
Abstract
Schizophrenia is a severe neuropsychiatric disorder with core features including hallucinations, delusions, and cognition deficits. Accumulating evidence has implicated abnormal DNA methylation in the development of schizophrenia. However, the mechanisms by which DNA methylation changes alter the risk for schizophrenia remain largely unknown. We recently carried out a DNA methylome study of peripheral blood samples from 469 first-episode patients with schizophrenia and 476 age- and gender-matched healthy controls of Han Chinese origin. Genomic DNA methylation patterns were quantified using an Illumina Infinium Human MethylationEPIC BeadChip. We identified multiple differentially methylated positions (DMPs) and regions between patients and controls. The most significant DMPs were annotated to genes C17orf53, THAP1 and KCNQ4 (KV7.4), with Bonferroni-adjusted P values of \({\mathrm{1}}{\mathrm{.34}} \times {\mathrm{10}}^{{\mathrm{ - 12}}}\), \({\mathrm{1}}{\mathrm{.15}} \times {\mathrm{10}}^{{\mathrm{ - 11}}}\), and \({\mathrm{3}}{\mathrm{.11}} \times {\mathrm{10}}^{{\mathrm{ - 11}}}\), respectively. In particular, KCNQ4 encodes a voltage-gated potassium channel of the KV7 family, which is linked to neuronal excitability. The genes associated with top-ranked DMPs also included many genes involved in nervous system development, such as LIMK2 and TMOD2. Gene ontology analysis of the differentially methylated genes further identified strong enrichment of neuronal networks, including neuron projection extension, axonogenesis and neuron apoptotic process. Finally, we provided evidence that schizophrenia-associated epigenetic alterations co-localize with genetic susceptibility loci. By focusing on first-episode schizophrenia patients, our investigation lends particularly strong support for an important role of DNA methylation in schizophrenia pathogenesis unconfounded by the effects of long-term antipsychotic medication or disease progression. The observed DNA methylation aberrations in schizophrenia patients could potentially provide a valuable resource for identifying diagnostic biomarkers and developing novel therapeutic targets to benefit schizophrenia patients.
Similar content being viewed by others


Introduction
Schizophrenia (SZ) is a serious mental disorder defined by the presence of symptoms including hallucinations, delusions, and cognitive impairments, which affects ~1% of the population worldwide [1, 2]. Genetic components, including single nucleotide polymorphisms (SNPs) [3, 4], structural variants [5], and gene expression patterns [6], are all likely to impact the risk of developing SZ. Notably, a genome-wide association study (GWAS) conducted by the Psychiatric Genomics Consortium (PGC) revealed 108 loci associated with SZ, highlighting the important role of common genetic variation in SZ susceptibility [3].
This GWAS found that most genetic variants associated with SZ tended to be enriched in regulatory domains of the genome [3], highlighting the importance of gene regulation in the etiology of SZ. Recently, mounting evidence has indicated that epigenetic modifications, which can be affected by environmental factors, might play a dynamic role in regulating gene expression [7, 8]. A wide range of environmental factors, including prenatal and perinatal events, urban environment, migration status, drug use, and social adversity [2], have been linked with SZ susceptibility and could be mediated by epigenetic changes. The presence of such epigenetic effects may explain the well-known phenotypic discordance of SZ in monozygotic twins [9]. In humans, as one of the major types of epigenetic modifications, and perhaps the most well-characterized, DNA methylation refers primarily to a biological process that adds a methyl group to the cytosines of CpG dinucleotides [10]. Recently, a few studies [11,12,13,14,15] have reported significant DNA methylation changes associated with SZ, which pointed to immune cells and neural signaling pathways as playing particularly pivotal roles. Therefore, DNA methylation may serve as a useful biomarker and important mediator to probe and gain insight into the links between genetic and environmental factors in the development of SZ.
However, the causal role of DNA methylation in the development of SZ remains largely unknown. One explanation is that most large-scale DNA methylation studies, either in postmortem brains or peripheral blood cells, were confounded by the cumulative effects of therapeutic intervention or disease progression. In this study, we examined DNA methylation profiles using a unique large cohort of first-episode schizophrenia (FESZ) patients and healthy volunteers from the Chinese Han population. By examining FESZ patients, we were able to make stronger inferences about the links between methylation changes and SZ which are unconfounded by the long-term effects of drugs or symptom progression. For the DNA methylation assay, we used Illumina Infinium Human MethylationEPIC BeadChip (Illumina, San Diego, CA, USA), a cost-effective platform with good coverage of methylation sites across the genome. Our analysis revealed a variety of differentially methylated patterns associated with FESZ. Notably, our work demonstrated significant DNA methylation changes in a subset of genes that participated in neuronal networks, including neuron projection extension, axonogenesis, and the neuron apoptotic process, supporting the neurodevelopmental origin hypothesis of SZ etiology [16]. Finally, we demonstrated that the DMPs identified in our study were located nearby known SZ risk loci. Results from our study provided strong evidence to support links between the blood DNA methylome and SZ pathogenesis.
Materials and methods
Study samples
In this study, a total of 499 first-episode patients with SZ of Han Chinese ancestry (25.4 ± 6.3 years; 207 males, 292 females) were recruited from three clinical sites, Beijing HuiLongGuan Hospital, Chongqing Three Gorges Central Hospital, and Zhumadian Psychiatry Hospital between 2017 and 2018. Diagnosis and blood sample collection were conducted by the clinical research physicians from Beijing HuiLongGuan Hospital using identical research protocols. SZ patients were included in the study if they met the following criteria: (a) they met the Structured Clinical Interview of DSM-IV diagnostic criteria for SZ; (b) they were aged between 14 and 50 years; (c) the total disease course was <3 years; (d) previous antipsychotic exposure did not exceed 2 weeks. Study participants were free of any diagnosis of mental deficiency, traumatic brain injury, or a history of illicit drug abuse or alcoholism. Patients were also screened for regular administration of neurotrophic agents and treatment with immune modulators or antioxidants in the preceding 8 weeks. A total of 500 healthy controls were enrolled from the local community and frequency-matched with the patients for age and gender. Only 497 controls (27.4 ± 5.2 years; 208 males, 289 females) were included for data analysis because three healthy volunteers were found to have a history of smoking. All healthy volunteers and their first-degree relatives had no history of any form of psychiatric disorders. All subjects were in good physical health, and they did not suffer from any neurological or other medical illness. Importantly, ethical approval for this project was obtained from the institutional research board committee of Beijing HuiLongGuan Hospital and all research activity was performed in concordance with their guidelines. Written informed consent was obtained from all study participants after a detailed explanation of the nature of this study was given to all study participants. If a study participant was unable to understand a particular question, their relative was asked to answer the relevant question. Using the online tool developed by Mansell et al. [17], we showed that our target sample provided over 80% power for 80% of CpG probes, even when adopting the relatively conservative Bonferroni correction and assuming a modest average case–control difference of 2%.
Genomic DNA was isolated from whole blood samples and then bisulfite-converted following standard procedures. The methylation status of bisulfite-converted DNA samples was assessed using the Illumina Infinium Human MethylationEPIC BeadChip (Illumina, San Diego, CA, USA), which measures DNA methylation levels across more than 850,000 probes at single-nucleotide resolution. The raw intensities were then scanned and DNA methylation measurements from 499 SZ patients and 500 controls obtained from the three previously mentioned hospitals were reported. A total of 39 patient samples were hybridized in duplicate. Thus, in total, 1038 (538 patient files and 500 control files) microarrays were scanned in 999 unique individuals.
Quality control
Supplementary Fig. S1 shows an overview of our methodological flow. Technically replicated samples and samples from smokers were removed from further analysis. Experimental quality control was conducted via BeadArray Controls Reporter software (https://support.illumina.com/downloads/beadarray-controls-reporter-installer.html), and a further 13 samples with low experimental quality were excluded. The statistical analysis started from raw intensity (.idat file) and was primarily performed using R software v3.6.1 (https://www.r-project.org/). Study participants’ gender status was checked by using the “minfi” R package [18]. Thirty-seven additional samples were excluded from subsequent analysis due to their ambiguous gender status. One sample was removed because at least 5% of the probes did not pass a 0.01 detection P value threshold. Background correction and dye-bias normalization were both performed by using the “Noob” method as described in [19]. Furthermore, functional normalization [20] was used to correct the unwanted between-array technical bias without removing any true biological signals. We then filtered the probes with >0.01 detection P value in more than 5% samples and the probes with <3 bead count in at least 5% samples. This step led to the removal of 3386 probes of poor quality. We then filtered probes (n = 98,222) with annotated SNPs as identified by Zhou et al. [21] together with probes (n = 16,868) located on sex chromosomes. Eleven probes aligned to multiple locations and were filtered using the recommended multi-hit list provided by Nordlund et al [22]. β values (ranging from 0 to 1) were then generated to represent methylation ratios at a given CpG site since the β value offers a more intuitive biological interpretation than the M value; higher β values indicate higher methylation levels. Technical differences between two different probe types were then normalized by “BMIQ” method [23] as implemented in the “ChAMP” R package [24]. Batch effect correction was then conducted by “ComBat” [25] using the “ENmix” R package [26]. Following these standard quality control procedures, data were available for 747,372 probes across 945 samples (469 patients and 476 controls) for subsequent analysis.
Cell-type composition estimation
Cell proportion differences between patients and controls constitute a critical aspect to be evaluated and controlled for DNA methylation analyses, because heterogeneous tissues such as blood are often used. Cell type heterogeneity was estimated by GLINT [27] software using the “ReFACTor” algorithm [28], which does not require any a priori knowledge of cell counts. Six “ReFACTor” components were incorporated as covariates in the association test to account for any cell type differences.
Statistical analysis
Identification of differentially methylated positions
To identify DMPs between SZ patients and healthy subjects, we used the “dmpFinder” function implemented in the “minfi” R package [18], adjusting for gender and age. In this case–control comparison, we did not account for cell type heterogeneity. Multiple testing was adjusted using a Bonferroni correction, with the significance threshold set at an adjusted P value < 0.05. To account for cell type heterogeneity, we performed an epigenome-wide association study (EWAS) to examine SZ-associated DMPs after regressing out the “ReFACTor” components along with gender and age. The EWAS was conducted using a logistic regression model, which is implemented in GLINT [27]. P values calculated by GLINT were then subjected to Bonferroni correction.
Identification of differentially methylated regions
We applied the “DMRcate” algorithm [29] to assess contiguous genomic regions showing DNA methylation differences in FESZ patients. Characterization of differentially methylated regions (DMRs), which is different from detection of a single genomic site that is differentially methylated, combines information from multiple nearby CpG sites. P values were corrected by using the Benjamini-Hochberg method with the threshold set at an adjusted P value < 0.05. For each DMR, the distance between two consecutive probes was constrained to <1000 bp and a minimum number of three consecutive CpG sites were required to constitute a DMR. All DMRs were annotated by their corresponding RefSeq gene using ANNOVAR software [30].
Gene ontology enrichment and network analysis
Functional properties of DMP-overlapping genes were characterized by gene ontology (GO) terms within the biological process domain using “clusterProfiler” R package [31]. GO terms were filtered to those that met an adjusted P value < 0.05 (Benjamini–Hochberg adjustment) threshold. The same procedure was conducted for all genes annotated to DMRs. Furthermore, unlike the GO enrichment analysis, which focuses only on sets of functionally similar genes, network analyses are capable of exploring more complex and detailed gene-gene interactions. Therefore, we selected genes according to biological function based on GO terms and constructed a protein-protein interaction network using STRING v11.0 [32] with a minimum required interaction score set at 0.9, indicating high confidence. The detailed interaction network graph was drawn using Cytoscape 3.8.0 [33].
Co-localization analysis of SZ GWAS loci and DMPs
We performed a co-localization analysis to identify overlapping signals between our differentially methylation results and previously reported SZ risk SNPs. In total, we used 3333 no-overlapping SZ risk SNPs identified as significant from previous GWAS conducted by PGC [3] and a study of east Asian and European population [34] along with the DMPs from our DNA methylation analysis. We considered DMPs within 200 kb of significant SNPs as co-localized.
Results
Identifying differentially methylated positions
We observed high concordance between the technical replicates (Supplementary Fig. S2A), which indicated good experimental quality of the generated data. Additionally, the false discovery rate was clearly reduced after correcting for experimental batch effects, including those from slides, plates, arrays and wells, along with gender and age, as shown in the Q–Q plot (Supplementary Fig. S2B). Notably, although the cell type heterogeneity correction was expected to strongly reduce false positive signals, the reduction was limited, suggesting that our results contained only a minor level of confounding effects from cellular composition (Supplementary Fig. S2B).
Of the 747,372 sites that passed the quality controls, a total of 4277 probes had an adjusted P value < 0.05 (Bonferroni adjustment), showing a significant difference in DNA methylation level between patients and controls. These DMPs were mapped to 3346 unique genes. Among these sites, 2534 were hypermethylated, whereas 1743 sites exhibited hypomethylation. Statistical significance, effect size, genomic location, and annotation of all DMPs ranked by Bonferroni-adjusted P values are documented in Supplementary Table S1 along with P values after controlling for cell type heterogeneity. The top 20-ranked DMPs are presented in Table 1 and highlighted in Fig. 1A. The most significant DMPs were annotated to genes C17orf53, THAP1 and KCNQ4 (Kv7.4), with Bonferroni-adjusted P values of \({\mathrm{1}}{\mathrm{.34}} \times {\mathrm{10}}^{{\mathrm{ - 12}}}\), \({\mathrm{1}}{\mathrm{.15}} \times {\mathrm{10}}^{{\mathrm{ - 11}}}\), and \({\mathrm{3}}{\mathrm{.11}} \times {\mathrm{10}}^{{\mathrm{ - 11}}}\), respectively. Figure 1B shows a volcano plot comparing the P values and \({\Delta}\beta\) values (the magnitude of methylation difference between patients and controls) for all CpG sites. As shown in Supplementary Fig. S3A, B, when \({\mathrm{|}}{\Delta}\beta {\mathrm{|}}\) thresholds were considered, more hypomethylated than hypermethylated DMPs were observed (\({\mathrm{|}}{\Delta}\beta {\mathrm{|}} \ge\) 0.01, 927 hypomethylated vs. 302 hypermethylated DMPs; \({\mathrm{|}}{\Delta}\beta {\mathrm{|}} \ge\) 0.02, 202 hypomethylated vs. 10 hypermethylated DMPs).

A Manhattan plot of all probes across the whole genome illustrating P values (y-axis, −log10 scale) against genomic location (x-axis). Chromosomes are distinguished by different colors. The red horizontal dashed line represents −log10(6.68 × 10−8), corresponding to the Bonferroni-adjusted P value = 0.05. B Volcano plot of all CpG sites. The X coordinate shows the ∆β, and the Y coordinate shows −log10(P value). Hypomethylated DMPs in SZ patients are labeled by using blue dots, whereas hypermethylated DMPs are represented by red dots. The horizontal gray dashed line stands for −log10(6.68 × 10−8), corresponding to Bonferroni-adjusted P values = 0.05. C Bar plot demonstrating the distribution of functional genomic regions. Colors represent different regions. TSS1500, 200–1500 bases upstream of the transcriptional start site; TSS200, 0–200 bases upstream of the transcriptional start site; 5′UTR, between the transcriptional start site and the ATG start site; 1stExon, first exon; body, between the ATG and stop codon; 3′UTR, between the stop codon and poly A signal. D Pie chart indicating the location of DMPs relative to CpG islands. Domains are labeled with different colors. N_Shelf, 2–4 kb upstream of island; N_Shore, 0–2 kb upstream of island; OpenSea, >4 kb from a CpG island; S_Shelf, 2–4 kb downstream of island; S_Shore, 0–2 kb downstream of island.
Insight into the functional genomic regions of SZ DMPs can be drawn from Fig. 1C. The majority of DMPs were located in regulatory regions, whereas fewer DMPs were aligned to the gene body. The same pattern was observed with the hypermethylated DMPs (Supplementary Fig. S4A); however, the opposite was seen in hypomethylated DMPs (Supplementary Fig. S4B). Most DMPs were found within or near CpG islands, while the minority of DMPs were scattered in open sea areas (located >4 kb from a CpG island), as shown in Fig. 1D. Again, a similar pattern was shown with hypermethylated DMPs (Supplementary Fig. S5A) in contrast to hypomethylated DMPs, which tended to occur in open sea areas (Supplementary Fig. S5B).
Among the top 20-ranked DMP list (Table 1), we identified several genes of particular interest because of their biological functions, including KCNQ4, a member of the voltage-gated potassium channels of the KV7 family related to neuronal excitability [35, 36], along with LIMK2 [37] and TMOD2 [38], which are associated with nervous system development. The patterns of DNA methylation changes for these genes are presented in Fig. 2A–C. In addition, we paid special attention to several DMP-related genes that were previously reported to confer clinical risk for SZ. Interestingly, the DMP cg16086782 was situated at SHANK2 (Supplementary Fig. S6A), a promising candidate risk gene for SZ [39]. Another interesting observation was that the significant DMP cg09392443 resided in the poised promoter of GAD1 (Supplementary Fig. S6B); alternative splicing of GAD1 and epigenetic state have been previously reported to confer SZ susceptibility and contribute to GABA dysfunction in the prefrontal cortex and hippocampus [40]. In addition, the DMP cg08388004 corresponded to BDNF (Supplementary Fig. S6C), a gene previously reported to be associated with SZ [41, 42]. The DNA methylation alterations of these DMPs in SZ patients are displayed in Fig. 2D–F.

SZ patients and healthy controls are distinguished by different colors. The Y-axis represents the beta value of each CpG site. Bonferroni-adjusted P values of DMPs are labeled with red.
Identifying differentially methylated regions
DMRs were identified by combining information from nearby CpG sites after adjusting for gender and age. We detected 6325 genomic regions (Supplementary Table S2), overlapping with 6264 unique genes. The largest differentially methylated segment was located on chromosome 6, spanning 94 probes and overlapping with exonal regions of the RGL2 gene. The most significant DMR was located on chromosome 2, spanning 19 CpG sites and corresponding to the HPCAL1 gene. The regional features of HPCAL1 are described in Fig. 3. These observations provided strong evidence that contiguous DNA methylation differences across specific genomic regions may be linked to SZ. A total of 2154 (64.38%) differentially methylated genes identified by the DMP analysis were also revealed by DMR analysis. Approximately 65.61% of the genes found through DMR analysis were not significant in the corresponding single-site DMP test.

In the first panel, the Y-axis shows P values (−log10 scale) calculated from the case–control analysis between FESZ patients and healthy individuals; the X-axis represents the genomic location of each CpG site on the chromosome. The index site was labeled using a black dot. Color reflects the magnitude of correlation between the reference site and all other CpGs in terms of methylation level, while red indicates a positive correlation and blue suggests a negative correlation. The middle plot shows the annotation tracks in a given genomic region. Chromatin annotation track color code: dark red, active promoter; pink, weak promoter; royal purple, poised promoter; orange: strong enhancer; light purple: strong enhancer; light yellow: weak enhancer; yellow: weak enhancer; dark blue: insulator; light green: transcriptional transition, green: transcriptional elongation; turquoise: weak transcribed; amaranth: repressed; brown: heterochromatin/low signal; gray: repetitive/CNV; light gray: repetitive/CNV. In the lower panel, co-methylation pattern based on Spearman correlation coefficients of selected CpGs is shown (red stands for positive correlation; blue stands for negative correlation). The depth of the color reflects the strength of the correlation.
GO enrichment profiling and network analysis
To describe common features of the DMPs, we performed GO enrichment analysis, identifying a total of 118 GO terms that passed a Benjamini–Hochberg adjusted P value < 0.05 threshold, as shown in Supplementary Table S3. The top-ranking enriched GO terms (Benjamini–Hochberg procedure adjusted P < 0.01) were involved in neuronal functions, such as neuron projection extension, axonogenesis, and the neuron apoptotic process (Fig. 4A), supporting the hypothesis of neurodevelopmental origin of SZ [16]. In addition, the Wnt signaling pathway, known to be important for neurodevelopment and nervous system regulation [43, 44], was also significantly enriched (Fig. 4A). Moreover, we found that genes in these neuronal function related GO terms displayed tight protein–protein interactions both in intra- and inter-GO terms (Fig. 4B), suggesting that these genes could potentially work together as a gene network and jointly contribute to SZ risk. Among the top-ranking enriched GO terms, a panel of genes were also found to be related to histone modification and covalent chromatin modification (Fig. 4A), indicating a potential link between DNA methylation and chromatin modifications, which has been supported by a recent study in human neurons [45].

A Bar plot of GO terms that passed a Benjamini-Hochberg-adjusted P value < 0.01. Colors are used to highlight the degree of significance (blue, lower significance; red, higher significance). The Y-axis represents the GO terms; the X-axis represents the fold of enrichment. Neural-related terms are labeled with red boxes. B Network of genes linked to DMPs. GO annotation was used to cluster the genes according to biological function. GO terms are indicated by various colors.
Consistent with the DMP results, DMR-related genes were also linked to neuron-related GO terms (e.g., neuron apoptotic process, axonogenesis, axon development, central nervous system neuron differentiation) as well as histone modification and covalent chromatin modification (Supplementary Table S4). Furthermore, we identified several GO terms related to the development of several brain regions, including the forebrain, telencephalon and cerebellar cortex. Taken together, these observations highlighted the importance of DNA methylation along with other epigenetic modifications in neurodevelopmental processes which have been previously implicated in SZ etiology. All significant GO terms (adjusted P value < 0.05) enriched for SZ DMRs are documented in Supplementary Table S4.
Co-localization of SZ-DMPs and SZ GWAS loci
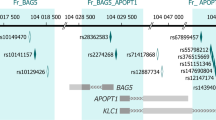
Our analysis revealed that 139 significant DMPs which were mapped to 126 genes, co-localized with 133 SZ risk SNPs (Supplementary Table S5). These 126 genes were then queried in PubMed (https://pubmed.ncbi.nlm.nih.gov/) along with the keyword “Schizophrenia”. Of the queried genes, 42 were found to have previous evidence of a link with SZ. As plotted in Fig. 5, on chromosome 7, both CpG site cg04894216 and SNP rs13230421 are located on GRM3 gene, which may confer risk for SZ by influencing glutamatergic neurotransmission and synaptic plasticity [3]. More co-location patterns are presented in Supplementary Fig. S7.

Red bars correspond to genomic locations of significant DMPs identified through our methylation study, with the height proportional to the −log (P value). Similarly, green bars indicate significant SNPs identified in published GWAS for SZ. Gene locations are labeled by horizontal yellow bars.
Discussion
In this study, we conducted a genome-wide DNA methylation analysis in peripheral blood cells from 469 FESZ patients and 476 matched controls of Han Chinese ancestry. We effectively controlled for confounding by batch effects, gender, age (Supplementary Fig. S2B) and smoking status. Despite the use of peripheral blood cells, many disease-associated DMPs and DMRs were linked to genes involved in neural function. Our gene set enrichment analyses strongly supported the possible involvement of aberrant DNA methylation of neurodevelopmental genes in SZ pathogenesis. Finally, the co-localization analysis showed the location of DMPs overlapped with previously reported SZ GWAS loci.
One of our top DMP findings was in the KCNQ4 (KV7.4) gene (Table 1), which encodes a component of voltage-gated potassium channels of the KV7 family (KV7.1–5) [35]. KCNQ4 could interact with KCNQ3 and mediate M-like currents that may modulate the excitability and synaptic transmission of prefrontal cortex neurons [36, 46]. In addition, Carment et al. demonstrated that abnormal regulation of cortical excitability and inhibition were exhibited in those with SZ [47]. Taken together, our findings suggested that abnormal DNA methylation of KCNQ4 may affect its expression, thereby contributing to SZ pathogenesis. We also identified LIMK2 as a gene potentially involved in SZ pathogenesis (Table 1). LIMK2, together with LIMK1 serve as the primary regulators of actin dynamics involved in structural plasticity by modulating cofilin protein [48]. LIMK-dependent actin reorganization is important for cortical development through its influence on neural progenitor cell proliferation and migration by the regulation of PAK1/Rho signaling [37]. Moreover, LIMK1 and LIMK2 gene expression were reported to be significantly altered in the brain tissue of SZ patients [49]. TMOD1 and TMOD2 (Supplementary Table S1), were also found to harbor significant DMPs. TMOD2 is critical for dendritic arborization, whereas TMOD1 is required for spine development and synapse formation [38]. Some genes with well-known neural functions also showed abnormal methylation in those with SZ, including SHANK2 [39], GAD1 [40], and BDNF [41, 42].
Multiple lines of evidence from our gene set enrichment analysis showed that differentially methylated genes were significantly enriched in neural pathways (Supplementary Table S3), which is consistent with the neurodevelopmental origin hypothesis of SZ [16]. In addition, several biological processes such as histone modification were also enriched for genes with abnormal methylation, warranting further investigation. In the DMR analysis, we found that regional DNA methylation changes likely contribute to the development of SZ. Expression of DMR-containing gene HPCAL1 (VILIP-3), which serves as a neuronal sensor of Ca2+, may alter signaling and synaptic function in GABA projection neurons [50]. Moreover, enrichment analyses of the annotated genes within DMRs were consistent with the results of the DMP-related gene analysis, suggesting that a network of neuronal-functioning genes contribute to SZ susceptibility (Supplementary Table S4).
Some DNA methylation studies have examined postmortem brain tissues, which are both costly and difficult to obtain. By comparison, peripheral blood is more readily accessible with a sizable sample, and CpGs from specific genomic regions have shown statistically significant correlations between brain and peripheral blood samples [51]. Notably, our observation of significantly enriched GO terms for the DMR-related genes revealed that the development of certain brain regions could be reflected in the DNA methylome of blood samples (Supplementary Table S4). This observation indicated that specific epigenetic markers in brain tissue can be mirrored by the corresponding sites in peripheral blood samples. Therefore, peripheral blood could serve as a valuable surrogate for brain tissue to meet the needs of large-scale or longitudinal studies.
Our results exhibited an interesting association between neuronal pathways and SZ in the blood DNA methylome which supported many previous findings. Hannon et al. have shown that neuronal proliferation and brain development were associated with SZ [12], while Jaffe et al. identified a panel of differentially methylated genes significantly enriched for nervous system differentiation in SZ [14]. In addition, Montano et al. identified multiple genes in SZ patients that were previously reported to be implicated in neuronal development and function [11]. In contrast to a few previous studies which found no such links, the neuronal function and development findings from pathway enrichment analysis were robust in our study. This may be because epigenetic alterations of FESZ patients are more closely linked to the onset of psychotic symptoms, rather than confounding factors, such as disease stage or antipsychotic medication use. Aberg et al. [13] reported enrichment of hypoxia in their methylome study, which may suggest this mechanism as a risk factor for SZ development. Nevertheless, we did not detect any association between hypoxia and SZ, which warrants further investigation. Future studies may clarify whether hypoxia is indeed a risk factor for SZ or whether it may be an effect of long-term antipsychotic use. A few studies [12, 13] have reported that immune related pathways might alter the risk for SZ. Interestingly, although we used peripheral blood cells, we did not identify any significant DNA methylation differences among genes in immune-related pathways; again, this might be due to our use of FESZ patients for the current study. Overall, our study provides a powerful examination of potential mechanisms which influence risk of FESZ, which are relatively unaffected by long-term treatment and progression effects as compared to non-first-episode SZ patients.
A host of research has demonstrated disease-related DNA methylation changes could be used as biomarkers to distinguish individuals into different subsets with respect to disease risk assessment, diagnosis, treatment monitoring and personalization [52,53,54,55]. Our results indicated that SZ pathogenesis could be linked to the DNA methylome, suggesting new avenues to develop biomarkers for early detection, accurate diagnosis and individualized treatment of SZ. The DNA methylome holds promise to identify patients from healthy controls or classify SZ patients into different subtypes with objective molecular markers, while also aiding in therapeutic target design. By comparing patients who respond to therapeutic regimens and those who do not, differentially methylated sites are an attractive source to select patients who will experience clinical benefit with that treatment. The stable, easy-to-access blood sample and cost-effective detection technology could meet the needs of real-time treatment response surveillance. In addition, converging evidence from DNA methylation analysis and GWAS at a given locus may represent more promising biomarkers for diagnosis and therapeutic targets for treatment of SZ. Despite the new findings coming out of peripheral blood cells, the biological mechanisms of the epigenetic aberrations contributing to SZ remain to be further investigated. Furthermore, although we have used a well-powered sample here, our findings need to be validated by future studies.
Conclusion
Focusing on FESZ patients, we completed a large-scale DNA methylation case–control study in a sample from the Chinese Han population. We identified a panel of DMPs and DMRs that were differentially methylated in FESZ patients relative to healthy controls. Notably, abnormal DNA methylation of KCNQ4, LIMK2, and TMOD2 exhibited a strong association with SZ. Gene enrichment analysis demonstrated that neurodevelopment, neurogenesis, and synaptic transmission contributes to SZ. Furthermore, our results provided evidence that SZ associated DMPs overlapped with known genetic risk loci. Taken together, our findings suggested that changes in the blood DNA methylome in FESZ patients provided a powerful approach to identify biomarkers and target genes that may facilitate an understanding of SZ biology underlying the genetic association with SZ and the development of novel strategies for diagnosis and more tailored treatments.
Data availability
Code used in the analyses is available to download from https://github.com/Mingrui-Li1992/SZ_methylation. All data can be viewed in NODE (http://www.biosino.org/node) by pasting the accession OEP001178 into the text search box or through the URL: http://www.biosino.org/node/project/detail/ OEP001178.
Change history
04 January 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41380-020-01013-w
References
James SL, Abate D, Abate KH, Abay SM, Abbafati C, Abbasi N, et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392:1789–858.
Kahn RS, Sommer IE, Murray RM, Meyer-Lindenberg A, Weinberger DR, Cannon TD, et al. Schizophrenia. Nat Rev Dis Prim. 2015;1:15067.
Ripke S, Neale BM, Corvin A, Walters JTR, Farh K-H, Holmans PA, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–7.
Bigdeli TB, Genovese G, Georgakopoulos P, Meyers JL, Peterson RE, Iyegbe CO, et al. Contributions of common genetic variants to risk of schizophrenia among individuals of African and Latino ancestry. Mol Psychiatry. 2020;25:2455–67.
Warland A, Kendall KM, Rees E, Kirov G, Caseras X. Schizophrenia-associated genomic copy number variants and subcortical brain volumes in the UK Biobank. Mol Psychiatry. 2020;25:854–62.
Jaffe AE, Straub RE, Shin JH, Tao R, Gao Y, Collado-Torres L, et al. Developmental and genetic regulation of the human cortex transcriptome illuminate schizophrenia pathogenesis. Nat Neurosci. 2018;21:1117–25.
Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33:245–54.
Cholewa-Waclaw J, Bird A, von Schimmelmann M, Schaefer A, Yu H, Song H, et al. The role of epigenetic mechanisms in the regulation of gene expression in the nervous system. J Neurosci. 2016;36:11427–34.
Tang J, Fan Y, Li H, Xiang Q, Zhang D-F, Li Z, et al. Whole-genome sequencing of monozygotic twins discordant for schizophrenia indicates multiple genetic risk factors for schizophrenia. J Genet Genom. 2017;44:295–306.
Greenberg MVC, Bourc’his D. The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol. 2019;20:590–607.
Montano C, Taub MA, Jaffe A, Briem E, Feinberg JI, Trygvadottir R, et al. Association of DNA methylation differences with schizophrenia in an epigenome-wide association study. JAMA Psychiatry. 2016;73:506–14.
Hannon E, Dempster E, Viana J, Burrage J, Smith AR, Macdonald R, et al. An integrated genetic-epigenetic analysis of schizophrenia: evidence for co-localization of genetic associations and differential DNA methylation. Genome Biol. 2016;17:176.
Aberg KA, McClay JL, Nerella S, Clark S, Kumar G, Chen W, et al. Methylome-wide association study of schizophrenia: identifying blood biomarker signatures of environmental insults. JAMA psychiatry. 2014;71:255–64.
Jaffe AE, Gao Y, Deep-Soboslay A, Tao R, Hyde TM, Weinberger DR, et al. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat Neurosci. 2016;19:40–7.
Duan J, Sanders AR, Gejman PV. From schizophrenia genetics to disease biology: harnessing new concepts and technologies. J Psychiatr Brain Sci. 2019;4:e190014.
Birnbaum R, Weinberger DR. Genetic insights into the neurodevelopmental origins of schizophrenia. Nat Rev Neurosci. 2017;18:727–40.
Mansell G, Gorrie-Stone TJ, Bao Y, Kumari M, Schalkwyk LS, Mill J, et al. Guidance for DNA methylation studies: statistical insights from the Illumina EPIC array. BMC Genom. 2019;20:366.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–9.
Triche TJ Jr., Weisenberger DJ, Van Den Berg D, Laird PW, Siegmund KD. Low-level processing of illumina infinium DNA methylation bead arrays. Nucleic Acids Res. 2013;41:e90–e90.
Fortin J-P, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ, et al. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014;15:503–503.
Zhou W, Laird PW, Shen H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 2016;45:e22–e22.
Nordlund J, Bäcklin CL, Wahlberg P, Busche S, Berglund EC, Eloranta M-L, et al. Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome Biol. 2013;14:r105.
Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29:189–96.
Tian Y, Morris TJ, Webster AP, Yang Z, Beck S, Feber A, et al. ChAMP: updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics. 2017;33:3982–4.
Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–27.
Xu Z, Niu L, Li L, Taylor JA. ENmix: a novel background correction method for Illumina HumanMethylation450 BeadChip. Nucleic Acids Res. 2016;44:e20–e20.
Rahmani E, Yedidim R, Shenhav L, Schweiger R, Weissbrod O, Zaitlen N, et al. GLINT: a user-friendly toolset for the analysis of high-throughput DNA-methylation array data. Bioinformatics. 2017;33:1870–2.
Rahmani E, Zaitlen N, Baran Y, Eng C, Hu D, Galanter J, et al. Sparse PCA corrects for cell type heterogeneity in epigenome-wide association studies. Nat Methods. 2016;13:443–5.
Peters TJ, Buckley MJ, Statham AL, Pidsley R, Samaras K, Lord VR, et al. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin. 2015;8:6.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164–e164.
Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7.
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504.
Lam M, Chen C-Y, Li Z, Martin AR, Bryois J, Ma X, et al. Comparative genetic architectures of schizophrenia in East Asian and European populations. Nat Genet. 2019;51:1670–8.
Wulff H, Castle NA, Pardo LA. Voltage-gated potassium channels as therapeutic targets. Nat Rev Drug Discov. 2009;8:982–1001.
Peng H, Bian X-L, Ma F-C, Wang K-W. Pharmacological modulation of the voltage-gated neuronal Kv7/KCNQ/M-channel alters the intrinsic excitability and synaptic responses of pyramidal neurons in rat prefrontal cortex slices. Acta Pharm Sin. 2017;38:1248–56.
Mao R, Deng R, Wei Y, Han L, Meng Y, Xie W, et al. LIMK1 and LIMK2 regulate cortical development through affecting neural progenitor cell proliferation and migration. Mol Brain. 2019;12:67.
Omotade OF, Rui Y, Lei W, Yu K, Hartzell HC, Fowler VM, et al. Tropomodulin isoform-specific regulation of dendrite development and synapse formation. J Neurosci. 2018;38:10271.
Peykov S, Berkel S, Schoen M, Weiss K, Degenhardt F, Strohmaier J, et al. Identification and functional characterization of rare SHANK2 variants in schizophrenia. Mol Psychiatry. 2015;20:1489–98.
Tao R, Davis KN, Li C, Shin JH, Gao Y, Jaffe AE, et al. GAD1 alternative transcripts and DNA methylation in human prefrontal cortex and hippocampus in brain development, schizophrenia. Mol Psychiatry. 2018;23:1496–505.
Zhang Y, Fang X, Fan W, Tang W, Cai J, Song L, et al. Interaction between BDNF and TNF-α genes in schizophrenia. Psychoneuroendocrinology. 2018;89:1–6.
Zhang XY, Tan YL, Chen DC, Tan SP, Yang FD, Wu HE, et al. Interaction of BDNF with cytokines in chronic schizophrenia. Brain Behav Immun. 2016;51:169–75.
Meffre D, Grenier J, Bernard S, Courtin F, Dudev T, Shackleford GG, et al. Wnt and lithium: a common destiny in the therapy of nervous system pathologies? Cell Mol Life Sci. 2014;71:1123–48.
Yu Z, Cheng C, Liu Y, Liu N, Lo EH, Wang X. Neuroglobin promotes neurogenesis through Wnt signaling pathway. Cell Death Dis. 2018;9:945–945.
Zhang S, Zhang H, Zhou Y, Qiao M, Zhao S, Kozlova A, et al. Allele-specific open chromatin in human iPSC neurons elucidates functional disease variants. Science. 2020;369:561.
Jentsch TJ. Neuronal KCNQ potassium channels:physislogy and role in disease. Nat Rev Neurosci. 2000;1:21–30.
Carment L, Dupin L, Guedj L, Térémetz M, Krebs M-O, Cuenca M, et al. Impaired attentional modulation of sensorimotor control and cortical excitability in schizophrenia. Brain. 2019;142:2149–64.
Cuberos H, Vallée B, Vourc’h P, Tastet J, Andres CR, Bénédetti H. Roles of LIM kinases in central nervous system function and dysfunction. FEBS Lett. 2015;589(24, Part B):3795–806.
Datta D, Arion D, Corradi JP, Lewis DA. Altered expression of CDC42 signaling pathway components in cortical layer 3 pyramidal cells in schizophrenia. Biol Psychiatry. 2015;78:775–85.
Sharp BM, Chen H, Gong S, Wu X, Liu Z, Hiler K, et al. Gene expression in accumbens GABA neurons from inbred rats with different drug-taking behavior. Genes Brain Behav. 2011;10:778–88.
Walton E, Hass J, Liu J, Roffman JL, Bernardoni F, Roessner V, et al. Correspondence of DNA methylation between blood and brain tissue and its application to schizophrenia research. Schizophr Bull. 2016;42:406–14.
Xu R-H, Wei W, Krawczyk M, Wang W, Luo H, Flagg K, et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat Mater. 2017;16:1155–61.
Taryma-Leśniak O, Sokolowska KE, Wojdacz TK. Current status of development of methylation biomarkers for in vitro diagnostic IVD applications. Clin Epigenetics. 2020;12:100.
Duruisseaux M, Martínez-Cardús A, Calleja-Cervantes ME, Moran S, Castro de Moura M, Davalos V, et al. Epigenetic prediction of response to anti-PD-1 treatment in non-small-cell lung cancer: a multicentre, retrospective analysis. Lancet Respir Med. 2018;6:771–81.
Zou D, Qiu Y, Li R, Meng Y, Wu Y. A novel schizophrenia diagnostic model based on statistically significant changes in gene methylation in specific brain regions. BioMed Res Int. 2020;2020:8047146.
Acknowledgements
We are grateful to the patients, healthy volunteers, and their families who contributed to this study. This study was funded by School of Life Sciences of Fudan University, as a start-up capital for Dr. Yin Yao, and was partially funded by Beijing HuiLongGuan Hospital. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
ML performed the DNA methylation analysis, result interpretation and discussion, and wrote the manuscript; YL contributed to sample recruitment and preparation; HQ participated in the data analysis pipeline construction, result discussion and interpretation; JDT helped revise the manuscript and provided insightful thoughts to the data interpretation; ML performed the co-localization analysis; CQ contributed to literature review and experimental procedure supervision; JL and QL contributed to literature review; FF, MG, JH, JT, and FY participated in the sample recruitment; YY and YT conceived the study, co-supervised study manuscript writing, interpreted the results and finalized the manuscript. All authors approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Li, M., Li, Y., Qin, H. et al. Genome-wide DNA methylation analysis of peripheral blood cells derived from patients with first-episode schizophrenia in the Chinese Han population. Mol Psychiatry 26, 4475–4485 (2021). https://doi.org/10.1038/s41380-020-00968-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-020-00968-0
- Springer Nature Limited
This article is cited by
-
DiseaseNet: a transfer learning approach to noncommunicable disease classification
BMC Bioinformatics (2024)
-
Immunophenotypes in psychosis: is it a premature inflamm-aging disorder?
Molecular Psychiatry (2024)
-
Genome-wide DNA methylation association study of recent and cumulative marijuana use in middle aged adults
Molecular Psychiatry (2023)
-
Genome-wide DNA methylation analysis in schizophrenia with tardive dyskinesia: a preliminary study
Genes & Genomics (2023)
-
Exploring the mediation of DNA methylation across the epigenome between childhood adversity and First Episode of Psychosis—findings from the EU-GEI study
Molecular Psychiatry (2023)