
Abstract
Major depressive disorder (MDD) is one of the most prevalent psychiatric disorders and a leading cause of disability worldwide. Though recent genome-wide association studies (GWAS) have identified multiple risk variants for MDD, how these variants confer MDD risk remains largely unknown. Here we systematically characterize the regulatory mechanism of MDD risk variants using a functional genomics approach. By integrating chromatin immunoprecipitation sequencing (ChIP-Seq) (from human brain tissues or neuronal cells) and position weight matrix (PWM) data, we identified 34 MDD risk SNPs that disrupt the binding of 15 transcription factors (TFs). We verified the regulatory effect of the TF binding–disrupting SNPs with reporter gene assays, allelic-specific expression analysis, and CRISPR-Cas9-mediated genome editing. Expression quantitative trait loci (eQTL) analysis identified the target genes that might be regulated by these regulatory risk SNPs. Finally, we found that NEGR1 (regulated by the TF binding–disrupting MDD risk SNP rs3101339) was dysregulated in the brains of MDD cases compared with controls, implying that rs3101339 may confer MDD risk by affecting NEGR1 expression. Our findings reveal how genetic variants contribute to MDD risk by affecting TF binding and gene regulation. More importantly, our study identifies the potential MDD causal variants and their target genes, thus providing pivotal candidates for future mechanistic study and drug development.
Similar content being viewed by others


Introduction
Major depressive disorder (MDD) is the most prevalent mental disorder [1] (the lifetime prevalence is ~16% [2]) and is characterized by loss of interest or pleasure, depressed mood, impaired cognitive function, hopelessness, anxiety, and sleep disturbance [3]. The high prevalence (~6% adults are affected by MDD [4]) and mortality (the suicide rate of MDD cases is much higher (~20-fold) than the general population [5]) make MDD a leading contributor to the global burden of disease [6,7,8] and a major leading cause of disability worldwide [9]. The economic burden of MDD is considerable (e.g., the costs of MDD in the USA were estimated to be $210.5 billion in 2010 [10]). Due to the high mortality, considerable morbidity, and substantial costs, MDD has become a major threat to global health.
To date, the etiology of MDD remains largely unknown. Accumulating evidence indicates that the causes of MDD are multifactorial [3]. Environmental factors, including stress, major life changes, and childhood physical or emotional abuse, have been reported to be associated with the risk of developing MDD [11, 12]. In addition, MDD also has a genetic component. The heritability of MDD was estimated to be ~37% [3, 13], indicating the important roles of genetic factors in MDD. Several genome-wide association studies (GWAS) have been conducted during the past decade, and multiple MDD risk loci have been identified [14,15,16,17,18]. Although over 100 risk loci have been reported, pinpointing the functional (or causal) variants at the reported risk loci and elucidating their biological effects remain major challenges. Considering that most of the reported risk variants are located in noncoding regions, it is likely that these identified risk variants confer risk of MDD by regulating gene expression. Expression quantitative trait loci (eQTL) analysis could link the identified risk variants to specific gene (or genes). However, as each risk locus identified by GWAS usually contains multiple SNPs that are in high linkage disequilibrium (LD), pinpointing the functional (or causal) SNPs from the reported risk loci remains a daunting task. Identifying the causal (or functional) variants from the reported risk loci and elucidating their functional consequences are pivotal for dissecting the genetic mechanisms of MDD and developing new therapeutic approaches.
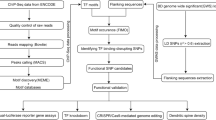
To systematically identify the functional variants from each of the risk loci and to explore how these functional risk variants exert their biological effects on MDD (i.e., the regulatory mechanisms of these functional variants), we performed functional genomics and eQTL analysis, followed by serial functional validations. We first processed 34 ChIP-Seq experiments conducted on neuronal cells or brain tissues and derived the binding motifs of 30 transcription factors (TFs). By comparing the derived motifs with the well-characterized binding motifs from the position weight matrix (PWM) databases (JASPAR, TRANSFAC, Uniprobe, and HI-SELEX) (compiled by Whitington et al. [19]), matched motifs were obtained. We then mapped the MDD risk SNPs (including the index (or lead) SNPs and SNPs in LD (r2 ≥ 0.3) with the index SNPs) to the matched motifs to investigate whether the MDD risk SNPs are located in the binding motif of a specific TF (or TFs) and if the different alleles of a risk SNP disrupt the binding of a specific TF (or TFs). We identified 34 regulatory SNPs that disrupt the binding of 15 TFs. We validated the regulatory effects of these TF binding–disrupting SNPs with reporter gene assays, allele-specific expression (ASE) analysis, short hairpin RNA (shRNA)-mediated knockdown of the corresponding TF, and CRISPR-Cas9-mediated genome editing. eQTL analysis identified the potential target genes regulated by these regulatory (i.e., TF binding–disrupting) SNPs. Finally, we found that NEGR1 (regulated by the TF binding–disrupting SNP rs3101339) was dysregulated in brain tissues (hippocampus and nucleus accumbens (NAc)) of MDD cases compared with controls, implying that the identified functional variant may confer MDD risk through modulating NEGR1 expression. Our study identified 34 potential functional SNPs (i.e., TF binding–disrupting SNPs) from the reported MDD risk loci and elucidated the regulatory mechanisms of MDD risk variants (including frequent disruption of CCCTC-binding factor (CTCF) binding). Our findings demonstrate the complex genetic regulatory architecture of MDD risk variants and provide new target genes. Further functional study and mechanistic investigation of these target genes will help understand the etiology of MDD and develop new therapeutic approaches.
Methods
Processing of ChIP-seq data
To identify the binding motifs of the selected TFs, we downloaded ChIP-seq data (raw data) of 34 TFs from ENCODE [20] (http://www.encodeproject.org). As previous studies have shown that MDD is mainly attributable to brain dysfunction [21, 22], only ChIP-seq assays conducted using human brain tissues or neuronal cells (including neuronal cell lines) were included in this study. More detailed information about the included TFs, ChIP-seq assays, and processing procedures can be found in our previous study [23]. Briefly, quality control and filtering of raw ChIP-seq reads were conducted with FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and Btrim [24]. Cutadapt was utilized to filter the overrepresented sequences, such as adapters, primers, and other sequences [25]. We then aligned the processed ChIP-seq reads to the human reference genome (GRCh37/hg19) using Bowtie [26], and peak calling was conducted with MACS [27]. We selected a control ChIP-seq assay that had the largest size (i.e., with the largest data content) of the bam file if there were biological replicates. If there were biological replicates in ChIP-seq experiments, we conducted peak calling by combining the bam files from different biological replicates.
Motif discovery and identification
The called peaks were used for identifying DNA binding motifs (i.e., PWM sequence) enriched (or overrepresented) in the genomic sequences surrounding the ChIP-seq signals. The top 500 ChIP-seq peaks (±20 bp, ranked by peak height) were used for motif discovery with MEME [28] (with following parameters: “-minw 6, -maxw 20” and -nmotifs 5) program. The ChIP-seq peaks of a specified TF were compared with the peaks of the corresponding control sample, and peaks that had FDR > 5% were excluded. In total, ChIP-seq peaks of 30 TFs (4 TFs were excluded due to low quality) were retained for further analysis. We then examined the binding specificity of the 30 included TFs by comparing the motifs from ChIP-seq with the PWM data from Whitington et al. [19]. By compiling PWMs from the Uniprobe, JASPAR, Hi-SELEX, and ChIP-seq data, Whitington et al. [19] collated a PWM database with 7699 high-quality PWMs. The DNA binding motifs (i.e., PWM) derived from the ChIP-seq assays were then compared with the PWMs from the PWM database compiled by Whitington et al. [19], and the matched PWMs (i.e., the one that matched the best (the most statistically significant)) were utilized to investigate whether the MDD risk SNPs were located in the binding motif of the studied TFs.
MDD risk SNPs used in this study
MDD risk SNPs identified by Wray et al. [17] were used in this study. Briefly, to identify risk variants for major depression, Wray et al. conducted a large-scale GWAS meta-analysis using 135,458 cases and 344,901 controls. By combining the subjects from PGC2 [14], deCODE [14], GenScotland [29], GERA [30], iPSYCH [31], UK Biobank [32], and 23andMe [16], Wray et al. identified 44 independent genome-wide significant (P < 5 × 10−8) risk loci for major depression. For each risk locus, a lead (or index) variant was selected to represent the association signal. Although 44 genome-wide significant loci (P < 5 × 10−8) were identified by Wray et al., only 40 lead SNPs were used in this study (as the lead variants were insertions/deletions (indels) for 4 risk loci). More detailed information about the MDD GWAS can be found in the study by Wray et al. [17].
Extraction of SNPs in LD with the reported index SNPs
Genotype data of 379 Europeans from the 1000 Genomes Project [33] (Phase I data, phase1_v3.20101123) were used to extract the SNPs that were in LD with the 40 index (or lead) SNPs (r2 > 0.3) reported by Wray et al. [17]. The LD values (r2) between the index SNPs and SNPs in LD with the index SNPs were calculated with PLINK [34]. In total, 8005 SNPs (including with the index SNPs) were obtained for downstream analysis.
Identification of risk SNPs that disrupt binding of TFs
We investigated whether the index SNPs (or SNPs in LD (r2 ≥ 0.3) the index SNPs) were located within ±50 bp of all processed ChIP-Seq peak summits (FDR ≤ 0.05). The genomic sequence containing a given SNP was scanned with the Find Individual Motif Occurrences (FIMO) software [35] (with the parameter “–thresh 0.001”) to identify the occurrences of a given PWM. The sequences surrounding (±20 bp) each studied SNP were extracted, and every genomic position in which the studied SNP overlapped with the matched PWM by at least one base pair was scored. If one or more alleles of a SNP have a FIMO log-likelihood ratio P value < 0.001, this SNP was defined as a TF binding–disrupting SNP (i.e., disrupts the PWM). More detailed information about defining and identifying the TF binding–disrupting SNPs and FIMO algorithm can be found in our previous study [23] and FIMO [35] website (http://mccb.umassmed.edu/meme/doc/fimo.html).
Brain eQTL annotation
To identify the target genes whose expression might be regulated by the TF binding–disrupting SNPs, we examined the associations between these TF binding–disrupting SNPs (i.e., with potential functional consequences) and gene expression in four brain eQTL datasets (including Common Mind Consortium (CMC) [36], Lieber Institute for Brain Development (LIBD) brain eQTL [37], brain quantitative trait locus (xQTL) [38], and Genotype-Tissue Expression Project (GTEx) [39]).
The CMC collected brain tissues from over 1150 individuals. Gene-level eQTLs (gene eQTL) used in this study were derived from the postmortem dorsolateral prefrontal cortex (DLPFC) of 467 Caucasian-inferred individuals [36] (including 209 schizophrenia cases, 206 controls, and 52 cases with affective disorder). Genotyping was conducted using the Illumina Infinium HumanOmniExpressExome array, and the gene expression level was measured with RNA sequencing (RNA-Seq). eQTL analysis was performed with the MatrixEQTL R package [40] (with the additive linear model). Detailed information about the included subjects, brain tissue collection, genotyping, gene expression quantification, quality control, and statistical analyses can be found in the original paper [36].
The LIBD brain eQTL dataset has been described in the study by Jaffe et al. [37]. Briefly, Jaffe et al. performed eQTL analyses using the DLPFC from 412 subjects (175 patients with schizophrenia and 237 unaffected controls). Genotyping was conducted using HumanHap650Y_V3, Human 1M-Duo_V3 and Omini5 BeadChips (Illumina). The gene expression level was quantified with RNA-Seq (Illumina HiSeq2000). eQTL analysis was performed using genotype data and RNA-Seq, adjusting for diagnosis, sex, ancestry (first three MDS components from the genotype data), and expression heterogeneity (principal components) [37]. Five types of transcripts were quantified, including transcripts, genes, exons, expressed regions, and junctions. Only gene-level eQTLs from LIBD were utilized in our study. Please refer to the original paper for further details on the sample collection, RNA extraction and sequencing, gene expression quantification, genotype data processing, and statistical analyses [37].
The xQTL presents a comprehensive resource by performing genome-wide xQTL analyses on multiomic data (including gene expression, DNA methylation, and histone acetylation data) derived from the DLPFC of up to 494 subjects (411 subjects have all three data types) [38]. Gene expression levels were measured with RNA-Seq, and genotyping was conducted using the Illumina OmniQuad Express platform (n = 384 individuals) and the Affymetrix Genome-Wide Human SNP Array 6.0 (n = 1709 subjects). After strict quality control, only 494 individuals were used for eQTL analysis. For detailed information, please refer to the original paper and xQTL online portal (http://mostafavilab.stat.ubc.ca/xQTLServe/) [38].
The GTEx project presents an analysis of RNA-Seq data derived from multiple tissues (including 13 brain tissues) of healthy subjects [39]. We only chose brain eQTL data in our study, and cis-eQTL analysis was carried out using FastQTL [41] with the following covariates: genotyping array platform, sex, and top three genotyping principal components. Detailed information on sample collection, genotyping, RNA-Seq, expression quantification, and statistical analyses was provided in the original paper and the GTEx website (https://gtexportal.org/home/) [39].
ASE analysis
In addition to eQTL analysis (which tests the association between gene expression level and genetic variants using multiple subjects, both genotypes and expression level data are needed for eQTL analysis), we also used ASE analysis to explore whether the TF binding–disrupting SNPs were associated with the expression level of the transcript containing different alleles of the TF binding–disrupting SNPs. By comparing the expression level of a specific transcript containing different alleles of the heterozygous site in a single individual, RNA-Seq data can be used to estimate ASE for genes (or transcripts) with transcribed polymorphic sites. Compared with eQTL analysis (which compares gene expression in individuals with different genotypes), ASE analysis is a within-individual comparison (it compares the expression level of the transcript with different alleles in a single individual).
RNA-Seq is usually used to quantify or measure the expression level of genes (or transcripts). In addition, RNA-Seq can also be used to identify heterozygous sites in the transcribed transcript (through mapping the RNA-Seq reads to the reference sequence) [42, 43]. If there is a polymorphic site in the transcript, then we can perform ASE (or allelic bias or imbalance) analysis by comparing the expression level of the transcript with different alleles at this polymorphic site (i.e., to see if one allele is expressed more highly than the other) in an individual. To detect ASE, we require a variant (usually a SNP) to be in the transcript [44]. Thus, only the heterozygous sites in the transcribed sequences (e.g., exonic regions) could be used for ASE analysis. For example, suppose that a heterozygous site (e.g., with A and C alleles) was detected (in a transcript) in an individual with SNP array or RNA-Seq. From the RNA-Seq data, we can quantify the expression level of the transcript with different alleles at this heterozygous site (e.g., the counts (i.e., RNA-Seq reads) of the transcript with the A allele were 400; however, the counts of the transcript with the C allele were only 20). Thus, the transcript with the A allele was preferentially expressed (or expressed more highly) compared with the C allele. To explore whether one allele of the TF binding–disrupting SNP was preferentially expressed compared with the other in the brain of a human individual, we downloaded ASE data from GTEx [39]. Only ASE data from brain tissues were analyzed in this study. For more detailed information on ASE analysis, please refer to a previous study [39] and the GTEx website (https://gtexportal.org/home/).
Spatio-temporal expression pattern analysis of target genes
To investigate the expression pattern of target genes in different human tissues, we downloaded the tissue RNA-Seq data from the GTEx data portal (http://gtexportal.org/) [39]. The expression values (reads per kilobase of transcript per million mapped reads (RPKM)) were used for analysis, and for a specific tissue, we calculated the median expression level of all genes in a gene set to represent the tissue expression level of the gene set.
To perform spatio-temporal expression analysis of the target gene sets, we downloaded the spatio-temporal expression RNA-Seq data of human brains across different developmental stages from BrainSpan [45] (http://www.brainspan.org/). The gene expression level was measured by RPKM. For each specific brain developmental stage, the median expression level of all the genes in a specific gene set represents the expression level of the gene set at this stage. Background gene sets were obtained from the study of Zhang et al. [46].
To explore the cell type-specific expression pattern of a gene set in single cells from brain tissues, we downloaded mouse brain single-cell data. This dataset includes single-cell expression data from multiple brain regions, such as the neocortex, hippocampus, striatum, and other brain regions, with a total of 9790 cells. We first converted our human genes into mouse orthologous genes using the R package “EWCE”. Then, we counted the number of genes with expression specificity >0.1 in a specific cell type. More details about the single-cell data and the calculation of expression specificity scores were described in the original paper of Skene et al. [47].
Reporter gene assays
DNA fragments (~400–700 bp) containing the target SNPs were inserted into the pGL4.11-basic (Promega, E6661) vector or the pGL3-promoter (Promega, E1761) vector based on the genomic locations (i.e., promoter or enhancer) of the TF binding–disrupting SNPs. If the target SNP was located in the promoter region, the pGL4.11-basic vector was used. Otherwise, the pGL3-promoter vector was used. The detailed procedures of vector construction have been described in our previous paper [23]. Briefly, the DNA fragments containing the target SNPs were amplified first using clone primers (Supplementary Table 1). Each pair of clone primers contained specific sequences (homologous to the sequence of the reporter vector, we called them homologous arms) at the 5′ end. After digesting the vectors (pGL4.11-basic vectors were digested with KpnI (Thermo Scientific, FD0524) and and HindIII (Thermo Scientific, FD0505), the pGL3-promoter vectors were digested with KpnI (Thermo Scientific, FD0524) and XhoI (Thermo Scientific, FD0694) with restriction enzymes, the PCR products containing the target SNPs were inserted into the vectors by using 2×SoSoo Mix (TSINGKE, TSV-S1). We then used the ligated vectors to transform DH5α competent cells, and Sanger sequencing was used to validate the sequence of inserted fragments. PCR-mediated mutation was utilized to obtain the vector carrying the alternative allele of the target SNP. All of the cloned sequences were validated by Sanger sequencing.
We conducted reporter gene assays using three cell lines, HEK293T, SH-SY5Y, and SK-N-SH. HEK293T, SH-SY5Y, and SK-N-SH cells were plated into 96-well plates at 3.5 × 104, 7.0 × 104, and 1.0 × 105 cells/well, respectively. After culturing for 48 h, the constructed vector (containing the test SNP) (100 ng for HEK293T, 150 ng for SH-SY5Y and SK-N-SH cells) and internal control plasmid pRL-TK (E2241, Promega) (20 ng for HEK293T, 30 ng for SH-SY5Y and SK-N-SH) were cotransfected into the tested cell lines by using Lipofectamine 3000 (Invitrogen, L3000-015). Forty-eight hours post transfection, the luciferase activity was measured by using the Dual-Luciferase Reporter Assay System (Promega, E1960) according to the manufacturer’s instructions. The fluorescence intensity (value) was read by the Luminoskan Ascent instrument (Thermo Scientific). The luciferase activities (values) were obtained from at least eight replicate wells. Student’s t test (two-tailed) was utilized to compare whether the luciferase activity in cells transfected with vectors containing different alleles of the target SNP was significantly different. The significance threshold value (for statistical inference) was set as P < 0.05.
Cell culture
The cell lines (HEK293T, SH-SY5Y, and SK-N-SH) used in this study were kindly provided by Dr ML’s laboratory (these cell lines were originally from Kunming Cell Bank, Kunming Institute of Zoology). HEK293T, SH-SY5Y, and SK-N-SH cells were cultured as described in our previous paper [23]. Briefly, high-glucose DMEM (Gibco, C11995500BT) containing 10% FBS (Gibco, 10091148) was used to culture HEK293T cells, and high-glucose DMEM (Gibco, C12430500BT) supplemented with 10% FBS, 10 mM sodium pyruvate solution (Gibco, 11360070), and 1×MEM nonessential amino acid solution (Gibco, 11140050) was used to culture SK-N-SH and SH-SY5Y cells. Antibiotics (penicillin (100 U/mL) and streptomycin (100 μg/mL) (Gibco, 15070063)) were added to the cell culture medium, and the antibiotics were withdrawn 48 h before assays. HEK293T, SH-SY5Y, and SK-N-SH cell lines were passaged at a ratio of 1:4 when their confluence reached ~90%, and the cell culture medium was changed every 2 days.
Knock down of REST
To verify whether REST regulates the expression of the target gene (i.e., NEGR1) of the identified TF binding–disrupting SNP rs3101339, we designed short hairpin RNA (shRNA) to knock down the expression level of REST. ThermoFisher shRNA design tools (http://rnaidesigner.thermofisher.com/rnaiexpress/setOption.do?designOption=shrna&pid=-3105315568901923019) were used to design shRNAs. qPCR was used to quantify REST mRNA expression levels. The targeting sequences were as follows: REST-shRNA#1, 5′-GCTGCTAATATCAACGAATCT-3′; REST-shRNA#2, and 5′-GCATCCTACTTGTCCTAATAA-3′. The 58 bp oligonucleotides (listed in Supplementary Table 2) were synthesized (by Sangon company) and annealed into double stranded DNAs. Then, the annealed shRNAs were inserted into the pLKO.1-EGFP-Puro vector by using the DNA Ligation Kit Ver2.1 (Takara, Cat. No: 6022). The DNA ligation products were used to transform Stbl3 supercompetent cells (produced by the Supercompetent Cell Preparation Kit (Beyotime, D0302) and Stbl3 (Beyotime, D0378)), and Sanger sequencing was used to validate the constructed shRNA vectors (extracted from a single colony). To obtain SH-SY5Y cells that stably expressed shRNAs, we generated lentiviruses by transfecting HEK293T cells with packaging vectors (pMD2.G (2 μg, Addgene, Cat. No: 12259) and psPAX2 (5 μg, Addgene, Cat. No: 12260)). The generated lentivirus particles were then used to infect SH-SY5Y cells, and 2 μg/mL puromycin was used to kill the cells that were not stably infected by lentiviruses for a week.
Knockout of genomic regions containing the target SNPs
To investigate whether the target genes (i.e., from eQTL analysis) of the identified regulatory SNPs (SNPs that disrupt TF binding) (i.e., rs3101339 and rs2050033) were regulated by the genomic regions containing the target SNPs, we deleted the DNA sequences containing the target SNPs using CRISPR-Cas9-mediated genome editing. For each target SNP, a pair of sgRNAs surrounding the target SNP (i.e., one sgRNA was located upstream of the target SNP (sgRNA1) and another sgRNA was located downstream of the target SNP (sgRNA2)) were designed using a CRISPR sgRNA Design Tool (https://zlab.bio/guide-design-resources) (Supplementary Table 3). The distance between the two sgRNAs was ~500 bp. The sgRNA1 and sgRNA2 were inserted into PX459M (provided by Mr Yan Ruiqing) vector and validated by Sanger sequencing. The recombinant PX459M-sgRNA1/2 vector can express the two sgRNAs and Cas9 protein simultaneously. All validated vectors used for this study were amplified by using DH5α competent cells (TaKaRa, Cat No: 9057) and extracted by using a plasmid extraction kit (TIANGEN, Cat No: DP107). HEK293T cells were used for genome editing. A total of 2.5 × 105 cells were plated into six-well plates containing 2 mL culture medium. After culturing for 18–20 h, 3 µg constructed PX459M-sgRNA1/2 vectors and control vectors were transfected into HEK293T cells by using Lipofectamine 3000 (Invitrogen, L3000-015). Forty-eight hours post transfection, 1 µg/mL puromycin was used to select the transfected cells for 3 days. The puromycin-resistant cells were then cultured for 10 days in 100 mm tissue culture-treated culture dishes at 200 cells/mL density to form monoclonal cell populations. Twelve monoclonal cell populations originating from single cells were selected and seeded into 24-well plates containing 500 µL culture medium. After culturing for 4 days, the cells were harvested and plated into 12-well plates containing 1 mL culture medium and cultured for 4 days. Then, the cells from the experimental and control groups were harvested for knockout efficiency validation and mRNA extraction. At least three independent monoclonal cell populations in each group were selected for qPCR assays.
Quantitative PCR (qPCR) analysis
Total RNA was extracted by using the TRIzol™ LS Reagent (Invitrogen, 10296028), and the PrimeScript™ RT Kit with gDNA Eraser (Takara, RR047A) was utilized to reverse transcribe 2 μg total RNA into cDNA. Reversely transcribed cDNA (1:10 dilution) was used as template to detect the expression level of the studied genes. We used TB Green™ Premix Ex Taq™ II (Tli RNaseH Plus) (Takara, RR820A) to measure the expression level of target genes. The qPCR primers utilized in this study are provided in Supplementary Table 4. We used the QuantStudio™12K Flex (Applied Biosystems) instrument and CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad) to conduct real-time qPCR. Genomic regions containing two TF binding–disrupting SNPs (rs3101339 and rs2050033) were deleted by CRISPR-Cas9-mediated genome editing. The eQTL genes of rs3101339 (NEGR1) and rs2050033 (MEI1, NHP2L1, CSDC2, and POLR3H) were quantified in normal and edited cells to investigate whether the target genes were regulated by the genomic sequences containing the studied SNPs. The ACTB gene was used to normalize the relative expression of the target gene, and the 2−ΔΔCt method was used to analyze the qPCR data [48]. All samples were run in triplicate, and the data are presented as the mea ± SD. Two-tailed Student’s t test was used to test whether the expression of the target gene was significantly different in normal and CRISPR-Cas9-edited cells. The specificity of qPCR primers was validated with RT-PCR and is shown in Supplementary Fig. 1. P < 0.05 was set as the significance threshold.
NEGR1 expression analysis in brains of MDD cases and controls
We examined NGER1 expression in the brains of MDD cases and controls using the expression data from Duric et al. [49] and Labonte et al. [50]. Briefly, Duric et al. [49] collected brain tissues from two brain regions (i.e., hippocampal tissues, the dentate gyrus and CA1 regions) of 21 MDD cases and 18 controls (matched for postmortem interval, age, and tissue pH). Gene expression was measured with MI Ready microarrays (Microarray, Inc.) (to detect human whole-genome expression), and expression data were analyzed with the Bioconductor package and R language. More detailed information about this study has been described in the study of Duric et al. [49]. Labonte et al. [50] collected brain tissues from 26 MDD cases and 22 controls. Six brain regions were isolated by Labonte et al., including the DLPFC, the ventromedial prefrontal cortex, the orbitofrontal cortex, the NAc, the anterior insula, and the ventral subiculum. Gene expression was quantified with RNA-Seq (Illumina HiSeq2500). The differentially expressed genes were identified for each region. To compare the differences between males and females, Labonte et al. [50] performed sex-specific analysis and identified the differentially expressed genes in male and female MDD cases and controls in each brain region. Detailed information about the study of Labonte et al. can be found in the original paper [50].
Results
Identification of TF binding–disrupting SNPs from the MDD risk loci
We carried out functional genomics analysis to identify the functional SNPs (or potential causal SNPs) from 40 of the 44 reported MDD risk loci [17] (as the index (or lead) variant for 4 loci was not SNP, we could not extract the SNPs in LD with these four lead variants) (Fig. 1). Briefly, we first processed 34 ChIP-seq data (as MDD is a psychiatric disorder that is mainly attributed to the dysfunction of the central nervous system, only ChIP-seq assays performed in neuronal cells or brain tissues were used in this study) to obtain the binding motifs of the included TFs (Fig. 1 and Supplementary Table 5). Through calling the ChIP-seq peaks (using MACS [27]) and running motif discovery on the called ChIP-seq peaks (MEME [28]), we derived binding motifs of 30 TFs after stringent quality control (peaks of four TFs were excluded due to the low quality). The derived binding motifs were then compared with the motifs from the well-characterized PWM database (compiled by Whitington et al. [19]), and the matched PWMs were used for further analysis. To pinpoint the functional SNPs (i.e., SNPs that disrupt the binding of the analyzed TFs) from the 40 of the 44 reported MDD risk loci [17], we extracted the SNPs that were in LD (r2 > 0.3) with the 40 index SNPs. A total of 8005 SNPs (including the 40 index SNPs and SNPs that were in LD with the 40 index SNPs) were obtained (Supplementary Table 6). We mapped these SNPs to the matched motifs to investigate whether these SNPs are located in the binding motif of the analyzed TFs and whether allelic differences at these SNPs affect the binding of the analyzed TFs [35]. In total, we identified 34 SNPs that disrupted the binding of 15 TFs (Fig. 1 and Supplementary Table 7). We found that 11 MDD risk SNPs disrupted CTCF binding (Fig. 2a). In addition, there were eight SNPs that disrupted the binding of two or more TFs (Fig. 2b). Of note, 75% of TF binding–disrupting SNPs reside in intronic and intergenic regions (Fig. 2c).

ChIP-seq assays performed using neuronal cell lines and brain tissues were used for peak calling and motif discovery. The identified motifs were then compared with the PWMs from the PWM database (see “Methods”), and the matched motifs were used for downstream analysis. SNPs in linkage disequilibrium (LD, r2 ≥ 0.3) with the index SNPs (identified by MDD GWAS, a total of 40 index SNPs) were extracted. A total of 8005 SNPs from 40 MDD risk loci (as the index (or lead) variant was not SNP for 4 loci, we could not extract the SNPs in LD with these four variants) were mapped to the identified motifs to investigate whether these SNPs disrupt the binding of transcription factors. In total, 34 TF binding–disrupting SNPs were identified.

a Left panel: the number of SNPs that disrupt binding of a specific transcription factor. Right panel: the distribution of the TF binding–disrupting SNPs in the human genome. Most (75%) of the TF binding–disrupting SNPs were located in intronic and intergenic regions. b Heatmap shows the number of SNPs that disrupt the binding of two or more TFs. c The distribution of TF binding–disrupting SNPs in different genomic regions.
Validation of the regulatory effects of the TF binding–disrupting SNPs with reporter gene assays
Our functional genomics identified 34 TF binding–disrupting MDD risk SNPs (these TF binding–disrupting SNPs were hereafter called regulatory SNPs). To verify the regulatory effects of these regulatory SNPs (i.e., TF binding–disrupting SNPs), we performed reporter gene assays. We tested whether the different alleles of the binding–disrupting SNPs affect luciferase activity in three cell lines (i.e., HEK293T, SH-SY5Y, and SK-N-SH). Among the 34 tested regulatory SNPs, we found that allelic differences in 29 regulatory SNPs altered the luciferase activity significantly in at least one tested cell line (Supplementary Table 8, Figs. 3, 4, and Supplementary Figs. 2, 3). Notably, different alleles of 13 regulatory SNPs significantly affected the luciferase activity in all three tested cell lines (Figs. 3, 4, and Supplementary Table 8). Taken together, these results demonstrated the regulatory effects of most of the identified TF binding–disrupting SNPs.

a The constructs containing the G allele of rs1467013 exhibited significantly higher luciferase activities than the constructs containing the A allele in all three tested cell lines. b The reporter vectors containing the T allele of rs159961 showed significantly higher luciferase activities than the A allele in all three tested cell lines. c The G allele of rs7554486 conferred significantly higher luciferase activity than the A allele in all three tested cell lines. d The constructs carrying the G allele of rs1950834 exhibited significantly higher luciferase activities than the constructs carrying the A allele in all three tested cell lines. e The reporter vectors containing the G allele of rs1111177 showed significantly higher luciferase activity than the A allele in all three tested cell lines. f The A allele of rs2924321 conferred significantly higher luciferase activity than the G allele in all three tested cell lines. Student’s t test (two-tailed) was used to detect if the differences were significant. n = 8 for the control group, n = 16 for each experimental group. *P < 0.05, **P < 0.01, ***P < 0.001.

Different alleles of the TF binding–disrupting SNPs listed in this figure conferred significant differences in luciferase activity in at least one tested cell line. a The C allele of rs10756277 conferred significantly higher luciferase activities than T allele in all three tested cell lines. b The reporter vectors containing the C allele of rs77498214 showed significantly higher luciferase activities than the A allele in HEK293T and SH-SY5Y cells. However, in SK-N-SH cells, the A allele of rs77498214 conferred significantly higher luciferase activities than C allele. c The A allele of rs301792 conferred significantly higher luciferase activity than the G allele in SH-SY5Y and SK-N-SH cell lines. d The constructs carrying the C allele of rs2403202 exhibited significantly higher luciferase activities than the constructs carrying the G allele in SH-SY5Y cells. e The reporter vectors containing the C allele of rs12607673 showed significantly higher luciferase activity than the T allele in SH-SY5Y and SK-N-SH cell lines. f The C allele of rs12605603 conferred significantly higher luciferase activity than the G allele in SK-N-SH cells. g The reporter vectors containing the T allele of rs12607674 showed significantly higher luciferase activity than the C allele in HEK293T and SH-SY5Y cells. h The reporter vectors containing the C allele of rs132792 showed significantly higher luciferase activity than the T allele in SH-SY5Y and SK-N-SH cells. Student’s t test (two-tailed) was used to compare if the differences were significant. n = 8 for the control group, n = 16 for each experimental group. *P < 0.05, **P < 0.01, ***P < 0.001.
Disruption of FOSL2, EP300, and JUND binding by rs9262142
Our functional genomics identified 34 TF binding–disrupting SNPs. We validated the regulatory effects of 29 SNPs with reporter gene assays, implying that these SNPs might have functional consequences by affecting the binding of TFs. To further characterize how the identified regulatory SNPs disrupt TF binding, we investigated a regulatory SNP (i.e., rs9212642) at 6p22.1. SNP rs9262142 is located in the binding motifs of three TFs, including FOSL2, EP300, and JUND (Fig. 5a–c). ChIP-seq data showed that the genomic sequence containing rs9262142 was marked by ChIP-seq peaks of FOSL2, EP300, and JUND (Fig. 5d), indicating that these three TFs could bind the genomic sequence containing rs9262142 in neuronal cells. In addition, we found that rs9262142 is located in a genomic region marked by DNase-seq signals (Fig. 5d), indicating that rs9262142 is located in a transcriptionally active region in neuronal cells. Notably, histone modification data also revealed that rs9262142 is located in a genomic region with high transcriptional activity in neuronal cells (Fig. 5d). Finally, reporter gene assays indicated that the constructed vectors carrying the G allele of rs9262142 exhibited significantly higher luciferase activities compared with the vectors carrying the A allele of rs9262142 in all three tested cell lines (P < 0.05, Fig. 5e–g). Taken together, these results indicate that rs9262142 is a functional SNP that disrupts the binding of FOSL2, EP300, and JUND.

a–c rs9262142 is located in the binding motifs of FOSL2, EP300, and JUND TFs. d rs9262142 is located in a genomic region marked by signals of DNase-seq, ChIP-Seq, and histone modifications, indicating that the genomic region containing rs9262142 was actively transcribed in neuronal cells. e–g Reporter gene assays validated the regulatory effect of rs9262142. The constructs containing the G allele of rs9262142 exhibited significantly higher luciferase activities in all three tested cell lines. The Two-tailed Student’s t test was used to detect if the differences were significant. n = 8 for the control group, n = 16 for each experimental group. *P < 0.05, ***P < 0.001.
Disruption of SMC3 and CTCF binding by rs3812986
In addition to rs9262142, we also investigated rs3812986, a SNP that disrupts the binding of SMC3 and CTCF (Fig. 6a, b). We found that rs3812986 is located in a genomic region marked by DNase-seq signal (Fig. 6c), indicating that the genomic region containing rs3812986 was actively transcribed in neuronal cells. ChIP-seq data showed that SMC3 and CTCF can bind the genomic region containing rs3812986 in neuronal cells. We further verified the regulatory effect of rs3812986 using reporter gene assays. Our reporter gene assays showed that the C allele of rs3812986 conferred significantly higher luciferase activity compared with the T allele in all three tested cell lines (Fig. 6d–f). These results demonstrated the regulatory effect of rs3812986.

a, b rs3812986 disrupts the binding of SMC3 and CTCF TFs. c rs3812986 is located in a region marked by signals of DNase-seq, indicating that rs3812986 is located in a genomic region with a high degree of transcription activity in neuronal cells. ChIP-seq data showed that the transcription factors SMC3 and CTCF bound to the genomic region containing rs3812986 in neuronal cells. d–f Reporter gene assays validated the regulatory effect of rs3812986. The luciferase activities of the constructs with the C allele of rs3812986 were significantly higher than those of the constructs with the T allele in all three tested cell lines. Student’s t test (two-tailed) was used to detect if the differences were significant. n = 8 for the control group, n = 16 for each experimental group. ***P < 0.001.
Disruption of RAD21 binding by rs2919451
We further studied rs2919451, a SNP that disrupts the binding of RAD21 (Fig. 7a). Similar to rs9262142 and rs3812986, we found that the genomic sequence surrounding rs2919451 was marked by DNase-seq and ChIP-seq signals, indicating that rs2919451 is located in a transcriptionally active region with RAD21 binding (Fig. 7b). We further verified the regulatory effect of rs2919451 with reporter gene assays. We found that the reporter constructs containing the T allele (of rs2919451) exhibited higher luciferase activities compared with the constructs carrying the C allele of rs2919451 in all three tested cell lines (Fig. 7c–e). These results indicated that rs2919451 is a functional SNP with a potential regulatory effect.

a rs2919451 is located in the binding motif of RAD21. b rs2919451 is located in a genomic region marked by signals of ChIP-seq (RAD21) and DNase-seq, indicating that rs2919451 is located in an actively transcribed genomic region in neuronal cells. c–e Reporter gene assays validated the regulatory effect of rs2919451. The constructs containing the T allele of rs2919451 exhibited significantly higher luciferase than the C allele in all three tested cell lines. Student’s t test (two-tailed) was used to detect if the differences were significant. n = 8 for the control group, n = 16 for each experimental group. ***P < 0.001. f–h Allele-specific expression (ASE) analysis showed that different alleles of rs8233, rs132792, and rs1054080 exhibited significant preferential expression in human brain tissues, further supporting the regulatory effects of these SNPs.
Validation of the regulatory effects of the TF binding–disrupting SNPs with ASE analysis
Our reporter gene assays validated the regulatory effects of most of the TF binding–disrupting SNPs (29 out of 34 SNPs). To further verify whether the identified TF binding–disrupting SNPs were associated with the expression level of transcripts, we used ASE data from the GTEx [39]. Among the 34 TF binding–disrupting SNPs, three SNPs had ASE data in brain tissues from GTEx (as ASE analysis requires that the TF binding–disrupting SNP was heterozygous and located in the transcribed region in a single individual, only a small percentage of the identified TF binding–disrupting SNPs can be used for ASE analysis). ASE analysis showed that three regulatory SNPs (rs8233, rs132792, and rs1054080) also exhibited significant ASE in brain tissues from the GTEx (Fig. 7f–h). The expression level (i.e., read counts from RNA-Seq) of the transcript with the A allele of rs8233 was higher than that of the transcript with the G allele (Fig. 7f). The expression level of the transcript with the T allele of rs132792 was significantly higher than that of the transcript carrying the C allele (Fig. 7g). For rs1054080, we noticed that all of the detected transcripts carried the C allele (though this individual was heterozygous at rs1054080), indicating that the transcript with the C allele was preferentially expressed compared with the A allele (Fig. 7h). Collectively, these results further supported the potential regulatory effect of these identified TF binding–disrupting SNPs.
Validation of the regulatory effects of rs3101339 with CRISPR-Cas9-mediated genome editing
Our reporter gene assays and ASE analysis revealed the regulatory effects of the identified TF binding–disrupting SNPs. To further investigate how the identified TF binding–disrupting SNPs regulate gene expression, we studied rs3101339, a SNP that disrupts the binding of the REST (RE1-silencing TF) TF (Fig. 8a). Our functional genomics analysis and PWM data showed that rs3101339 was located in the binding motif of REST (Fig. 8a), a neuron-restrictive silencing factor. DNase-seq data revealed that rs3101339 was located in a transcriptionally active region in neuronal cells (Fig. 8b). ChIP-seq data showed that REST can bind to the genomic region containing rs3101339 (Fig. 8b). Consistent with the DNase-seq and ChIP-seq data, histone modification data showed that the genomic region containing rs3101339 was enriched in H3K4me3 signals (Fig. 8b), further supporting that rs3101339 was located in a genomic region with a high level of transcriptional activity. We further verified the regulatory effect of rs3101339 with reporter gene assays (Fig. 8c–e). The reporter vector containing the C allele of rs3101339 exhibited significantly higher luciferase activity than the vector containing the A allele in all three tested cell lines (Fig. 8c–e). These results indicate that rs3101339 is a functional SNP.

a rs3101339 is located in the binding motif of the REST transcription factor, and different alleles of rs3101339 disrupt the binding of REST. b rs3101339 is located in a genomic region marked by signals of DNase-seq, ChIP-seq (REST), and histone modifications, indicating that rs3101339 is located in a genomic region with a high degree of transcriptional activity in neuronal cells. c–e Reporter gene assays validated the regulatory effect of rs3101339. The C allele of rs3101339 conferred significantly higher luciferase activity than the A allele in all three tested cell lines. f eQTL analysis showed that rs3101339 was significantly associated with NEGR1 expression in human brain tissues. Consistent with the results of reporter gene assays, NEGR1 expression in individuals with the CC genotype was significantly higher than that in individuals with the AA genotype. g, h REST knockdown downregulated NEGR1 expression, indicating that NEGR1 expression was regulated by REST. i, j CRISPR-Cas9-mediated knockout of the genomic region containing rs3101339 resulted in significant upregulation of NEGR1 expression, suggesting that the genomic region containing rs3101339 regulates NEGR1 expression. i Electrophoresis showed the successful deletion of the genomic sequence containing rs3101339. WT indicates the length of PCR products (690 bp) containing rs3101339 in wild-type cells. KO indicates the length of DNA fragments (126 bp) containing rs3101339 in edited cells (as the genomic sequence (564 bp) containing rs3101339 was deleted, the length of PCR products was decreased compared with wild-type cells). RNA extracted from cells marked by red arrowheads was used for qPCR analysis. n = 8 for the control group in c–e, n = 16 for each experimental group in c–e, n = 3 for g, h, n = 3 for the control group in j and n = 4 for the knockout group in j. Student’s t test (two-tailed) was used to detect if the differences were significant. **P < 0.01, ***P < 0.001.
Our above data revealed that rs3101339 is a functional SNP with a potential regulatory effect. To further identify the gene (or genes) regulated by rs3101339, we performed eQTL analysis. We found that rs3101339 was significantly associated with NGER1 expression in the xQTL (P = 3.46 × 10−19) and GTEx (P = 4.19 × 10−7) datasets (Fig. 8f). The eQTL data showed that NERG1 expression in individuals with the AA genotype was significantly lower than that in individuals with the CC genotype, which was consistent with the reporter gene assays (Fig. 8c–e). Notably, rs3101339 is located in the promoter region of NGER1 (447 bp upstream of the NEGR1 transcription start site, based on the UCSC genome browser) (Supplementary Fig. 4), a region with multiple SNPs showed a significant association with MDD (Supplementary Fig. 5). These results (including PWM, ChIP-Seq, DNase-Seq, histone modification, eQTL analysis and SNP location) collectively suggest that rs3101339 may regulate NEGR1 by affecting REST binding and NEGR1 promoter activity. To investigate whether NEGR1 was regulated by REST, we knocked down REST using shRNAs. We found that REST knockdown resulted in significant downregulation of NEGR1 in SH-SY5Y cells (Fig. 8g, h), indicating that the expression of NEGR1 was regulated by the REST TF. We further validated the regulation of NEGR1 by rs3101339 using CRISPR-Cas9-mediated genome editing. The genomic sequence (586 bp) containing rs3101339 was knocked out by a pair of sgRNAs (Fig. 8i and Supplementary Fig. 4). Compared with the control cells, we found that NEGR1 expression was significantly upregulated in rs3101339 knockout cells (Fig. 8j), indicating that the expression of NEGR1 was regulated by the genomic sequence containing rs3101339. Taken together, these results indicate that rs3101339 is a functional SNP that regulates the expression of the NEGR1 gene.
Validation of the regulatory effects of rs2050033 with CRISPR-Cas9-mediated genome editing
In addition to rs3101339, we also validated the regulatory effect of rs2050033 using CRISPR-Cas9-mediated genome editing. The reasons that we selected rs2050033 were as follows: first, our motif analysis and PWM data showed that rs2050033 is located in the binding motif of the CTCF TF, and allelic differences at rs2050033 disrupt the binding of CTCF (Fig. 9a). Second, DNase-seq data revealed that rs2050033 is located in a genomic region with high transcription activity in neuronal cells (Fig. 9b). Third, ChIP-seq data showed that CTCF bound to the genomic sequence containing rs2050033 (Fig. 9b). Fourth, reporter gene assays supported the regulatory effects of rs2050033. We found that the G allele of rs2050033 conferred significantly higher luciferase activity than the T allele in all of the tested cell lines (P < 0.05, Fig. 9c–e). Fifth, to identify the target gene (or genes) that might be regulated by rs2050033, we conducted eQTL analysis using the data from the CMC [36] and LIBD [37]. We found that rs2050033 was significantly associated with the expression levels of CDCS2, POLR3H, NPH2L1, TOB2, MEI1, and CENPM (Fig. 9f–m), implying that these genes were potential target genes of rs2050033. These convergent lines of evidence suggest that rs2050033 is a functional SNP.

a rs2050033 disrupts the binding of the CTCF transcription factor. b CTCF ChIP-seq and DNase-seq data showed that rs2050033 is located in a genomic region marked by ChIP-seq and DNase-seq signals, indicating that rs2050033 is located in an actively transcribed region and CTCF bound to the genomic sequence containing rs2050033 in neuronal cells. c–e Reporter gene assays verified the regulatory effect of rs2050033. In all three tested cell lines, the constructs containing the G allele of rs2050033 exhibited significantly higher luciferase activities compared with the T allele. f–m eQTL analysis showed that rs2050033 was significantly associated with the expression of CSDC2, POLR3H, NPH2L1, TOB2, MEI1, and CENPM in human brain tissues. f–i eQTL data were from the CMC brain eQTL dataset. j–m eQTL data were from the LIBD brain eQTL dataset. n–r Deletion of the DNA sequence containing rs2050033 led to significant expression changes in CSDC2, POLR3H, MEI1, and NPH2L1, suggesting that the genomic region containing rs2050033 regulates the expression of these genes. i Electrophoresis showed the successful deletion of the genomic sequence containing rs2050033. WT indicates the length of PCR fragments (878 bp) containing rs2050033 in wild-type cells. KO indicates the length of DNA fragments (375 bp) containing rs2050033 in edited cells. RNA extracted from cells marked by red arrowheads was used for qPCR analysis. Student’s t test (two-tailed) was used to detect if the differences were significant. *P < 0. 05, ***P < 0.001. n = 8 for the control group in reporter gene assays (i.e., c–e), n = 16 for each experimental group in reporter gene assays (i.e., c–e), n = 4 for o–r.
To further verify whether these genes are regulated by rs2050033, we knocked out the genomic region containing rs2050033 using CRIPSR-Cas9-mediated genome editing (Fig. 9n). We found that the expression of CSDC2, POLR3H, MEI1, and NPH2L1 was significantly changed in rs2050033 knockout cells compared with control cells (Fig. 9o–r), indicating that these genes were regulated by the genomic region containing rs2050033. Collectively, these results validated that rs2050033 is a functional SNP.
Identification of the potential target genes of the TF binding–disrupting SNPs
Our serial experiments showed that the identified regulatory SNPs (i.e., TF binding–disrupting SNPs) were functional. To further explore the target genes that might be regulated by these regulatory SNPs, we examined the associations between these regulatory SNPs and gene expression in four independent brain eQTL datasets (i.e., CMC [36], LIBD [37], xQTL [38], and GTEx [51]. Detailed information on sample information, brain tissue isolation, RNA extraction and quantification, genotyping, and eQTL analysis can be found in “Methods” and original papers). Among the 34 TF binding–disrupting SNPs, we found that 29 SNPs showed significant association with gene expression in human brains in at least one brain eQTL dataset (Supplementary Table 9). Of note, we noticed that 17 identified regulatory SNPs were associated with the expression of the same gene in at least two independent brain eQTL datasets (Table 1), implying that these genes might be regulated by these TF binding–disrupting SNPs. Collectively, these results suggest that the identified TF binding–disrupting SNPs may confer MDD risk by regulating these target genes.
Spatio-temporal expression pattern of target genes
We explored the spatio-temporal expression pattern of target genes (whose expression levels were associated with the identified TF binding–disrupting SNPs). Two gene sets were used. Genes from Supplementary Table 9 were defined as gene set 1, and genes from Table 1 were defined as gene set 2. We found that the expression levels of target genes (including gene sets 1 and 2) were higher than those of background genes in human brain tissues (Supplementary Fig. 6). In addition, the expression levels of target genes (including gene sets 1 and 2) were higher than background genes across different developmental stages of the human brain (Supplementary Fig. 7). Further expression analysis using the single-cell RNA-Seq results revealed that target genes were highly expressed in pyramidal cells (Supplementary Fig. 8). These results suggest that the target genes may play a role in pyramidal cells.
Dysregulation of NEGR1 in MDD
Our functional genomics showed that the functional SNP rs3101339 may confer risk of MDD by regulating NEGR1 expression (Fig. 8). To further explore the role of NGER1 in MDD, we examined NEGR1 expression in the brains of MDD cases and controls using expression data from studies by Duric et al. [49] and Labonte et al. [50]. We found that NEGR1 was significantly downregulated in the CA1 (reduced by 35%, P = 0.004) and dentate gyrus regions (reduced by 76%, P = 0.002) of MDD cases compared with controls in the study of Durin et al. [49] (N = 21 MDD cases and N = 18 controls). Consistently, we found that NEGR1 was also significantly downregulated in the NAc of female MDD cases compared with controls in the study of Labonte et al. (N = 12 female MDD cases and N = 9 female controls) (reduced by 15%, P = 0.038) (Supplementary Fig. 9). NEGR1 expression was not changed in other brain regions in the study of Labonte et al. These expression results suggested that dysregulation of NGER1 may have a role in MDD. Interestingly, Szczurkowska et al. [52] found that downregulation of NEGR1 affected neuronal migration and spine density, implying that NEGR1 may confer risk of MDD by affecting dendritic spines.
Discussion
We have witnessed the rapid progress of genetic studies of MDD during the past five years. Since the identification of two genome-wide significant MDD risk loci by whole-genome sequencing in 2015 [15], over 100 risk loci for MDD have been identified by GWAS [16,17,18] during the past 5 years. Despite the fact that new risk loci are identified at an unprecedented rate, pinpointing the causal risk variants from the reported risk loci and elucidating the role of causal variants in the pathophysiology of MDD remain major challenges in the post-GWAS era. In this study, we systematically investigated the regulatory mechanisms of MDD risk variants. We identified 34 SNPs that disrupt the binding of 15 TFs using a functional genomics approach. Our reporter gene assays validated that most of the identified TF binding–disrupting SNPs were functional. ASE analysis and CRISPR-Cas9-mediated genome editing further supported the regulatory effects of the identified TF binding–disrupting SNPs. Interestingly, we found that most of the identified TF binding–disrupting SNPs were located in intronic and intergenic regions. The ChIP-seq, PWM, DNase-seq signal, and histone modification profiling of the 34 TF binding–disrupting SNPs are provided in Supplementary Figs. 10–43. These lines of convergent evidence support that these identified regulatory SNPs may contribute to MDD risk by regulating gene expression.
In addition to the discovery of 34 regulatory SNPs, we also identified the target genes that might be regulated by these regulatory SNPs using eQTL analysis. Our eQTL analysis showed that most of the identified regulatory SNPs (29 out of 34 TF binding–disrupting SNPs) were associated with gene expression in human brain tissues. Considering the regulatory effects of these TF binding–disrupting SNPs, it is likely that the identified regulatory SNPs exert their biological effects by regulating these target genes. More work is needed to elucidate the roles and mechanisms of these target genes in MDD pathophysiology.
We noticed that approximately one-third (11 out of 34) of TF binding–disrupting SNPs were located in the CTCF binding motif, implying that disruption of CFCT binding may represent a common mechanism of MDD risk variants. CTCF is a conserved zinc-finger protein that plays pivotal roles in transcriptional regulation [53, 54]. Recent studies have shown the important role of CTCT in 3D genome organization [54,55,56,57].
Notably, expression analysis showed that NEGR1 (regulated by the REST binding–disrupting SNP rs3101339) was significantly downregulated in MDD cases compared with controls. NEGR1 encodes neural cell adhesion molecule neuronal growth regulator 1, a protein that has pivotal roles in regulating cortical development, dendritic spine density, and neuronal morphological maturation [52, 58]. Increasing evidence suggests the dysfunction of dendritic spines in MDD [59, 60]. Decreased expression of synapse-related genes and loss of synapses were observed in MDD [60]. Interestingly, sustained rescue of lost synapses could relieve depression-like behavioral states [61]. These findings suggest that NEGR1 may play pivotal roles in MDD by regulating the density and morphology of dendritic spines. Intriguingly, recent studies also showed that Negr1 deficiency resulted in behavioral abnormalities related to psychiatric disorders [52, 62], further supporting the potential role of NEGR1 in MDD pathogenesis. Collectively, these lines of evidence suggest that rs3101339 may confer risk of MDD by regulating NGER1 expression.
We noticed that the risk allele (i.e., C allele) of rs3101339 was associated with higher reporter gene activity and NEGR1 expression (Fig. 8c–f). Based on these data, NEGR1 expression was predicted to be upregulated in MDD cases compared with controls. However, we found that NEGR1 expression was downregulated in MDD cases compared with controls. A possible reason for this inconsistency is that rs3101339 and other functional variants act synergistically to regulate NEGR1 expression. In addition, considering that the samples used for eQTL analysis and gene expression analysis were not the same, and the sample size used for differential expression analysis was relatively small, the number of individuals with the rs3101339 AA genotype in MDD cases might be more than in controls (which may also lead to the observation of significant downregulation of NEGR1 in MDD cases). Finally, treatment with antidepressants might also change gene expression. Thus, the expression level of NEGR1 observed in MDD cases might not exactly reflect the genetic effects of rs3101339. More work is needed to investigate how rs3101339 confers MDD risk by modulating NEGR1 expression.
In summary, we pinpointed 34 functional SNPs (i.e., TF binding–disrupting SNPs) from the 44 reported MDD risk loci [17], and we elucidated the regulatory mechanisms of some functional SNPs. These regulatory SNPs may represent potential causal variants for MDD, as they disrupt the binding of corresponding TFs. More importantly, we verified the regulatory effects of these regulatory SNPs and identified the potential target genes regulated by these SNPs. Further functional characterization of these functional SNPs and target genes will help elucidate the genetic mechanisms and pathogenesis of MDD. Finally, our findings also provide potential targets for the development of new drugs for MDD.
URLs
MDD GWAS data from PGC2, http://www.med.unc.edu/pgc/; CMC, http://www.synapse.org/CMC; GTEx, https://gtexportal.org/home/; LIBD brain eQTL browser, http://eqtl.brainseq.org/phase1/eqtl/; ENCODE, https://www.encodeproject.org/; FastQC, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/; Cutadapt, https://cutadapt.readthedocs.io/en/stable/index.html; FIMO, http://meme-suite.org/tools/fimo; MACS, http://liulab.dfci.harvard.edu/MACS/; Bowtie, http://bowtie-bio.sourceforge.net/index.shtml; MEME, http://meme-suite.org/tools/meme; BrainSpan, http://www.brainspan.org/; The 1000 Genomes Project, http://www.1000genomes.org/; PLINK, http://zzz.bwh.harvard.edu/plink/; UCSC Genome Browser, http://genome.ucsc.edu/
References
Sullivan PF, Daly MJ, O’Donovan M. Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat Rev Genet. 2012;13:537–51.
Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA. 2003;289:3095–105.
Otte C, Gold SM, Penninx BW, Pariante CM, Etkin A, Fava M, et al. Major depressive disorder. Nat Rev Dis Prim. 2016;2:16065.
Bromet E, Andrade LH, Hwang I, Sampson NA, Alonso J, de Girolamo G, et al. Cross-national epidemiology of DSM-IV major depressive episode. BMC Med. 2011;9:90.
Chesney E, Goodwin GM, Fazel S. Risks of all-cause and suicide mortality in mental disorders: a meta-review. World Psychiatry. 2014;13:153–60.
Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJ. Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data. Lancet. 2006;367:1747–57.
Ferrari AJ, Charlson FJ, Norman RE, Patten SB, Freedman G, Murray CJ, et al. Burden of depressive disorders by country, sex, age, and year: findings from the global burden of disease study 2010. PLoS Med. 2013;10:e1001547.
Global Burden of Disease Study 2013 Collaborators Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;386:743–800.
Vos T, Flaxman AD, Naghavi M, Lozano R, Michaud C, Ezzati M, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2010;380:2163–96.
Greenberg PE, Fournier AA, Sisitsky T, Pike CT, Kessler RC. The economic burden of adults with major depressive disorder in the United States (2005 and 2010). J Clin Psychiatry. 2015;76:155–62.
Li M, D’Arcy C, Meng X. Maltreatment in childhood substantially increases the risk of adult depression and anxiety in prospective cohort studies: systematic review, meta-analysis, and proportional attributable fractions. Psychol Med. 2016;46:717–30.
American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. Arlington: American Psychiatric Publishing; 2013.
Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry. 2000;157:1552–62.
Ripke S, Wray NR, Lewis CM, Hamilton SP, Weissman MM, Breen G, et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry. 2013;18:497–511.
CONVERGE consortium*. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature. 2015;523:588–91.
Hyde CL, Nagle MW, Tian C, Chen X, Paciga SA, Wendland JR, et al. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat Genet. 2016;48:1031–6.
Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet. 2018;50:668–81.
Howard DM, Adams MJ, Clarke TK, Hafferty JD, Gibson J, Shirali M, et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat Neurosci. 2019;22:343–52.
Whitington T, Gao P, Song W, Ross-Adams H, Lamb AD, Yang Y, et al. Gene regulatory mechanisms underpinning prostate cancer susceptibility. Nat Genet. 2016;48:387–97.
The ENCODE Project Consortium*. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74.
Kim YK, Won E. The influence of stress on neuroinflammation and alterations in brain structure and function in major depressive disorder. Behav Brain Res. 2017;329:6–11.
Won E, Kim YK. Stress, the autonomic nervous system, and the immune-kynurenine pathway in the etiology of depression. Curr Neuropharmacol. 2016;14:665–73.
Huo Y, Li S, Liu J, Li X, Luo XJ. Functional genomics reveal gene regulatory mechanisms underlying schizophrenia risk. Nat Commun. 2019;10:670.
Kong Y. Btrim: a fast, lightweight adapter and quality trimming program for next-generation sequencing technologies. Genomics. 2011;98:152–3.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–12.
Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25.
Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-seq (MACS). Genome Biol. 2008;9:R137.
Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:28–36.
Fernandez-Pujals AM, Adams MJ, Thomson P, McKechanie AG, Blackwood DH, Smith BH, et al. Epidemiology and heritability of major depressive disorder, stratified by age of onset, sex, and illness course in generation Scotland: Scottish Family Health Study (GS:SFHS). PLoS ONE. 2015;10:e0142197.
Banda Y, Kvale MN, Hoffmann TJ, Hesselson SE, Ranatunga D, Tang H, et al. Characterizing race/ethnicity and genetic ancestry for 100,000 subjects in the genetic epidemiology research on adult health and aging (GERA) cohort. Genetics. 2015;200:1285–95.
Pedersen CB, Bybjerg-Grauholm J, Pedersen MG, Grove J, Agerbo E, Baekvad-Hansen M, et al. The iPSYCH2012 case-cohort sample: new directions for unravelling genetic and environmental architectures of severe mental disorders. Mol Psychiatry. 2018;23:6–14.
Allen NE, Sudlow C, Peakman T, Collins R. UK biobank data: come and get it. Sci Transl Med. 2014;6:224ed224.
The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526:68–74.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011;27:1017–8.
Fromer M, Roussos P, Sieberts SK, Johnson JS, Kavanagh DH, Perumal TM, et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat Neurosci. 2016;19:1442–53.
Jaffe AE, Straub RE, Shin JH, Tao R, Gao Y, Collado-Torres L, et al. Developmental and genetic regulation of the human cortex transcriptome illuminate schizophrenia pathogenesis. Nat Neurosci. 2018;21:1117–25.
Ng B, White CC, Klein HU, Sieberts SK, McCabe C, Patrick E, et al. An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nat Neurosci. 2017;20:1418–26.
The GTEx Consortium. The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–60.
Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012;28:1353–8.
Ongen H, Buil A, Brown AA, Dermitzakis ET, Delaneau O. Fast and efficient QTL mapper for thousands of molecular phenotypes. Bioinformatics. 2016;32:1479–85.
Piskol R, Ramaswami G, Li JB. Reliable identification of genomic variants from RNA-seq data. Am J Hum Genet. 2013;93:641–51.
Castel SE, Levy-Moonshine A, Mohammadi P, Banks E, Lappalainen T. Tools and best practices for data processing in allelic expression analysis. Genome Biol. 2015;16:195.
Khansefid M, Pryce JE, Bolormaa S, Chen Y, Millen CA, Chamberlain AJ, et al. Comparing allele specific expression and local expression quantitative trait loci and the influence of gene expression on complex trait variation in cattle. BMC Genomics. 2018;19:793.
Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–9.
Zhang Q, Nogales-Cadenas R, Lin JR, Zhang W, Cai Y, Vijg J, et al. Systems-level analysis of human aging genes shed new light on mechanisms of aging. Hum Mol Genet. 2016;25:2934–47.
Skene NG, Bryois J, Bakken TE, Breen G, Crowley JJ, Gaspar HA, et al. Genetic identification of brain cell types underlying schizophrenia. Nat Genet. 2018;50:825–33.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–8.
Duric V, Banasr M, Licznerski P, Schmidt HD, Stockmeier CA, Simen AA, et al. A negative regulator of MAP kinase causes depressive behavior. Nat Med. 2010;16:1328–32.
Labonte B, Engmann O, Purushothaman I, Menard C, Wang J, Tan C, et al. Sex-specific transcriptional signatures in human depression. Nat Med. 2017;23:1102–11.
The GTEx Consortium. The genotype-tissue expression (GTEx) project. Nat Genet. 2013;45:580–5.
Szczurkowska J, Pischedda F, Pinto B, Manago F, Haas CA, Summa M, et al. NEGR1 and FGFR2 cooperatively regulate cortical development and core behaviours related to autism disorders in mice. Brain. 2018;141:2772–94.
Merkenschlager M, Nora EP. CTCF and cohesin in genome folding and transcriptional gene regulation. Annu Rev Genom Hum Genet. 2016;17:17–43.
Zheng H, Xie W. The role of 3D genome organization in development and cell differentiation. Nat Rev Mol Cell Biol. 2019;20:535–50.
Dekker J, Mirny L. The 3D genome as moderator of chromosomal communication. Cell. 2016;164:1110–21.
Heinz S, Texari L, Hayes MGB, Urbanowski M, Chang MW, Givarkes N, et al. Transcription elongation can affect genome 3D structure. Cell. 2018;174:1522–36.
Rowley MJ, Corces VG. Organizational principles of 3D genome architecture. Nat Rev Genet. 2018;19:789–800.
Pischedda F, Piccoli G. The IgLON family member Negr1 promotes neuronal arborization acting as soluble factor via FGFR2. Front Mol Neurosci. 2016;8:89.
Ren Z, Sahir N, Murakami S, Luellen BA, Earnheart JC, Lal R, et al. Defects in dendrite and spine maturation and synaptogenesis associated with an anxious-depressive-like phenotype of GABAA receptor-deficient mice. Neuropharmacology. 2015;88:171–9.
Kang HJ, Voleti B, Hajszan T, Rajkowska G, Stockmeier CA, Licznerski P, et al. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat Med. 2012;18:1413–7.
Moda-Sava RN, Murdock MH, Parekh PK, Fetcho RN, Huang BS, Huynh TN, et al. Sustained rescue of prefrontal circuit dysfunction by antidepressant-induced spine formation. Science. 2019;364:eaat8078.
Singh K, Jayaram M, Kaare M, Leidmaa E, Jagomae T, Heinla I, et al. Neural cell adhesion molecule Negr1 deficiency in mouse results in structural brain endophenotypes and behavioral deviations related to psychiatric disorders. Sci Rep. 2019;9:5457.
Acknowledgements
This work was equally supported by the National Key R&D Program of China (2018YFC1314600) and the National Key Research and Development Program of China (Stem Cell and Translational Research) (2016YFA0100900). This study was also supported by the Innovative Research Team of Science and Technology Department of Yunnan Province (2019HC004) and the Key Research Project of Yunnan Province (2017FA008 to X-JL). One of the brain eQTL datasets used in this study was generated as part of the Common Mind Consortium supported by funding from Takeda Pharmaceuticals Company Limited, F. Hoffman-La Roche Ltd, and NIH grants R01MH085542, R01MH093725, P50MH066392, P50MH080405, R01MH097276, RO1-MH-075916, P50M096891, P50MH084053S1, R37MH057881 and R37MH057881S1, HHSN271201300031C, AG02219, AG05138, and MH06692. Brain tissue for the study was obtained from the following brain bank collections: the Mount Sinai NIH Brain and Tissue Repository, the University of Pennsylvania Alzheimer’s Disease Core Center, the University of Pittsburgh NeuroBioBank and Brain and Tissue Repositories, and the NIMH Human Brain Collection Core. CMC leadership: Pamela Sklar, Joseph Buxbaum (Icahn School of Medicine at Mount Sinai), Bernie Devlin, David Lewis (University of Pittsburgh), Raquel Gur, Chang-Gyu Hahn (University of Pennsylvania), Keisuke Hirai, Hiroyoshi Toyoshiba (Takeda Pharmaceuticals Company Limited), Enrico Domenici, Laurent Essioux (F. Hoffman-La Roche Ltd), Lara Mangravite, Mette Peters (Sage Bionetworks), and Thomas Lehner, Barbara Lipska (NIMH). The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS.
Author information
Authors and Affiliations
Contributions
X-JL conceived and designed the study. YH performed most of the bioinformatic analyses, including the processing of the raw ChIP-seq data, the identification of PWMs from the ChIP-seq peaks, and the identification of TF binding–disrupting SNPs. SL, YL, and JW performed the reporter gene assays, knock down of transcription factors, and CRISPR-Cas9 mediated genome editing. XL carried out the eQTL analysis. JL conducted spatio-temporal expression pattern and cell type-specific expression analysis. SL, YL, XL, JL, JW, ZL, ML, and X-JL contributed to this work in data generation and analysis, results interpretation, and manuscript writing. X-JL oversaw the project and drafted the first version of the manuscript. All authors revised the manuscript critically and approved the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Li, S., Li, Y., Li, X. et al. Regulatory mechanisms of major depressive disorder risk variants. Mol Psychiatry 25, 1926–1945 (2020). https://doi.org/10.1038/s41380-020-0715-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-020-0715-7
- Springer Nature Limited
This article is cited by
-
LncRNA NPTN-IT1-201 Ameliorates Depressive-like Behavior by Targeting miR-142-5p and Regulating Inflammation and Apoptosis via BDNF
Current Medical Science (2024)
-
Genome-wide meta-analysis, functional genomics and integrative analyses implicate new risk genes and therapeutic targets for anxiety disorders
Nature Human Behaviour (2023)
-
Disentangling the complexity of psoriasis in the post-genome-wide association era
Genes & Immunity (2023)
-
Functional genomics elucidates regulatory mechanisms of Parkinson’s disease-associated variants
BMC Medicine (2022)
-
Functional genomic analysis delineates regulatory mechanisms of GWAS-identified bipolar disorder risk variants
Genome Medicine (2022)