brain injury from cardiac arrest: results of a pilot study. Resuscitation. 2013;84:351–6.
Mielke MM, Hagen CE, Xu J, Chai X, Vemuri P, Lowe VJ, et al. Plasma phospho-tau181 increases with Alzheimer’s disease clinical severity and is associated with tau- and amyloid-positron emission tomography. Alzheimers Dement. 2018;14:989–97.
Thijssen EH, La Joie R, Wolf A, Strom A, Wang P, Iaccarino L, et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer’s disease and frontotemporal lobar degeneration. Nat Med. 2020;26:387–97.
Janelidze S, Mattsson N, Palmqvist S, Insel P, Smith R, Eichenlaub U, et al. Plasma P-tau181 in Alzheimer’s disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat Med. 2020;26:379–86.
Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59.
Saint-Aubert L, Lemoine L, Chiotis K, Leuzy A, Rodriguez-Vieitez E, Nordberg A, et al. imaging: present and future directions. Mol Neurodegener. 2017;12:19.
Ng KP, Pascoal TA, Mathotaarachchi S, Therriault J, Kang MS, Shin M, et al. Monoamine oxidase B inhibitor, selegiline, reduces (18)F-THK5351 uptake in the human brain. Alzheimers Res Ther. 2017;9:25.
Betthauser TJ. AD molecular: imaging tau aggregates with positron emissions tomography. Prog Mol Biol Transl Sci. 2019;165:107–38.
Marquie M, Siao Tick Chong M, Anton-Fernandez A, Verwer EE, Saez-Calveras N, Meltzer AC. et al. [F-18]-AV-1451 binding correlates with postmortem neurofibrillary tangle Braak staging. Acta Neuropathol.2017;134:619–28.
Marquie M, Normandin MD, Meltzer AC, Siao Tick Chong M, Andrea NV, Anton-Fernandez A. et al. Pathological correlations of [F-18]-AV-1451 imaging in non-alzheimer tauopathies. Ann Neurol.2017;81:117–28.
Betthauser TJ, Cody KA, Zammit MD, Murali D, Converse AK, Barnhart TE, et al. In vivo characterization and quantification of neurofibrillary tau PET radioligand (18)F-MK-6240 in humans from Alzheimer disease dementia to young controls. J Nucl Med. 2019;60:93–9.
Kuwabara H, Comley RA, Borroni E, Honer M, Kitmiller K, Roberts J, et al. Evaluation of (18)F-RO-948 PET for quantitative assessment of tau accumulation in the human brain. J Nucl Med. 2018;59:1877–84.
Mueller A, Bullich S, Barret O, Madonia J, Berndt M, Papin C, et al. Tau PET imaging with (18)F-PI-2620 in patients with Alzheimer’s disease and healthy controls: a first-in-human study. J Nucl Med. 2019. https://doi.org/10.2967/jnumed.119.236224. [Online ahead of print].
Sanabria Bohorquez S, Marik J, Ogasawara A, Tinianow JN, Gill HS, Barret O, et al. [(18)F]GTP1 (Genentech Tau Probe 1), a radioligand for detecting neurofibrillary tangle tau pathology in Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2019;46:2077–89.
Scholl M, Maass A, Mattsson N, Ashton NJ, Blennow K, Zetterberg H, et al. Biomarkers for tau pathology. Mol Cell Neurosci. 2019;97:18–33.
Frisoni GB, Fox NC, Jack CR Jr., Scheltens P, Thompson PM. The clinical use of structural MRI in Alzheimer disease. Nat Rev Neurol. 2010;6:67–77.
Fox NC, Warrington EK, Rossor MN. Serial magnetic resonance imaging of cerebral atrophy in preclinical Alzheimer’s disease. Lancet. 1999;353:2125.
Jack CR Jr., Petersen RC, O’Brien PC, Tangalos EG. MR-based hippocampal volumetry in the diagnosis of Alzheimer’s disease. Neurology. 1992;42:183–8.
Dickerson BC, Stoub TR, Shah RC, Sperling RA, Killiany RJ, Albert MS, et al. Alzheimer-signature MRI biomarker predicts AD dementia in cognitively normal adults. Neurology. 2011;76:1395–402.
Vogt NM, Hunt JF, Adluru N, Dean DC, Johnson SC, Asthana S, et al. Cortical microstructural alterations in mild cognitive impairment and Alzheimer’s disease dementia. Cereb Cortex. 2019. https://doi.org/10.1093/cercor/bhz286. [Online ahead of print].
Khalil M, Teunissen CE, Otto M, Piehl F, Sormani MP, Gattringer T, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol. 2018;14:577–89.
Gaetani L, Blennow K, Calabresi P, Di Filippo M, Parnetti L, Zetterberg H. Neurofilament light chain as a biomarker in neurological disorders. J Neurol Neurosurg Psychiatry. 2019;90:870–81.
Bridel C, van Wieringen WN, Zetterberg H, Tijms BM, Teunissen CE, and the NFLG, et al. Diagnostic value of cerebrospinal fluid neurofilament light protein in neurology: a systematic review and meta-analysis. JAMA Neurol. 2019;76:1035–48.
Weston PSJ, Poole T, O’Connor A, Heslegrave A, Ryan NS, Liang Y, et al. Longitudinal measurement of serum neurofilament light in presymptomatic familial Alzheimer’s disease. Alzheimers Res Ther. 2019;11:19.
Preische O, Schultz SA, Apel A, Kuhle J, Kaeser SA, Barro C, et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer’s disease. Nat Med. 2019;25:277–83.
Mattsson N, Cullen NC, Andreasson U, Zetterberg H, Blennow K. Association between longitudinal plasma neurofilament light and neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 2019;76:791–9.
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–80.
Alexander GE, Chen K, Pietrini P, Rapoport SI, Reiman EM. Longitudinal PET evaluation of cerebral metabolic decline in dementia: a potential outcome measure in Alzheimer’s disease treatment studies. Am J Psychiatry. 2002;159:738–45.
Reiman EM. Fluorodeoxyglucose positron emission tomography: emerging roles in the evaluation of putative Alzheimer’s disease-modifying treatments. Neurobiol Aging. 2011;32 Suppl 1:S44–7.
Zimmer ER, Parent MJ, Souza DG, Leuzy A, Lecrux C, Kim HI, et al. [(18)F]FDG PET signal is driven by astroglial glutamate transport. Nat Neurosci. 2017;20:393–5.
Chen MK, Mecca AP, Naganawa M, Finnema SJ, Toyonaga T, Lin SF, et al. Assessing synaptic density in Alzheimer disease with synaptic vesicle glycoprotein 2A positron emission tomographic imaging. JAMA Neurol. 2018;75:1215–24.
Constantinescu CC, Tresse C, Zheng M, Gouasmat A, Carroll VM, Mistico L, et al. Development and in vivo preclinical imaging of fluorine-18-labeled synaptic vesicle protein 2A (SV2A) PET tracers. Mol Imaging Biol. 2019;21:509–18.
Li S, Cai Z, Zhang W, Holden D, Lin SF, Finnema SJ, et al. Synthesis and in vivo evaluation of [(18)F]UCB-J for PET imaging of synaptic vesicle glycoprotein 2A (SV2A). Eur J Nucl Med Mol Imaging. 2019;46:1952–65.
Kvartsberg H, Duits FH, Ingelsson M, Andreasen N, Ohrfelt A, Andersson K, et al. Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer’s disease. Alzheimers Dement. 2015;11:1180–90.
Wellington H, Paterson RW, Portelius E, Tornqvist U, Magdalinou N, Fox NC, et al. Increased CSF neurogranin concentration is specific to Alzheimer disease. Neurology. 2016;86:829–35.
Portelius E, Olsson B, Hoglund K, Cullen NC, Kvartsberg H, Andreasson U, et al. Cerebrospinal fluid neurogranin concentration in neurodegeneration: relation to clinical phenotypes and neuropathology. Acta Neuropathol. 2018;136:363–76.
Casaletto KB, Elahi FM, Bettcher BM, Neuhaus J, Bendlin BB, Asthana S, et al. Neurogranin, a synaptic protein, is associated with memory independent of Alzheimer biomarkers. Neurology. 2017;89:1782–8.
De Vos A, Jacobs D, Struyfs H, Fransen E, Andersson K, Portelius E, et al. C-terminal neurogranin is increased in cerebrospinal fluid but unchanged in plasma in Alzheimer’s disease. Alzheimers Dement. 2015;11:1461–9.
Gratuze M, Leyns CEG, Holtzman DM. New insights into the role of TREM2 in Alzheimer’s disease. Mol Neurodegener. 2018;13:66.
Molinuevo JL, Ayton S, Batrla R, Bednar MM, Bittner T, Cummings J, et al. Current state of Alzheimer’s fluid biomarkers. Acta Neuropathol. 2018;136:821–53.
Suarez-Calvet M, Kleinberger G, Araque Caballero MA, Brendel M, Rominger A, Alcolea D, et al. sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol Med. 2016;8:466–76.
Elahi FM, Casaletto KB, La Joie R, Walters SM, Harvey D, Wolf A, et al. Plasma biomarkers of astrocytic and neuronal dysfunction in early- and late-onset Alzheimer’s disease. Alzheimers Dement. 2019. https://doi.org/10.1016/j.jalz.2019.09.004. [Online ahead of print].
Scarf AM, Kassiou M. The translocator protein. J Nucl Med. 2011;52:677–80.
Parbo P, Ismail R, Hansen KV, Amidi A, Marup FH, Gottrup H, et al. Brain inflammation accompanies amyloid in the majority of mild cognitive impairment cases due to Alzheimer’s disease. Brain. 2017;140:2002–11.
Edison P, Archer HA, Hinz R, Hammers A, Pavese N, Tai YF, et al. Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F]FDG PET study. Neurology. 2007;68:501–8.
Owen DR, Yeo AJ, Gunn RN, Song K, Wadsworth G, Lewis A, et al. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metab. 2012;32:1–5.
Shen XN, Niu LD, Wang YJ, Cao XP, Liu Q, Tan L, et al. Inflammatory markers in Alzheimer’s disease and mild cognitive impairment: a meta-analysis and systematic review of 170 studies. J Neurol Neurosurg Psychiatry. 2019;90:590–8.
Robinson JL, Lee EB, Xie SX, Rennert L, Suh E, Bredenberg C, et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain. 2018;141:2181–93.
James BD, Wilson RS, Boyle PA, Trojanowski JQ, Bennett DA, Schneider JA. TDP-43 stage, mixed pathologies, and clinical Alzheimer’s-type dementia. Brain. 2016;139:2983–93.
Mollenhauer B. Quantification of alpha-synuclein in cerebrospinal fluid: how ideal is this biomarker for Parkinson’s disease? Parkinsonism Relat Disord. 2014;20(Suppl 1):S76–9.
Paciotti S, Bellomo G, Gatticchi L, Parnetti L. Are we ready for detecting alpha-synuclein prone to aggregation in patients? the case of “protein-misfolding cyclic amplification” and “real-time quaking-induced conversion” as diagnostic tools. Front Neurol. 2018;9:415.
Shahnawaz M, Tokuda T, Waragai M, Mendez N, Ishii R, Trenkwalder C, et al. Development of a biochemical diagnosis of parkinson disease by detection of alpha-synuclein misfolded aggregates in cerebrospinal fluid. JAMA Neurol. 2017;74:163–72.
Fairfoul G, McGuire LI, Pal S, Ironside JW, Neumann J, Christie S, et al. Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann Clin Transl Neurol. 2016;3:812–8.
Groveman BR, Orru CD, Hughson AG, Raymond LD, Zanusso G, Ghetti B, et al. Rapid and ultra-sensitive quantitation of disease-associated alpha-synuclein seeds in brain and cerebrospinal fluid by alphaSyn RT-QuIC. Acta Neuropathol Commun. 2018;6:7.
Jack CR Jr., Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207–16.
Suarez-Calvet M, Araque Caballero MA, Kleinberger G, Bateman RJ, Fagan AM, Morris JC, et al. Early changes in CSF sTREM2 in dominantly inherited Alzheimer’s disease occur after amyloid deposition and neuronal injury. Sci Transl Med. 2016;8:369ra178.
Brookmeyer R, Abdalla N. Estimation of lifetime risks of Alzheimer’s disease dementia using biomarkers for preclinical disease. Alzheimers Dement. 2018;14:981–8.
Elobeid A, Libard S, Leino M, Popova SN, Alafuzoff I. Altered proteins in the aging brain. J Neuropathol Exp Neurol. 2016;75:316–25.
Mattsson N, Rosen E, Hansson O, Andreasen N, Parnetti L, Jonsson M, et al. Age and diagnostic performance of Alzheimer disease CSF biomarkers. Neurology. 2012;78:468–76.
Tang Y, Whitman GT, Lopez I, Baloh RW. Brain volume changes on longitudinal magnetic resonance imaging in normal older people. J Neuroimaging. 2001;11:393–400.
Zhang Y, Qiu C, Lindberg O, Bronge L, Aspelin P, Backman L, et al. Acceleration of hippocampal atrophy in a non-demented elderly population: the SNAC-K study. Int Psychogeriatr. 2010;22:14–25.
Haeberlein SB, von Hehn C, Tian Y, Chalkias S, Muralidharan KK, Chen T, et al. Topline results from phase 3 aducanumab studies clinical trials on Alzheimer’s disease. Abstract and presentation at CTAD2019, San Diego; 2019.
Acknowledgements
HZ is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018-02532), the European Research Council (#681712), Swedish State Support for Clinical Research (#ALFGBG-720931) and the UK Dementia Research Institute at UCL. BBB is supported by grants from the National Institute on Aging (P30AG062715, R01AG037639, RF1AG059312, R01AG062285, RF1AG057784, UF1AG051216).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
HZ has served at scientific advisory boards for Denali, Roche Diagnostics, Wave, Samumed and CogRx, has given lectures in symposia sponsored by Fujirebio, Alzecure and Biogen, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB, a GU Ventures-based platform company at the University of Gothenburg. BBB has received precursors and imaging agents from Avid Radiopharmaceuticals.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zetterberg, H., Bendlin, B.B. Biomarkers for Alzheimer’s disease—preparing for a new era of disease-modifying therapies. Mol Psychiatry 26, 296–308 (2021). https://doi.org/10.1038/s41380-020-0721-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-020-0721-9
- Springer Nature Limited
This article is cited by
-
Phytoactive drugs used in the treatment of Alzheimer’s disease and dementia
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)
-
Distinct tau and alpha-synuclein molecular signatures in Alzheimer’s disease with and without Lewy bodies and Parkinson’s disease with dementia
Acta Neuropathologica (2024)
-
Cognitive and Neuropathophysiological Outcomes of Gamma-tACS in Dementia: A Systematic Review
Neuropsychology Review (2024)
-
Single-cell RNA-seq reveals alterations in peripheral CX3CR1 and nonclassical monocytes in familial tauopathy
Genome Medicine (2023)
-
Predicting cognitive dysfunction and regional hubs using Braak staging amyloid-beta biomarkers and machine learning
Brain Informatics (2023)