
Abstract
Traumatic brain injury (TBI) is a pervasive problem in the United States and worldwide, as the number of diagnosed individuals is increasing yearly and there are no efficacious therapeutic interventions. A large number of patients suffer with cognitive disabilities and psychiatric conditions after TBI, especially anxiety and depression. The constellation of post-injury cognitive and behavioral symptoms suggest permanent effects of injury on neurotransmission. Guided in part by preclinical studies, clinical trials have focused on high-yield pathophysiologic mechanisms, including protein aggregation, inflammation, metabolic disruption, cell generation, physiology, and alterations in neurotransmitter signaling. Despite successful treatment of experimental TBI in animal models, clinical studies based on these findings have failed to translate to humans. The current international effort to reshape TBI research is focusing on redefining the taxonomy and characterization of TBI. In addition, as the next round of clinical trials is pending, there is a pressing need to consider what the field has learned over the past two decades of research, and how we can best capitalize on this knowledge to inform the hypotheses for future innovations. Thus, it is critically important to extend our understanding of the pathophysiology of TBI, particularly to mechanisms that are associated with recovery versus development of chronic symptoms. In this review, we focus on the pathology of neurotransmission after TBI, reflecting on what has been learned from both the preclinical and clinical studies, and we discuss new directions and opportunities for future work.
Similar content being viewed by others


Introduction
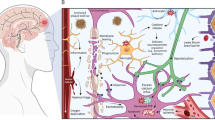
Traumatic brain injury (TBI) affects 2.8 million people annually in the U.S. [1], and over 10 million people worldwide [2]. The economic burden of TBI, both in direct healthcare costs and lost revenue, is enormous, as injured persons often fail to return to work and incur significant morbidity and mortality [3]. The past two decades have produced over 200 Phase II or higher interventional trials for TBI. The pathological mechanisms targeted in TBI clinical trials parallels trends in preclinical research, falling into several general categories, including mitigating damage-related tauopathy [4], acute intervention for physiologic stabilization [5], cell replacement strategies to re-establish plasticity [6, 7], attenuating acute (minutes to days), subacute (days to weeks) and chronic (months to years) inflammation [8, 9], restoring cell and tissue metabolism [10], and modulating neurotransmission [11, 12] (Fig. 1).

Strategies to treat acute and chronic TBI currently under investigation (counterclockwise from top left). Molecular targeting of damage-related tauopathy to delay or prevent chronic cognitive effects of TBI. Improving treatments for elevated intracranial pressure and new diagnostics to guide measured improvements in acute patient management. Pharmacological manipulation of transmitter systems to alleviate cognitive and behavioral symptoms. Molecular targeting of acute and chronic inflammatory responses, to mitigate secondary damage. Pharmacological and dietary interventions to support cell and tissue metabolism and alleviate endocrine dysfunction. Manipulating endogenous repair mechanisms to boost neurogenesis or otherwise replace or repopulate damaged tissue and re-establish plasticity. In this review, we focus on the pathophysiological changes on neurotransmitter systems and the studies targeting neurotransmission
A common theme among TBI clinical trials is their failure. While some failed due to low enrollment or change in sponsor priorities, many were terminated for futility. Twenty-four percent of the interventional clinical trials in the last decade address neurotransmitter systems (Fig. 2); the small successes in TBI clinical research thus far originate from trials that fix on neurotransmitter dysfunction, restore electrical activity, or treat post-traumatic depression [12,13,14,15,16]. There are over 40 ongoing or completed registered clinical trials targeting major neurotransmitter systems, encompassing more than 4000 patients; the published, completed trials are summarized below and in Table 1. Inspired by this theme, this review focuses on preclinical and clinical studies of neurotransmitter systems after TBI, the lessons learned from this work, and consideration of new leads for the field.

Completed and ongoing clinical trials for traumatic brain injury. Of 203 interventional clinical trials, 3% are targeting Tau aggregation or other tauopathy, 34% acute physiological disruption or intracranial pressure, 23.6% neurotransmitter systems, 9.9% metabolic interventions, 7.4% inflammation or immune function, 4.9% neurogenesis or stem cell therapy, and 17.2% employ a variety of other strategies including behavioral interventions, transcranial stimulation, and/or neuromodulation
Pathophysiology of TBI: state of the field
TBI severity is graded by a clinical assessment of the extent of coma, the Glasgow Coma Scale (GCS), which classifies patients using a minimum score of 3 to a maximum score of 15 [17, 18]. This score was designed as a clinical assessment tool, but is routinely used to categorize patients for inclusion in clinical trials. Using this nomenclature, the majority of TBIs are categorized as mild (GCS 13−15), and may include a brief loss of consciousness, confusion, disorientation, and memory dysfunction, frequently with no abnormalities on conventional diagnostic imaging [17]. Mild TBI typically results from mechanical or rotational force to the head resulting in a period of altered sensorium, including sports-related concussions. In contrast, moderate (GCS 9−12) and severe (GCS ≤ 8) TBIs are more often associated with high-velocity impacts (motor vehicle accidents) or significant acceleration−deceleration injury. This nomenclature for categorizing TBI, although universally used, lacks an appreciation of the pathophysiology behind injury [19].
Despite the static TBI taxonomy, much has been learned about the pathophysiology of TBI. TBI involves both the primary physical injury to brain tissues and molecular cascades that propagate injury into surrounding tissue, a phenomenon known as secondary injury (reviewed in refs. [20, 21]). These cascades include unregulated neurotransmitter and ion release, cell swelling, diffuse axonal injury, free radical production, mitochondrial dysfunction, inflammatory cytokines, and altered gene transcription [20]. Secondary events further exacerbate the direct consequences of primary injury [20, 22]. As a result, TBI pathophysiology evolves over weeks to months, making behavioral outcomes, particularly in mild injuries without focal lesions, difficult to predict [23, 24]. Despite the large gains in our understanding of the molecular events occurring in the brain following injury, efforts to translate these findings to clinical practice have almost universally failed. The reasons for this are numerous, not the least of which is crossing the wide gap from relatively homogeneous research population to a highly diverse clinical population [25].
Methodologies for studying TBI in humans include behavioral and cognitive evaluations, ante-mortem imaging, accessible tissue samples (including cerebrospinal fluid in severe TBI), or postmortem analysis of structural and molecular changes. However, research in humans is challenging due to heterogeneity in injury severity and physiology, covarying factors such as time from injury, age, sex, lifestyle factors, comorbid illness, patient availability for follow-up, and technical limitations [26,27,28]. Animal models overcome some of these obstacles, but have limitations as well. Preclinical TBI is performed under anesthesia and some models require directly accessing the brain [29]. Behavioral evaluations of many aspects of executive function including attention, processing speed, motivation, and response inhibition in rodents require labor-intensive training of the rodents prior to testing [30]. While there are inherent challenges in interpreting behavioral changes in animal models, fundamental brain structures and cellular organization are similar between rodent and human and many behavioral endophenotypes are well validated [30, 31].
There are four primary in vivo injury models in rodents: fluid percussion injury, controlled cortical impact, weight-drop acceleration impact, and blast injury [29]. These models vary in the biomechanics of damage and progression of pathology, but produce similar cognitive and behavioral deficits and changes in brain biochemistry [29]. Ultimately, different mechanisms of injury, i.e. direct cortical impact versus blast, may converge on defects of neurotransmission resulting in a similar spectrum of cognitive and behavioral changes [32]. There is no single in vivo model of TBI that is objectively superior to other models at producing robust behavioral and cognitive effects, and decisions regarding the selection of a preclinical model may depend on injury mechanisms and study objectives. In vitro models of TBI, including slice culture, primary cell culture, and immortalized cell lines, permit a more precise focus on specific aspects of injury mechanics and cellular mechanisms [33]. In combination, these models provide powerful tools for mechanistic studies of initial damage responses, restoration of homeostasis, and long-term neurochemical plasticity after TBI that are impossible in human subjects.
Neurotransmission shapes behavior and cognition after TBI
The strength and timing of excitatory and inhibitory transmission (E/I balance) within neural circuits shapes activity-dependent plasticity required for behavioral adaptation to environmental stimuli [34]. Repetitive activation at excitatory synapses increases synaptic strength through long-term potentiation (LTP). The balance of LTP and the related phenomenon long-term depression (LTD) coordinate changes in synaptic strength linked to learning and memory. Neuroplastic function requires coordination of pre- and postsynaptic neurons in cohesive circuits, as well as diffusional signals from surrounding glia [11]. Driven by preclinical findings, a fundamental theme in TBI research is modulating neurotransmission to support and restore adaptive plasticity, E/I balance, and the overall capacity of the injured brain to respond appropriately to incoming stimuli [35, 36].
Even TBIs categorized as mild may profoundly affect learning and memory, concentration, and processing speed [37, 38]. TBI may subtly impact multiple domains of executive functioning, including goal identification, planning, behavioral flexibility, problem solving, self-awareness, and insight [39]. These changes frequently result in impaired decision making, low frustration tolerance, and difficulty returning to premorbid levels of function [39]. The effects of TBI on cognition and executive function can be immediate and permanent [40].
Additionally, anxiety and depression frequently develop after TBI [41, 42], sometimes months or years after injury [41]. Post-TBI depression and anxiety are associated with poor treatment response [43, 44], and frequently coincide with cognitive symptoms [45]. The mechanisms underlying post-TBI depression are unknown, but are not tied to injury severity [46]. However, secondary processes after TBI are independently implicated in depression, including impaired neurogenesis [47], inflammation and microglial activation [48], and glutamate system dysfunction [49]. As with many chronic neurodegenerative conditions, post-TBI neuropsychiatric and cognitive symptoms likely involve damage to homeostatic mechanisms, ultimately resulting in deterioration of the molecular machinery underlying effective neurotransmission in response to environmental demands.
As damage and recovery process are ongoing, the landscape of neurotransmission continues to evolve well into chronic stages, months to years after injury (Fig. 3). Understanding how regulation of transmitter systems is altered after TBI, the progression of these changes over time, and the cumulative impact of damage to transmitter systems that work together to coordinate behavioral responses will be instrumental in developing effective treatment strategies. Here, we describe the current understanding of how TBI impacts major transmitter systems, strategies implemented to mitigate the effects of TBI, and lingering knowledge gaps within the field.

Summary of temporal changes in neurotransmitter systems after experimental TBI. In acute TBI, there are increases in extracellular glutamate (green panel) and decreases in glutamate uptake due to changes in excitatory amino acid transporter (EAAT) isoform expression. Within hours after experimental TBI, there are decreases in NMDA receptor subunits, and changes in AMPA receptor subunit composition. Chronically, there is sustained depression of glutamate signaling including changes in postsynaptic receptor composition, decreased astroglial (red process) EAAT expression, and a shift in the relative expression of neuronal and glial transporters. Changes in GABA signaling (red panel) include an acute shift in the balance of synaptic and extrasynaptic receptors acutely after injury. There is chronic dysregulation of GABAergic tone with mechanisms that vary by brain region, illustrated here as decreases in GABA receptor binding. In cortex, excessive inhibitory control results from structural change in pyramidal neurons. In subcortical regions, there is a change in the composition and localization of GABA receptors that may contribute to epileptogenic potential. For acetylcholine (Ach) (blue panel), there is an initial unregulated release of ACh and decreases in transporter density and receptor binding beginning as early as 1 h after TBI. However there is chronic cholinergic hypofunction associated with decreased evoked release of Ach and changes in acetylcholinesterase activity. With norepinephrine (NE) (pink panel) there is an immediate accelerated turnover of NE and concurrent downregulation of receptors. The accelerated turnover is quickly reversed and remains suppressed weeks after injury, leaving chronic downregulation of NE receptors and associated decreased signaling. TBI initiates changes in both receptor and transporter expression that regulate dopamine (DA) (orange panel) function. Acutely, elevated tissue dopamine levels can remain elevated for weeks. However, there is chronic dopaminergic hypofunction illustrated by changes in presynaptic terminal dopamine recycling and decreases in DA tonic and evoked release. Serotonin (purple panel) changes are characterized by sustained decreases in both transporters and receptors after TBI
Glutamatergic neurotransmission in TBI
Glutamate, the predominant excitatory neurotransmitter in the brain, signals through metabotropic and ionotropic receptors localized to synaptic and extrasynaptic membranes on neurons and glial. Extracellular glutamate levels are regulated by a family of membrane-bound excitatory amino acid transporters (EAATs) found on postsynaptic neurons and astrocytes [50]. Glutamate release and reuptake is tightly regulated to optimize synaptic transmission and prevent excitotoxicity from excessive extracellular glutamate [51]. Alterations in glutamate buffering and reuptake modulate synaptic function, leading to neuroplastic changes in learning and memory [52]. Thus, if acute deficits in glutamate regulation become persistent, they may contribute to the long-term cognitive and emotional deficits common after TBI.
Data from both humans and animal models indicate that in the initial minutes and hours, post-TBI, release of glutamate and ions plays an important role in initiating secondary injury cascades [53,54,55,56]. Elevated extracellular potassium impairs astrocytic potassium conductance essential for glutamate uptake and transporter-mediated buffering [57, 58]. In severe injuries, high levels of extracellular glutamate and impaired glial uptake contribute to propagating waves of regional electrophysiological hyperactivity followed by depolarization (cortical spreading depolarizations, CSDs), that temporarily silences synaptic activity [5, 59,60,61]. CSDs are often repetitive and associate with increases in extracellular glutamate and lactate [59]. In severe injuries, high or intermittently high levels of extracellular glutamate may continue for a week or longer [54, 55], with the initial unregulated release of glutamate followed by sustained depression of glutamate signaling [62, 63] (Fig. 3).
Preclinical findings
In preclinical models of moderate-severe TBI, N-methyl-d-aspartic acid (NMDA) glutamate receptor subunit expression is decreased within hours as NMDAR hyperactivity is followed by receptor hypofunction [64, 65]. However, a study of severe injury using a PET ligand selective for open NMDA channels, 18F-GE-179, found an increase in open NMDA channels within 5−6 days and an even more widespread increase in NMDA channel activation 6 weeks after injury [66]. After severe injury there is an initial increase in expression of calcium-permeable AMPA receptors, which may reverse later [67, 68]. Activity and expression of glutamate transporters is also disrupted in acute, mild-moderate injury, impairing uptake capacity [58, 69, 70] and glutamate cycling between astrocytes and neurons. It is unknown whether changes in the cellular patterns of EAAT expression, such as increased expression of typically astroglial EAAT isoforms on neurons and microglia, are pathological, compensatory, or both [71,72,73]. In human cortex, expression of EAAT2 on glial cells was decreased within a day after TBI, while reduced expression of EAAT2 was still evident in postmortem samples with post-injury survival times of months or years [74].
Clinical findings
Clinical attempts to prevent acute neurotoxicity in moderate-severe TBI by blocking NMDAR signaling failed to improve outcomes [75, 76]. Although there are a number of technical and methodological challenges associated with these trials, they did not account for two key biological factors. First, the rapid reversal from glutamatergic hyperexcitability to hypoexcitability possibly resulted in drug treatments further exacerbating glutamatergic hypofunction [62, 77, 78]. Second, is a failure to appreciate the biology of synaptic versus extrasynaptic NMDA receptors [76, 79]. Blocking extrasynaptic NMDA signaling may initiate neuronal apoptosis [76, 80], amplifying secondary damage after injury. Ultimately, exclusive targeting of NMDA-mediated glutamate signaling failed to re-establish an appropriate excitatory (glutamate) and inhibitory (GABA) environment that could positively impact recovery trajectory.
GABAergic neurotransmission in TBI
Inhibitory Gamma aminobutyric acid (GABA) transmission balances excitatory glutamatergic signaling. E/I balance integrates inhibitory and excitatory signaling through feed-back (direct response to excitatory activity) and feed-forward (stimulation of other GABAergic neurons) mechanisms [34]. E/I balance represents a fundamental biological process found in circuits throughout the CNS, and perturbations of this factor typically yield dysfunction of synapses and circuits, leading to alterations in behavior and function [81].
Preclinical findings
GABA signals through ionotropic GABAA or GABAC receptors or metabotropic GABAB receptors. GABAA receptors are the most well studied of the GABA receptors and are heteromeric pentamers derived from at least 19 separate proteins classified by sequence homology into six classes [82]. GABAA receptors are targeted to synaptic or extrasynaptic membranes by their subunit composition, and synaptic or extrasynaptic localization of GABAA receptors determines tonic and phasic inhibitory tone, respectively [83, 84]. Increased expression of extrasynaptic GABAA receptors increases GABAergic tone, but concurrently reduces the phasic response [83]. Within a week after TBI, there is a shift in the balance of synaptic GABAA versus extrasynaptic GABA receptors [85, 86], summarized in Fig. 3. This suggests a change in E/I balance or synaptic scaling due to the differences in downstream signaling following activation of these differentially localized receptors. Twelve hours after moderate-severe TBI, GABAR binding was decreased [87], while another group found increased GABA receptor binding near the lesion center several days after severe injury [66].
Taken together, these findings indicate a role for alterations of GABA receptor expression and function in TBI. Acute changes in the expression of genes regulating GABA, glutamate, and E/I balance as part of the initial response to TBI may stabilize through epigenetic mechanisms, leading to long-lasting changes in homeostatic control [88]. Damage to mechanisms governing E/I balance and an inability to either repair or compensate may ultimately lead to inappropriate neural response and maladaptive plasticity [11, 89].
E/I balance: a moving target
Preclinical findings
Changes in the cellular machinery regulating glutamatergic and GABAergic signaling only provide indirect evidence of altered E/I balance after TBI, while preclinical models afford access to direct measurements of excitation and inhibition in neural circuits after experimental injury. Within hours of mild injury, hippocampal LTP is suppressed, consistent with overall glutamatergic hypofunction reported early in TBI [90,91,92,93]. Notably, changes in the brain following injury are not limited to the lesion center, or even adjacent areas, but may include widely distributed brain circuits. For example, in disparate cortical regions, decreased firing frequency, increased latency of evoked response, and elevated action potential thresholds persist at least several days after mild or moderate injury to the sensory cortex [94,95,96]. Concurrently, there are differential shifts in E/I balance within the hippocampal circuit and impairment of LTP and NMDA-evoked currents in CA1 [97, 98]. Also within this timeframe, inhibitory signaling in the amygdala is decreased, suggesting hyperexcitability in subcortical structures regulating emotion [99].
Disruption of E/I balance continues to change over weeks, months, and years, as TBI progresses through subacute and chronic phases, although mechanisms underlying these changes are not uniform across or within brain regions. Two weeks after moderate-severe experimental injury, exaggerated GABAergic inhibition in frontal cortex is reversible by GABA inhibitors, but over time GABAergic tone is restored and cortical hypo-excitation reflects hypotrophy in deep-layer cortical pyramidal neurons [100, 101]. In the dentate gyrus there is a gradual loss of phasic GABAergic inhibition over 6 months ipsilateral to severe injury, becoming bilateral over time [102]. Evoked responses to extrasynaptic GABAR agonist are diminished both early after moderate-severe TBI and chronically [103]. Also in severe injury, tonic GABAergic inhibition in the dentate gyrus may increase over time [104]. Remodeling of hippocampal circuits in severe TBI increases excitatory inputs from CA3 and granule cells into hilar interneurons 10 weeks after injury. In contrast, inhibitory inputs to the dentate gyrus are decreased, leaving the overall balance of signaling excitatory [105]. After severe injury, spontaneous epileptiform electrical activity increases in hippocampus within weeks of injury, along with the stimulus threshold to initiate LTP [106], while after mild injury LTP can be initiated, but not sustained [107]. These changes highlight the complexity of the post-injury milieu, where individual circuits, cells and/or synapses may differentially respond to pharmacological interventions.
Clinical findings
Thought to reflect changes in E/I balance, post-traumatic seizures have a cumulative incidence of up to 20% by 5 years [108]. The latency period associated with the development of post-traumatic epilepsy further supports progressive pathological changes in E/I balance after injury. Clinical trials to prevent post-traumatic seizures after moderate to severe TBI effectively prevented early (within 7 days) but not later seizures [13, 14, 109]. Phenytoin and leviteracetam are anti-epileptic medications whose mechanisms of action are thought to involve diffuse blockage of voltage-gated channels. The clinical effectiveness of these drugs for subacute but not chronic seizure is additional evidence that E/I balance evolves over the course of injury.
Emerging shifts in E/I balance suggest that modulating glutamate and/or GABA neurotransmission could restore neuroplastic function and improve cognitive symptoms in chronic TBI. The NMDAR antagonist/dopamine agonist, amantadine, 100 mg twice daily, alleviated aggressive behavior after TBI [110], but the evidence for efficacy in other behavioral domains was equivocal [111,112,113]. In a multicenter study of 184 patients, amantadine (maximum dose 200 mg twice daily) administered subacutely (initiated 4−16 weeks after injury) improved functional recovery in patients with disorders of consciousness [12]. A similar drug, the uncompetitive NMDA receptor antagonist memantine [114, 115], was approved for clinical trials but small sample size (n = 11) and early termination due to lack of enrollment provided uninterpretable results (NCT00462228).
Taken together, these data suggest a central role for abnormalities of glutamate and GABA neurotransmission in acute and chronic TBIs of varying severity. Changes encompassing receptors, transporters, release mechanisms, and signaling cascades clearly establish that defects of neuroplasticity emerge following brain injury. However, the glutamate and GABA neurotransmitter systems do not operate in isolation. Excitatory and inhibitory tone is modulated by inputs from local and widely distributed diverse subtypes of neurons.
Acetylcholine neurotransmission in TBI
Cholinergic inputs into frontal cortex and hippocampus regulate attention and memory consolidation. Acetylcholine signals through two classes of receptor expressed on both neurons and glia, G-protein-coupled muscarinic receptors which can be excitatory or inhibitory, and ionotropic (nicotinic) receptors which are excitatory [116]. Acetylcholine synthesis, release, and degradation are regulated by presynaptic choline acetyltransferase (ChAT), the vesicular transporter vAChT, and postsynaptic acetylcholinesterase (AChE), respectively [116]. Acetylcholine release can increase spontaneous activity, facilitate evoked responses, or inhibit evoked responses [117]; however, the overall effect of acetylcholine signaling in cortex is to enhance NMDA-mediated currents [116, 117]. Acetylcholine release exhibits both cue-evoked spikes in acetylcholine receptor activity, as well as slower increases in activity corresponding to attentional processes [118].
Preclinical findings
After TBI, there is acute release of acetylcholine that increases extracellular acetylcholine concentration followed by long-term suppression of acetylcholine signaling [119, 120], illustrated in Fig. 3. Receptor binding and vChAT transporter density decrease in preclinical models of moderate injury as early as 1 h after TBI, and may persist for at least 3 days [87, 119, 121]. Two weeks after injury, evoked release of acetylcholine is impaired in moderate TBI animals [122]. In mild TBI patients, acetylcholine hypofunction and decreased AChE activity can be evident more than a year after injury [120].
Preclinical and postmortem evidence indicate cholinergic projections from basal forebrain are susceptible to damage after TBI [119, 123]. Cortical projections from the basal forebrain may be particularly vulnerable to accumulation of neurofibrillary tangles and tau protein aggregation, potentially contributing to chronic, cholinergic hypofunction after TBI [123].
Clinical findings
A variety of pro-cholinergic agents, including receptor agonists [124], cholinesterase inhibitors [125], and mechanisms activating acetylcholine release [126], showed promise in preclinical TBI models. Acetycholinesterase inhibitors, notably donepezil and rivastigmine, went to randomized controlled trials (RCTs) [127,128,129]. Both drugs showed mildly beneficial effects in specific cognitive domains of attention and short-term memory [127,128,129]. In chronic (>1 year) mild TBI subjects recruited from a previous rivastigmine trial, patients who responded to a minimum daily dose of 3 mg daily of rivastigmine had significantly lower initial acetylcholinesterase activity than nonresponders [130]. Although hampered by the small sample sizes common to most human imaging studies, additional studies of this kind, specifically identifying biologically distinct subpopulations of TBI subjects, may provide important insight into tailoring treatment strategies.
Catecholamines in TBI
The principal source of forebrain the catecholamines dopamine (DA) and norepinephrine (NE) is dorsal midbrain (DA) and brainstem (NE) [131]. Changes in DA and NE after TBI are summarized in Fig. 3. Catecholaminergic nuclei are vulnerable to direct brainstem damage and shearing of projections to forebrain, and are particularly susceptible to metabolic stress due to intrinsically high energy requirements [131]. Catecholaminergic nuclei receive reciprocal projections from cortex, and damage to these inputs from forebrain after TBI will affect catecholaminergic regulation. Tonic NE signaling mediates arousal and wakefulness, while phasic firing is associated with attentional focus and vigilance [131, 132].
Preclinical findings
The most consistent preclinical finding for noradrenergic signaling is turnover of norepinephrine [131]. The initial accelerated turnover of cortical NE reverses by 6 h, and suppression of NE turnover is still evident 8 weeks after moderate-severe injury [133,134,135]. Concurrent downregulation of α1 adrenoreceptors is evident within 30 min after TBI [136, 137]. Atomoxetine (80 mg/day for 14 days), an NE uptake inhibitor, provided no significant improvement in attention or in self-reported post-injury depressive symptoms [138].
Dopamine signals through G-protein-coupled receptors [131] and is removed from the synapse by the neuronal dopamine transporter DAT or enzymatic degradation [139]. In preclinical models of moderate TBI, there is a progressive loss of dopaminergic neurons in the substantia nigra [140, 141], evident from 2 weeks and continuing for months post injury. Similar to other neurotransmitters, there is an acute rise in extracellular dopamine evident 1 h after moderate-severe injury, which is sustained in some regions for a day or more [131, 142]. Regional increases in tissue dopamine levels potentially last considerably longer, up to several weeks in severe injury [143]. However, other studies support an initial increase in tissue dopamine followed closely by prolonged dopaminergic hypofunction in severe injury [144, 145]. Preclinical studies suggest dopaminergic dysfunction after TBI is driven by defects in tonic and evoked dopamine release, as well as impaired dopamine reuptake into neurons, rather than through alterations in dopamine receptor expression [144, 145]. PET imaging indicates regional changes in both DAT activity and D2 receptor binding in human TBI [131, 146].
Clinical findings
Dopamine has potent modulatory effects on glutamate transmission suggesting that dopaminergic drugs have potential to improve cognitive domain symptoms after TBI. The D2-dopamine receptor agonist bromocriptine, titrated to a dose of 5 mg twice daily for 6 weeks, performed no better than placebo in resolving cognitive symptoms [147]. Methylphenidate and amphetamines, typically used to treat disorders of attention, produced equivocal effects on cognition, although interpretation of study results is limited by small sample sizes [148, 149]. Armodafinil (50−150 mg/12 weeks), an indirect dopamine agonist approved for treating narcolepsy, was effective in increasing wakefulness and sleep latency in TBI patients with excessive sleepiness in a dose-dependent manner [15]. Similar to the amantadine trial [12], dopamine modulation demonstrates efficacy for improving wakefulness, but long-term positive effects on cognition are unproven.
Serotonin neurotransmission in TBI
Serotonin originates in the raphe nucleus and is distributed throughout the forebrain. There are at least 16 serotonin receptors expressed on both excitatory and inhibitory neurons [81] and once released, serotonin is taken up by presynaptic serotonin transporters [150].
Preclinical findings
After moderate-severe TBI, serotonin transporters are downregulated within a day and remain downregulated for at least 2 weeks [151] (Fig. 3). In preclinical models of moderate-severe injury, the tricyclic antidepressant imipramine (20 mg/kg for 2 or 4 weeks) and the selective serotonin reuptake inhibitor (SSRI), fluoxetine (10 mg/kg for 4 weeks), increased neurogenesis measured 4 weeks after TBI; only the tricyclic improved recognition memory [6, 152]. Neither of these medications have gone into clinical trials for reversal of cognitive defects in TBI.
Clinical findings
As depression and anxiety develops in many patients after injury, and post-TBI depression has clinical features that mirror noninjury depression, it has been presumed that SSRIs could be useful for prevention and treatment of post-TBI depression. Over 25 RCTs, open label, or case studies have tested SSRIs as treatment for post-TBI depression and cognitive dysfunction, with varying results [153]. Disruption of the serotonergic system likely begins soon after injury. Subacute (initiated on average at 3 weeks post-TBI) administration of the SSRI sertraline for 12 weeks, at a dose of up to 100 mg/day mitigated depressive symptoms during the course of treatment, but did not provide lasting protection against development of post-TBI depression [154,155,156]. Sertraline did not prevent or improve deficits in attention, memory, or executive function [16, 157]. A recent trial, however, showed improvement in information processing in doses up to 200 mg/day, but suggested the dopaminergic activity of sertraline as the mechanism [158]. In summary, the SSRIs are promising interventions for improving post-TBI depression in chronic TBI, but improvement in cognitive symptoms after TBI needs further study.
Adenosine neurotransmission in TBI
Adenosine signals through G-protein-coupled A1,A2A, A2B and A3 receptors or through a myriad of metabotropic P2X and P2Y receptors, expressed throughout the brain [159, 160]. Adenosine accumulates rapidly after injury due to breakdown of adenosine triphosphate [159]. Adenosine levels are regulated by 5′-nucleotidases, which generate adenosine from adenosine phosphates, adenosine deaminase, which converts adenosine to inosine, adenosine kinase which phosphorylates adenosine to become adenosine monophosphate, and nucleoside transporters, none of which are well studied after TBI [159].
Adenosine acts presynaptically to suppress excitatory transmitter release and postsynaptically to maintain hyperpolarization [159, 161]. Adenosine triphosphate released from astrocytes stimulates hippocampal interneurons as part of homeostatic regulation of E/I balance [162, 163]. However, adenosine itself can suppress tonic GABAergic signaling and astroglial glutamate uptake [164, 165]. Signaling through A1 and A2A receptors has opposing effects on excitation; A1R is primarily inhibitory, while A2AR facilitates synaptic signaling [161]. Local glutamate levels regulate adenosine A2AR signaling [166], while imbalance in adenosine signaling affects working and short-term memory, and goal directed or effortful behavior [161].
Preclinical findings
While the adenosine neurotransmitter machinery is only recently gaining attention in experimental TBI, extracellular concentrations of adenosine, and its metabolites inosine and hypoxanthine are known to increase within minutes after injury [167]. Extracellular adenosine returns to basal levels within an hour, but inosine and hypoxanthine remain elevated [167]. Genetic knockout of the A1 receptor results in lethal status epilepticus after experimental TBI [168]. Conversely, attenuation of A2A receptor signaling improves aspects of TBI symptomology [169, 170]. The modulatory roles of the adenosine neurotransmitter system suggests this system as a high-yield substrate for pharmacological intervention. Supporting this hypothesis, polymorphisms of adenosine regulatory genes are associated with an increased risk for developing post-traumatic epilepsy [171,172,173].
In summary, a single neurotransmitter-focused approach has not (so far) delivered targets yielding efficacious therapies for the cognitive effects of TBI. A more integrated approach accounting for diverse neurotransmitter systems is needed to develop novel therapeutics to modulate and/or restore function to premorbid states.
Integration of diverse neurotransmitter systems: modulation of E/I balance
Effective neurotransmission requires constant rebalancing of excitatory and inhibitory signaling. Collectively and with varying impact, diverse neurotransmitter systems work cooperatively in healthy brain to fine-tune E/I balance [174]. Hypofunctional D1-dopamine receptor signaling impairs LTP in hippocampus [175], and interactions between dopamine and acetylcholine facilitate LTP in frontal cortex [176]. The D2-dopamine receptor directly interacts with both adenosine and metabotropic glutamate receptors to modulate excitatory transmission [175]. Cholinergic and dopaminergic neurons contact both glutamatergic neurons and GABAergic interneurons, tuning the response of the neuronal ensemble [176]. Comodulation of synaptic plasticity by dopamine and serotonin also cooperatively shifts E/I balance towards either LTP or LTD [81]. NE sensitizes cortical circuits reflecting its role in vigilance and risk assessment [174], while decreasing serotonin signaling through 5-HT1A receptors potentiates circuit inhibition [177].
Concerted actions of multiple transmitter systems coordinate neurotransmission at individual synapses and within neural circuits across multiple temporal scales. The effects of injury on the coordination between neurotransmitter systems to regulate cognitive and emotional processing, at any stage of injury, is poorly understood. The complex nature of neurotransmission, including the intricate interactions between neurotransmitter systems, suggests that less-specific drugs may be more efficacious than highly selective compounds. This notion is supported by the relatively promiscuous receptor binding profiles of the TCAs and amantadine, drugs that appear to provide improvement in a few symptom domains of TBI [178,179,180].
Opportunities for future work
Despite intense interest in identifying the underpinnings of cognitive and behavioral symptoms after TBI, there are still significant gaps in our understanding of how injury impacts neurotransmission. For example, very little is known about extrasynaptic signaling mechanisms after TBI. In contrast to synaptic NMDAR signaling that promotes cell survival, extrasynaptic NMDAR signaling activates cell death pathways and likely contributes to early cell loss and excitotoxicity after TBI [79]. Extrasynaptic NMDAR signaling also antagonizes coupling of glutamate signaling to protein transcription, blocking processes required for memory consolidation and potentially contributing to cognitive dysfunction well after acute stages of injury [181].
Extrasynaptic glutamate comes from several sources, including astrocytic release and spillover from neuronal synapses. The cystine-glutamate antiporter, xCT, is a significant source of extrasynaptic glutamate [182]. Glutamate released by astrocytes into the extrasynaptic space via system xC‾ can stimulate metabotropic glutamate receptors peripheral to the synapse as well as extrasynaptic ionotropic receptors [182]. System xc‾ also contributes to the physiologic roles of extrasynaptic glutamate in synaptic scaling [183]. Expression and activity of xCT and system xc‾ is regulated by cytokine signaling (notably IL-1β and TNFα), NO− and other reactive species, growth factors, and neuronal activity, linking inflammation and metabolic stress to regulation of synaptic strength and excitatory transmission [182]. Pharmacologically blocking system xc‾ produces defects in working memory and long-term memory; however hyperactivity of system xc‾ is potentially even more damaging due to excitotoxic activation of extrasynaptic NMDARs [182]. Despite the recognized role of system xc‾ in tuning excitatory transmission, it has not been studied in TBI.
Recent studies indicate that lactate can function as a neurotransmitter [184, 185], through a G-protein-coupled receptor, hydrocarboxylic receptor 1 HCA1 (GPR81). HCA1 is expressed on pyramidal neurons in cerebellum, hippocampus, and cortex, and to a lesser extent astrocytes [184, 186]. HCA1 activation suppresses neuronal activity [187] and HCA1 signaling requires high extracellular lactate concentrations. These findings suggest HCA1 is activated in response to injury or intense neural activity [184]. Interestingly, an acute (minutes to hours) increase in brain lactate after severe TBI [10, 188] is associated with poor outcomes [189]. Paradoxically, sodium lactate infusion shows some beneficial effect and is being trialed to prevent acute increases in intracranial pressure in severe injury [190]. Whether HCA1 activation contributes to either the beneficial or negative effects of acutely elevated lactate levels after TBI is unknown. It is also unknown whether the metabolic switch to aerobic glycolysis and subsequent increase in lactate production in activated glia influences HCA1 signaling in TBI or other inflammatory neurodegenerative disorders.
Metabolic imaging approaches such as magnetic resonance spectroscopy (MRS) and positron emission tomography (PET) have great potential to provide insight into the regulation and function of brain activity and amino acid neurotransmitter systems after TBI, particularly when combined with other imaging modalities. However, these technologies are not yet being used to maximal effect in TBI research. MRS studies report both increased [191, 192] and decreased [193, 194] tissue concentrations of glx, the combined glutamate/glutamine signal. Inconsistencies may have to do with timing, ROI, subject populations, or methodological differences. In general, interpretation of imaging studies in TBI is hindered by small sample size. Despite available PET ligands for all of the major transmitter systems, it has been only minimally utilized [130, 146, 195, 196].
Conclusion
Pathological processes after brain injury continuously evolve (Fig. 4) [197], and we are only now beginning to understand compensatory and recovery mechanisms that could be enhanced [198,199,200]. There are abundant opportunities for improvement in study design and outcomes reporting in TBI clinical drug trials [112, 201]. Most previous clinical trials included patients with similar injury severities graded by GCS, but overlooked key clinical features and commonalities in the pathophysiology of injury [19, 202]. The next big clinical trials will involve identifying the appropriate patient populations for targeted therapeutic interventions. Currently an international effort is underway to advance innovation in TBI therapeutics via comparative effectiveness research and open source data sharing. The international initiative for TBI research (InTBIR), a collaborative between the European Commission, the National Institutes of Health, and the Canadian Institute of Health Research, is tasked with changing the face of TBI research by 2020 [203, 204]. A crucial part of the new infrastructure for TBI research includes creation and utilization of Common Data Elements for TBI [205]. The Transforming Research and Clinical Knowledge in TBI (TRACK-TBI) [205] and the Collaborative European Neurotrauma Effectiveness Research in TBI (CENTER-TBI) [206] prospective longitudinal observational studies are critical to this effort. Data collected will be publically available for research through the Federal Interagency Traumatic Brain Injury Research (FITBIR) informatics system [207, 208]. These data will allow more nuanced approaches in selecting study populations and stratifying treatment responses. Identifying commonalities in study subpopulations is a promising focus for future tailoring of TBI treatment. Finally, focusing treatment strategies on individual neurotransmitters has not been successful. Identifying pathophysiological measures that reflect circuit level changes in plasticity, such as CSDs, may present an opportunity to integrate findings from diverse neurotransmitter systems and provide new targeting strategies for this difficult to treat condition.

Temporal evolution of TBI pathophysiology. In response to mechanical damage, there is a rapid unregulated release of ions and transmitters and mitochondrial damage that can result in edema, and transient cell membrane depolarizations. Unregulated transmission quickly leads to metabolic depression and hypofunction. Mechanisms of damage and repair, including changes in gene expression, induction of inflammatory glial responses, structural remodeling of proteins, cells, and circuits, and proliferation of neurons and glia are ongoing from acute, through subacute and into chronic stages of injury. In response to these secondary damage signals, regional differences in E/I balance can develop leading to regional hypofunction or hyperexcitability and chronic neurological, cognitive, and behavioral symptoms
References
Taylor CA, Bell JM, Breiding MJ, Xu L. Traumatic brain injury–related emergency department visits, hospitalizations, and deaths—United States, 2007 and 2013. Morb Mortal Wkly Rep Surveill Summ. 2017;66:1–16.
Roozenbeek B, Maas AI, Menon DK. Changing patterns in the epidemiology of traumatic brain injury. Nat Rev Neurol. 2013;9:231–6.
Griesbach GS, Kreber LA, Harrington D, Ashley MJ. Post-acute traumatic brain injury rehabilitation: effects on outcome measures and life care costs. J Neurotrauma. 2015;32:704–11.
Kondo A, Shahpasand K, Mannix R, Qiu J, Moncaster J, Chen CH, et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature. 2015;523:431–6.
Hartings JA, Strong AJ, Fabricius M, Manning A, Bhatia R, Dreier JP, et al. Spreading depolarizations and late secondary insults after traumatic brain injury. J Neurotrauma. 2009;26:1857–66.
Han X, Tong J, Zhang J, Farahvar A, Wang E, Yang J, et al. Imipramine treatment improves cognitive outcome associated with enhanced hippocampal neurogenesis after traumatic brain injury in mice. J Neurotrauma. 2011;28:995–1007.
Ngwenya LB, Mazumder S, Porter ZR, Minnema A, Oswald DJ, Farhadi HF. Implantation of neuronal stem cells enhances object recognition without increasing neurogenesis after lateral fluid percussion injury in mice. Stem Cells Int. 2018;2018:4209821.
Kovesdi E, Kamnaksh A, Wingo D, Ahmed F, Grunberg NE, Long JB, et al. Acute minocycline treatment mitigates the symptoms of mild blast-induced traumatic brain injury. Front Neurol. 2012;3:111.
Siopi E, Llufriu-Daben G, Fanucchi F, Plotkine M, Marchand-Leroux C, Jafarian-Tehrani M. Evaluation of late cognitive impairment and anxiety states following traumatic brain injury in mice: the effect of minocycline. Neurosci Lett. 2012;511:110–5.
Lama S, Auer RN, Tyson R, Gallagher CN, Tomanek B, Sutherland GR. Lactate storm marks cerebral metabolism following brain trauma. J Biol Chem. 2014;289:20200–8.
Dorsett CR, McGuire JL, DePasquale EA, Gardner AE, Floyd CL, McCullumsmith RE. Glutamate neurotransmission in rodent models of traumatic brain injury. J Neurotrauma. 2017;34:263–72.
Giacino JT, Whyte J, Bagiella E, Kalmar K, Childs N, Khademi A, et al. Placebo-controlled trial of amantadine for severe traumatic brain injury. N Eng J Med. 2012;366:819–26.
Klein P, Herr D, Pearl PL, Natale J, Levine Z, Nogay C, et al. Results of phase 2 safety and feasibility study of treatment with levetiracetam for prevention of posttraumatic epilepsy. Arch Neurol. 2012;69:1290–5.
Szaflarski JP, Sangha KS, Lindsell CJ, Shutter LA. Prospective, randomized, single-blinded comparative trial of intravenous levetiracetam versus phenytoin for seizure prophylaxis. Neurocrit Care. 2010;12:165–72.
Menn SJ, Yang R, Lankford A. Armodafinil for the treatment of excessive sleepiness associated with mild or moderate closed traumatic brain injury: a 12-week, randomized, double-blind study followed by a 12-month open-label extension. J Clin Sleep Med. 2014;10:1181–91.
Jorge RE, Acion L, Burin DI, Robinson RG. Sertraline for preventing mood disorders following traumatic brain injury: a randomized clinical trial. JAMA Psychiatry. 2016;73:1041–7.
Choe MC, Giza CC. Diagnosis and management of acute concussion. Semin Neurol. 2015;35:29–41.
Teasdale G, Maas A, Lecky F, Manley G, Stocchetti N, Murray G. The Glasgow Coma Scale at 40 years: standing the test of time. Lancet Neurol. 2014;13:844–54.
Saatman KE, Duhaime AC, Bullock R, Maas AI, Valadka A, Manley GT. Classification of traumatic brain injury for targeted therapies. J Neurotrauma. 2008;25:719–38.
Farkas O, Povlishock JT. Cellular and subcellular change evoked by diffuse traumatic brain injury: a complex web of change extending far beyond focal damage. Prog Brain Res. 2007;161:43–59.
Barkhoudarian G, Hovda DA, Giza CC. The molecular pathophysiology of concussive brain injury—an update. Phys Med Rehabil Clin N Am. 2016;27:373–93.
Gaetz M. The neurophysiology of brain injury. Clin Neurophysiol. 2004;115:4–18.
Bramlett HM, Deitrich WD. Long-term consequences of traumatic brain injury: current status of 10.1038/s41380-018-0239-6 potential mechanisms of injury and neurological outcomes. J Neurotrauma. 2015;32:1834–48.
Ruff RM, Crouch JA, Troster AI, Marshall LF, Buchsbaum MS, Lottenberg S, et al. Selected cases of poor outcome following a minor brain trauma: comparing neuropsychological and positron emission tomography assessment. Brain Inj. 1994;8:297–308.
Tortella FC. Challenging the paradigms of experimental TBI models: from preclinical to clinical practice. In: Kobeissy FH, Dixon CE, Hayes RL, Mondello S, editors. Injury models of the central nervous system: methods and protocols. New York, NY: Springer New York; 2016. p. 735−40.
Bolouri H, Zetterberg H. Frontiers in neuroengineering animal models for concussion: molecular and cognitive assessments—relevance to sport and military concussions. In: Kobeissy FH, editor. Brain neurotrauma: molecular, neuropsychological, and rehabilitation aspects. Boca Raton, FL: CRC Press/Taylor & Francis; 2015.
Wong VS, Langley B. Epigenetic changes following traumatic brain injury and their implications for outcome, recovery and therapy. Neurosci Lett. 2016;625:26–33.
Corrigan JD, Harrison-Felix C, Bogner J, Dijkers M, Terrill MS, Whiteneck G. Systematic bias in traumatic brain injury outcome studies because of loss to follow-up. Arch Phys Med Rehabil. 2003;84:153–60.
Xiong Y, Mahmood A, Chopp M. Animal models of traumatic brain injury. Nat Rev Neurosci. 2013;14:128–42.
Dewitt DS, Perez-Polo R, Hulsebosch CE, Dash PK, Robertson CS. Challenges in the development of rodent models of mild traumatic brain injury. J Neurotrauma. 2013;30:688–701.
Blanchard RJ, Blanchard DC. Attack and defense in rodents as ethoexperimental models for the study of emotion. Prog Neuropsychopharmacol Biol Psychiatry. 1989;13(Suppl):S3–14.
Belanger HG, Proctor-Weber Z, Kretzmer T, Kim M, French LM, Vanderploeg RD. Symptom complaints following reports of blast versus non-blast mild TBI: does mechanism of injury matter? Clin Neuropsychol. 2011;25:702–15.
Morrison B 3rd, Elkin BS, Dolle JP, Yarmush ML. In vitro models of traumatic brain injury. Annu Rev Biomed Eng. 2011;13:91–126.
Isaacson JS, Scanziani M. How inhibition shapes cortical activity. Neuron. 2011;72:231–43.
Alwis DS, Rajan R. Environmental enrichment and the sensory brain: the role of enrichment in remediating brain injury. Front Syst Neurosci. 2014;8:156.
Bach-y-Rita P. Theoretical basis for brain plasticity after a TBI. Brain Inj. 2003;17:643–51.
Dymowski AR, Owens JA, Ponsford JL, Willmott C. Speed of processing and strategic control of attention after traumatic brain injury. J Clin Exp Neuropsychol. 2015;37:1024–35.
Nelson LD, Ranson J, Ferguson AR, Giacino J, Okonkwo DO, Valadka A, et al. Validating multidimensional outcome assessment using the TBI common data elements: an analysis of the TRACK-TBI pilot sample. J Neurotrauma. 2017;34:3158–72.
McDonald BC, Flashman LA, Saykin AJ. Executive dysfunction following traumatic brain injury: neural substrates and treatment strategies. NeuroRehabilitation. 2002;17:333–44.
Draper K, Ponsford J. Cognitive functioning ten years following traumatic brain injury and rehabilitation. Neuropsychology. 2008;22:618–25.
Whelan-Goodinson R, Ponsford J, Johnston L, Grant F. Psychiatric disorders following traumatic brain injury: their nature and frequency. J Head Trauma Rehabil. 2009;24:324–32.
Guillamondegui OD, Montgomery SA, Phibbs FT, McPheeters ML, Alexander PT, Jerome RN, et al. AHRQ comparative effectiveness reviews. Traumatic brain injury and depression. Rockville, MD: Agency for Healthcare Research and Quality (US); 2011.
Perry DC, Sturm VE, Peterson MJ, Pieper CF, Bullock T, Boeve BF, et al. Association of traumatic brain injury with subsequent neurological and psychiatric disease: a meta-analysis. J Neurosurg. 2016;124:511–26.
Vaishnavi S, Rao V, Fann JR. Neuropsychiatric problems after traumatic brain injury: unraveling the silent epidemic. Psychosomatics. 2009;50:198–205.
Gould KR, Ponsford JL, Spitz G. Association between cognitive impairments and anxiety disorders following traumatic brain injury. J Clin Exp Neuropsychol. 2014;36:1–14.
Osborn AJ, Mathias JL, Fairweather-Schmidt AK. Depression following adult, non-penetrating traumatic brain injury: a meta-analysis examining methodological variables and sample characteristics. Neurosci Biobehav Rev. 2014;47:1–15.
Jesulola E, Micalos P, Baguley IJ. Understanding the pathophysiology of depression: from monoamines to the neurogenesis hypothesis model—are we there yet? Behav Brain Res. 2017;341:79–90.
Bhattacharya A, Drevets WC. Role of neuro-immunological factors in the pathophysiology of mood disorders: implications for novel therapeutics for treatment resistant depression. Curr Top Behav Neurosci. 2017;31:339–56.
Sanacora G, Treccani G, Popoli M. Towards a glutamate hypothesis of depression: an emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology. 2012;62:63–77.
Danbolt NC, Chaudhry FA, Dehenes Y, Lehre KP, Ullensvang K, Storm-Mathisen J. Properties and localization of glutamate transporters. Prog Brain Res. 1998;116:23–43.
O’Donovan SM, Sullivan CR, McCullumsmith RE. The role of glutamate transporters in the pathophysiology of neuropsychiatric disorders. NPJ Schizophr. 2017;3:32.
Levenson J, Weeber E, Selcher JC, Kategaya LS, Sweatt JD, Eskin A. Long-term potentiation and contextual fear conditioning increase neuronal glutamate uptake. Nat Neurosci. 2002;5:155–61.
Katayama Y, Becker DP, Tamura T, Hovda DA. Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J Neurosurg. 1990;73:889–900.
Zhang H, Zhang X, Zhang T, Chen L. Excitatory amino acids in cerebrospinal fluid of patients with acute head injuries. Clin Chem. 2001;47:1458–62.
Yamamoto T, Rossi S, Stiefel M, Doppenberg E, Zauner A, Bullock R, et al. CSF and ECF glutamate concentrations in head injured patients. Acta Neurochir Suppl. 1999;75:17–9.
Faden AI, Demediuk P, Panter SS, Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:798–800.
Rimmele TS, Rocher AB, Wellbourne-Wood J, Chatton JY. Control of glutamate transport by extracellular potassium: basis for a negative feedback on synaptic transmission. Cereb Cortex. 2017;27:3272–83.
Dorsett CR, McGuire JL, Niedzielko TL, DePasquale EA, Meller J, Floyd CL, et al. Traumatic brain injury induces alterations in cortical glutamate uptake without a reduction in glutamate dransporter-1 protein expression. J Neurotrauma. 2017;34:220–34.
Hinzman JM, Wilson JA, Mazzeo AT, Bullock MR, Hartings JA. Excitotoxicity and metabolic crisis are associated with spreading depolarizations in severe traumatic brain injury patients. J Neurotrauma. 2016;33:1775–83.
Torrente D, Cabezas R, Avila MF, Garcia-Segura LM, Barreto GE, Guedes RC. Cortical spreading depression in traumatic brain injuries: is there a role for astrocytes? Neurosci Lett. 2014;565:2–6.
Hartings JA. Spreading depolarization monitoring in neurocritical care of acute brain injury. Curr Opin Crit Care. 2017;23:94–102.
Shohami E, Biegon A. Novel approach to the role of NMDA receptors in traumatic brain injury. CNS Neurol Disord Drug Targets. 2014;13:567–73.
Henry LC, Tremblay S, Boulanger Y, Ellemberg D, Lassonde M. Neurometabolic changes in the acute phase after sports concussions correlate with symptom severity. J Neurotrauma. 2010;27:65–76.
Biegon A, Fry PA, Paden CM, Alexandrovich A, Tsenter J, Shohami E. Dynamic changes in N-methyl-d-aspartate receptors after closed head injury in mice: Implications for treatment of neurological and cognitive deficits. Proc Natl Acad Sci USA. 2004;101:5117–22.
Miller LP, Lyeth BG, Jenkins LW, Oleniak L, Panchision D, Hamm RJ, et al. Excitatory amino acid receptor subtype binding following traumatic brain injury. Brain Res. 1990;526:103–7.
Lopez-Picon F, Snellman A, Shatillo O, Lehtiniemi P, Gronroos TJ, Marjamaki P, et al. Ex vivo tracing of NMDA and GABA-A receptors in rat brain after traumatic brain injury using 18F-GE-179 and 18F-GE-194 autoradiography. J Nucl Med. 2016;57:1442–7.
Kharlamov EA, Lepsveridze E, Meparishvili M, Solomonia RO, Lu B, Miller ER, et al. Alterations of GABA(A) and glutamate receptor subunits and heat shock protein in rat hippocampus following traumatic brain injury and in posttraumatic epilepsy. Epilepsy Res. 2011;95:20–34.
Spaethling JM, Klein DM, Singh P, Meaney DF. Calcium-permeable AMPA receptors appear in cortical neurons after traumatic mechanical injury and contribute to neuronal fate. J Neurotrauma. 2008;25:1207–16.
Rao VL, Baskaya MK, Dogan A, Rothstein JD, Dempsey RJ. Traumatic brain injury down-regulates glial glutamate transporter (GLT-1 and GLAST) proteins in rat brain. J Neurochem. 1998;70:2020–7.
Yi JH, Pow DV, Hazell AS. Early loss of the glutamate transporter splice-variant GLT-1v in rat cerebral cortex following lateral fluid-percussion injury. Glia. 2005;49:121–33.
Yi JH, Herrero R, Chen G, Hazell AS. Glutamate transporter EAAT4 is increased in hippocampal astrocytes following lateral fluid-percussion injury in the rat. Brain Res. 2007;1154:200–5.
van Landeghem FK, Stover JF, Bechmann I, Bruck W, Unterberg A, Buhrer C, et al. Early expression of glutamate transporter proteins in ramified microglia after controlled cortical impact injury in the rat. Glia. 2001;35:167–79.
Beschorner R, Dietz K, Schauer N, Mittelbronn M, Schluesener HJ, Trautmann K, et al. Expression of EAAT1 reflects a possible neuroprotective function of reactive astrocytes and activated microglia following human traumatic brain injury. Histol Histopathol. 2007;22:515–26.
van Landeghem FK, Weiss T, Oehmichen M, von Deimling A. Decreased expression of glutamate transporters in astrocytes after human traumatic brain injury. J Neurotrauma. 2006;23:1518–28.
Maas AI, Roozenbeek B, Manley GT. Clinical trials in traumatic brain injury: past experience and current developments. Neurotherapeutics. 2010;7:115–26.
Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002;1:383–6.
Muir KW. Glutamate-based therapeutic approaches: clinical trials with NMDA antagonists. Curr Opin Pharmacol. 2006;6:53–60.
Temkin NR, Anderson GD, Winn HR, Ellenbogen RG, Britz GW, Schuster J, et al. Magnesium sulfate for neuroprotection after traumatic brain injury: a randomised controlled trial. Lancet Neurol. 2007;6:29–38.
Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–14.
Ikonomidou C, Stefovska V, Turski L. Neuronal death enhanced by N-methyl-d-aspartate antagonists. Proc Natl Acad Sci USA. 2000;97:12885–90.
Meunier CN, Chameau P, Fossier PM. Modulation of synaptic plasticity in the cortex needs to understand all the players. Front Synaptic Neurosci. 2017;9:2.
Nutt D. GABAA receptors: subtypes, regional distribution, and function. J Clin Sleep Med. 2006;2:S7–11.
Wu X, Huang L, Wu Z, Zhang C, Jiang D, Bai Y, et al. Homeostatic competition between phasic and tonic inhibition. J Biol Chem. 2013;288:25053–65.
Wu X, Wu Z, Ning G, Guo Y, Ali R, Macdonald RL, et al. gamma-Aminobutyric acid type A (GABAA) receptor alpha subunits play a direct role in synaptic versus extrasynaptic targeting. J Biol Chem. 2012;287:27417–30.
Guerriero RM, Giza CC, Rotenberg A. Glutamate and GABA imbalance following traumatic brain injury. Curr Neurol Neurosci Rep. 2015;15:545.
Raible DJ, Frey LC, Cruz Del Angel Y, Russek SJ, Brooks-Kayal AR. GABA(A) receptor regulation after experimental traumatic brain injury. J Neurotrauma. 2012;29:2548–54.
Sihver S, Marklund N, Hillered L, Langstrom B, Watanabe Y, Bergstrom M. Changes in mACh, NMDA and GABA(A) receptor binding after lateral fluid-percussion injury: in vitro autoradiography of rat brain frozen sections. J Neurochem. 2001;78:417–23.
Wong VS, Langley B. Epigenetic changes following traumatic brain injury and their implications for outcome, recovery and therapy. Neurosci Lett. 2016;625:26–33.
Tomaszczyk JC, Green NL, Frasca D, Colella B, Turner GR, Christensen BK, et al. Negative neuroplasticity in chronic traumatic brain injury and implications for neurorehabilitation. Neuropsychol Rev. 2014;24:409–27.
Miyazaki S, Katayama Y, Lyeth BG, Jenkins LW, DeWitt DS, Goldberg SJ, et al. Enduring suppression of hippocampal long-term potentiation following traumatic brain injury in rat. Brain Res. 1992;585:335–9.
Sick TJ, Perez-Pinzon MA, Feng ZZ. Impaired expression of long-term potentiation in hippocampal slices 4 and 48 h following mild fluid-percussion brain injury in vivo. Brain Res. 1998;785:287–92.
Reeves TM, Lyeth BG, Povlishock JT. Long-term potentiation deficits and excitability changes following traumatic brain injury. Exp Brain Res. 1995;106:248–56.
Reeves TM, Kao CQ, Phillips LL, Bullock MR, Povlishock JT. Presynaptic excitability changes following traumatic brain injury in the rat. J Neurosci Res. 2000;60:370–9.
Sanders MJ, Dietrich WD, Green EJ. Behavioral, electrophysiological, and histopathological consequences of mild fluid-percussion injury in the rat. Brain Res. 2001;904:141–4.
Smith CJ, Xiong G, Elkind JA, Putnam B, Cohen AS. Brain injury impairs working memory and prefrontal circuit function. Front Neurol. 2015;6:240.
Ping X, Jin X. Transition from initial hypoactivity to hyperactivity in cortical layer V pyramidal neurons after traumatic brain injury in vivo. J Neurotrauma. 2016;33:354–61.
Witgen BM, Lifshitz J, Smith ML, Schwarzbach E, Liang SL, Grady MS, et al. Regional hippocampal alteration associated with cognitive deficit following experimental brain injury: a systems, network and cellular evaluation. Neuroscience. 2005;133:1–15.
Schwarzbach E, Bonislawski DP, Xiong G, Cohen AS. Mechanisms underlying the inability to induce area CA1 LTP in the mouse after traumatic brain injury. Hippocampus. 2006;16:541–50.
Almeida-Suhett CP, Prager EM, Pidoplichko V, Figueiredo TH, Marini AM, Li Z, et al. Reduced GABAergic inhibition in the basolateral amygdala and the development of anxiety-like behaviors after mild traumatic brain injury. PLoS ONE. 2014;9:e102627.
Hoskison MM, Moore AN, Hu B, Orsi S, Kobori N, Dash PK. Persistent working memory dysfunction following traumatic brain injury: evidence for a time-dependent mechanism. Neuroscience. 2009;159:483–91.
Allitt BJ, Iva P, Yan EB, Rajan R. Hypo-excitation across all cortical laminae defines intermediate stages of cortical neuronal dysfunction in diffuse traumatic brain injury. Neuroscience. 2016;334:290–308.
Pavlov I, Huusko N, Drexel M, Kirchmair E, Sperk G, Pitkanen A, et al. Progressive loss of phasic, but not tonic, GABAA receptor-mediated inhibition in dentate granule cells in a model of post-traumatic epilepsy in rats. Neuroscience. 2011;194:208–19.
Boychuk JA, Butler CR, Halmos KC, Smith BN. Enduring changes in tonic GABAA receptor signaling in dentate granule cells after controlled cortical impact brain injury in mice. Exp Neurol. 2016;277:178–89.
Mtchedlishvili Z, Lepsveridze E, Xu H, Kharlamov EA, Lu B, Kelly KM. Increase of GABAA receptor-mediated tonic inhibition in dentate granule cells after traumatic brain injury. Neurobiol Dis. 2010;38:464–75.
Hunt RF, Scheff SW, Smith BN. Synaptic reorganization of inhibitory hilar interneuron circuitry after traumatic brain injury in mice. J Neurosci. 2011;31:6880–90.
Cantu D, Walker K, Andresen L, Taylor-Weiner A, Hampton D, Tesco G, et al. Traumatic brain injury increases cortical glutamate network activity by compromising GABAergic control. Cereb Cortex. 2015;25:2306–20.
Sanders MJ, Sick TJ, Perez-Pinzon MA, Dietrich WD, Green EJ. Chronic failure in the maintenance of long-term potentiation following fluid percussion injury in the rat. Brain Res. 2000;861:69–76.
Ritter AC, Kammerer CM, Brooks MM, Conley YP, Wagner AK. Genetic variation in neuronal glutamate transport genes and associations with posttraumatic seizure. Epilepsia. 2016;57:984–93.
Temkin NR, Dikmen SS, Wilensky AJ, Keihm J, Chabal S, Winn HR. A randomized, double-blind study of phenytoin for the prevention of post-traumatic seizures. N Eng J Med. 1990;323:497–502.
Hammond FM, Bickett AK, Norton JH, Pershad R. Effectiveness of amantadine hydrochloride in the reduction of chronic traumatic brain injury irritability and aggression. J Head Trauma Rehabil. 2014;29:391–9.
Hammond FM, Sherer M, Malec JF, Zafonte RD, Whitney M, Bell K, et al. Amantadine effect on perceptions of irritability after traumatic brain injury: results of the Amantadine Irritability Multisite Study. J Neurotrauma. 2015;32:1230–8.
Dougall D, Poole N, Agrawal N. Pharmacotherapy for chronic cognitive impairment in traumatic brain injury. Cochrane Database Syst Rev. 2015:Cd009221.
Spritzer SD, Kinney CL, Condie J, Wellik KE, Hoffman-Snyder CR, Wingerchuk DM, et al. Amantadine for patients with severe traumatic brain injury: a critically appraised topic. Neurologist. 2015;19:61–4.
Xia P, Chen HS, Zhang D, Lipton SA. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J Neurosci. 2010;30:11246–50.
Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat Rev Drug Discov. 2006;5:160–70.
Ma S, Hangya B, Leonard CS, Wisden W, Gundlach AL. Dual-transmitter systems regulating arousal, attention, learning and memory. Neurosci Biobehav Rev. 2018;85:21–33.
Gu Q. Neuromodulatory transmitter systems in the cortex and their role in cortical plasticity. Neuroscience. 2002;111:815–35.
Parikh V, Kozak R, Martinez V, Sarter M. Prefrontal acetylcholine release controls cue detection on multiple timescales. Neuron. 2007;56:141–54.
Shin SS, Dixon CE. Alterations in cholinergic pathways and therapeutic strategies targeting cholinergic system after traumatic brain Injury. J Neurotrauma. 2015;32:1429–40.
Ostberg A, Virta J, Rinne JO, Oikonen V, Luoto P, Nagren K, et al. Cholinergic dysfunction after traumatic brain injury: preliminary findings from a PET study. Neurology. 2011;76:1046–50.
Donat CK, Schuhmann MU, Voigt C, Nieber K, Deuther-Conrad W, Brust P. Time-dependent alterations of cholinergic markers after experimental traumatic brain injury. Brain Res. 2008;1246:167–77.
Dixon CE, Bao J, Long DA, Hayes RL. Reduced evoked release of acetylcholine in the rodent hippocampus following traumatic brain injury. Pharmacol Biochem Behav. 1996;53:679–86.
Mufson EJ, Perez SE, Nadeem M, Mahady L, Kanaan NM, Abrahamson EE, et al. Progression of tau pathology within cholinergic nucleus basalis neurons in chronic traumatic encephalopathy: a chronic effects of neurotrauma consortium study. Brain. 2016;30:1399–413.
Pike BR, Hamm RJ. Post-injury administration of BIBN 99, a selective muscarinic M2 receptor antagonist, improves cognitive performance following traumatic brain injury in rats. Brain Res. 1995;686:37–43.
Pike BR, Hamm RJ. Activating the posttraumatic cholinergic system for the treatment of cognitive impairment following traumatic brain injury. Pharmacol Biochem Behav. 1997;57:785–91.
Dixon CE, Ma X, Marion DW. Effects of CDP-choline treatment on neurobehavioral deficits after TBI and on hippocampal and neocortical acetylcholine release. J Neurotrauma. 1997;14:161–9.
Zhang L, Plotkin RC, Wang G, Sandel ME, Lee S. Cholinergic augmentation with donepezil enhances recovery in short-term memory and sustained attention after traumatic brain injury. Arch Phys Med Rehabil. 2004;85:1050–5.
Tenovuo O. Central acetylcholinesterase inhibitors in the treatment of chronic traumatic brain injury-clinical experience in 111 patients. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:61–7.
Silver JM, Koumaras B, Chen M, Mirski D, Potkin SG, Reyes P, et al. Effects of rivastigmine on cognitive function in patients with traumatic brain injury. Neurology. 2006;67:748–55.
Ostberg A, Virta J, Rinne JO, Oikonen V, Luoto P, Nagren K, et al. Brain cholinergic function and response to rivastigmine in patients with chronic sequels of traumatic brain injury: a PET study. J Head Trauma Rehabil. 2018;33:25–32.
Jenkins PO, Mehta MA, Sharp DJ. Catecholamines and cognition after traumatic brain injury. Brain. 2016;139(Pt 9):2345–71.
Aston-Jones G, Rajkowski J, Cohen J. Role of locus coeruleus in attention and behavioral flexibility. Biol Psychiatry. 1999;46:1309–20.
Dunn-Meynell A, Pan S, Levin BE. Focal traumatic brain injury causes widespread reductions in rat brain norepinephrine turnover from 6 to 24 h. Brain Res. 1994;660:88–95.
Fujinaka T, Kohmura E, Yuguchi T, Yoshimine T. The morphological and neurochemical effects of diffuse brain injury on rat central noradrenergic system. Neurol Res. 2003;25:35–41.
Levin BE, Brown KL, Pawar G, Dunn-Meynell A. Widespread and lateralization effects of acute traumatic brain injury on norepinephrine turnover in the rat brain. Brain Res. 1995;674:307–13.
Levin BE, Pan S, Dunn-Meynell A. Chronic alterations in rat brain alpha-adrenoceptors following traumatic brain injury. Restor Neurol Neurosci. 1994;7:5–12.
Prasad MR, Tzigaret CM, Smith D, Soares H, McIntosh TK. Decreased alpha 1-adrenergic receptors after experimental brain injury. J Neurotrauma. 1992;9:269–79.
Ripley DL, Morey CE, Gerber D, Harrison-Felix C, Brenner LA, Pretz CR, et al. Atomoxetine for attention deficits following traumatic brain injury: results from a randomized controlled trial. Brain. 2014;28:1514–22.
Meiser J, Weindl D, Hiller K. Complexity of dopamine metabolism. Cell Commun Signal. 2013;11:34.
Hutson CB, Lazo CR, Mortazavi F, Giza CC, Hovda D, Chesselet MF. Traumatic brain injury in adult rats causes progressive nigrostriatal dopaminergic cell loss and enhanced vulnerability to the pesticide paraquat. J Neurotrauma. 2011;28:1783–801.
van Bregt DR, Thomas TC, Hinzman JM, Cao T, Liu M, Bing G, et al. Substantia nigra vulnerability after a single moderate diffuse brain injury in the rat. Exp Neurol. 2012;234:8–19.
Massucci JL, Kline AE, Ma X, Zafonte RD, Dixon CE. Time dependent alterations in dopamine tissue levels and metabolism after experimental traumatic brain injury in rats. Neurosci Lett. 2004;372:127–31.
Kobori N, Clifton GL, Dash PK. Enhanced catecholamine synthesis in the prefrontal cortex after traumatic brain injury: implications for prefrontal dysfunction. J Neurotrauma. 2006;23:1094–102.
Huang EY, Tsui PF, Kuo TT, Tsai JJ, Chou YC, Ma HI, et al. Amantadine ameliorates dopamine-releasing deficits and behavioral deficits in rats after fluid percussion injury. PLoS ONE. 2014;9:e86354.
Wagner AK, Sokoloski JE, Ren D, Chen X, Khan AS, Zafonte RD, et al. Controlled cortical impact injury affects dopaminergic transmission in the rat striatum. J Neurochem. 2005;95:457–65.
Wagner AK, Scanlon JM, Becker CR, Ritter AC, Niyonkuru C, Dixon CE, et al. The influence of genetic variants on striatal dopamine transporter and D2 receptor binding after TBI. J Cereb Blood Flow Metab. 2014;34:1328–39.
Whyte J, Vaccaro M, Grieb-Neff P, Hart T, Polansky M, Coslett HB. The effects of bromocriptine on attention deficits after traumatic brain injury: a placebo-controlled pilot study. Am J Phys Med Rehabil/Assoc Acad Physiatr. 2008;87:85–99.
Siddall OM. Use of methylphenidate in traumatic brain injury. Ann Pharmacother. 2005;39:1309–13.
Jin C, Schachar R. Methylphenidate treatment of attention-deficit/hyperactivity disorder secondary to traumatic brain injury: a critical appraisal of treatment studies. CNS Spectr. 2004;9:217–26.
De Felice LJ. A current review of serotonin transporters. F1000Res 2016, 5(F1000 Faculty Rev):1884. the article is 7 pages long so presumably it is 1884–90.
Abe K, Shimada R, Okada Y, Kibayashi K. Traumatic brain injury decreases serotonin transporter expression in the rat cerebrum. Neurol Res. 2016;38:358–63.
Wang Y, Neumann M, Hansen K, Hong SM, Kim S, Noble-Haeusslein LJ, et al. Fluoxetine increases hippocampal neurogenesis and induces epigenetic factors but does not improve functional recovery after traumatic brain injury. J Neurotrauma. 2011;28:259–68.
Yue JK, Burke JF, Upadhyayula PS, Winkler EA, Deng H, Robinson CK, et al. Selective serotonin reuptake inhibitors for treating neurocognitive and neuropsychiatric disorders following traumatic brain injury: an evaluation of current evidence. Brain Sci. 2017;7:93–120.
Novack TA, Banos JH, Brunner R, Renfroe S, Meythaler JM. Impact of early administration of sertraline on depressive symptoms in the first year after traumatic brain injury. J Neurotrauma. 2009;26:1921–8.
Ashman TA, Cantor JB, Gordon WA, Spielman L, Flanagan S, Ginsberg A, et al. A randomized controlled trial of sertraline for the treatment of depression in persons with traumatic brain injury. Arch Phys Med Rehabil. 2009;90:733–40.
Rapoport MJ, Mitchell RA, McCullagh S, Herrmann N, Chan F, Kiss A, et al. A randomized controlled trial of antidepressant continuation for major depression following traumatic brain injury. J Clin Psychiatry. 2010;71:1125–30.
Banos JH, Novack TA, Brunner R, Renfroe S, Lin HY, Meythaler J. Impact of early administration of sertraline on cognitive and behavioral recovery in the first year after moderate to severe traumatic brain injury. J Head Trauma Rehabil. 2010;25:357–61.
Fann JR, Bombardier CH, Temkin N, Esselman P, Warms C, Barber J, et al. Sertraline for major depression during the year following traumatic brain injury: a randomized controlled trial. J Head Trauma Rehabil. 2017;32:332–42.
Lusardi TA. Adenosine neuromodulation and traumatic brain injury. Curr Neuropharmacol. 2009;7:228–37.
Krugel U. Purinergic receptors in psychiatric disorders. Neuropharmacology. 2016;104:212–25.
Chen JF, Lee CF, Chern Y. Adenosine receptor neurobiology: overview. Int Rev Neurobiol. 2014;119:1–49.
Gundersen V, Storm-Mathisen J, Bergersen LH. Neuroglial transmission. Physiol Rev. 2015;95:695–726.
Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, et al. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–6.
Rombo DM, Ribeiro JA, Sebastiao AM. Hippocampal GABAergic transmission: a new target for adenosine control of excitability. J Neurochem. 2016;139:1056–70.
Matos M, Augusto E, Santos-Rodrigues AD, Schwarzschild MA, Chen JF, Cunha RA, et al. Adenosine A2A receptors modulate glutamate uptake in cultured astrocytes and gliosomes. Glia. 2012;60:702–16.
Dai SS, Zhou YG, Li W, An JH, Li P, Yang N, et al. Local glutamate level dictates adenosine A2A receptor regulation of neuroinflammation and traumatic brain injury. J Neurosci. 2010;30:5802–10.
Bell MJ, Kochanek PM, Carcillo JA, Mi Z, Schiding JK, Wisniewski SR, et al. Interstitial adenosine, inosine, and hypoxanthine are increased after experimental traumatic brain injury in the rat. J Neurotrauma. 1998;15:163–70.
Kochanek PM, Vagni VA, Janesko KL, Washington CB, Crumrine PK, Garman RH, et al. Adenosine A1 receptor knockout mice develop lethal status epilepticus after experimental traumatic brain injury. J Cereb Blood Flow Metab. 2006;26:565–75.
Ning YL, Yang N, Chen X, Xiong RP, Zhang XZ, Li P, et al. Adenosine A2A receptor deficiency alleviates blast-induced cognitive dysfunction. J Cereb Blood Flow Metab. 2013;33:1789–98.
Zhao ZA, Zhao Y, Ning YL, Yang N, Peng Y, Li P, et al. Adenosine A2A receptor inactivation alleviates early-onset cognitive dysfunction after traumatic brain injury involving an inhibition of tau hyperphosphorylation. Transl Psychiatry. 2017;7:e1123.
Diamond ML, Ritter AC, Jackson EK, Conley YP, Kochanek PM, Boison D, et al. Genetic variation in the adenosine regulatory cycle is associated with posttraumatic epilepsy development. Epilepsia. 2015;56:1198–206.
Wagner AK, Miller MA, Scanlon J, Ren D, Kochanek PM, Conley YP. Adenosine A1 receptor gene variants associated with post-traumatic seizures after severe TBI. Epilepsy Res. 2010;90:259–72.
Cotter D, Kelso A, Neligan A. Genetic biomarkers of posttraumatic epilepsy: a systematic review. Seizure. 2017;46:53–8.
Froemke RC. Plasticity of cortical excitatory−inhibitory balance. Annu Rev Neurosci. 2015;38:195–219.
Bales JW, Wagner AK, Kline AE, Dixon CE. Persistent cognitive dysfunction after traumatic brain injury: a dopamine hypothesis. Neurosci Biobehav Rev. 2009;33:981–1003.
Froudist-Walsh S, Lopez-Barroso D, Jose Torres-Prioris M, Croxson PL, Berthier ML. Plasticity in the working memory system: life span changes and response to injury. Neuroscientist. 2018;24:261–76.
Moreau AW, Amar M, Le Roux N, Morel N, Fossier P. Serotoninergic fine-tuning of the excitation-inhibition balance in rat visual cortical networks. Cereb cortex (New Y, NY: 1991). 2010;20:456–67.
Sommerauer C, Rebernik P, Reither H, Nanoff C, Pifl C. The noradrenaline transporter as site of action for the anti-Parkinson drug amantadine. Neuropharmacology. 2012;62:1708–16.
Huber TJ, Dietrich DE, Emrich HM. Possible use of amantadine in depression. Pharmacopsychiatry. 1999;32:47–55.
Cottingham C, Ferryman CJ, Wang Q. Chapter nine—α2 adrenergic receptor trafficking as a therapeutic target in antidepressant drug action. In: Wu G, editor. Progress in molecular biology and translational science, vol. 132. Academic Press: New York; 2015. p. 207−25.
Bading H. Therapeutic targeting of the pathological triad of extrasynaptic NMDA receptor signaling in neurodegenerations. J Exp Med. 2017;214:569–78.
Massie A, Boillee S, Hewett S, Knackstedt L, Lewerenz J. Main path and byways: non-vesicular glutamate release by system x(c)(−) as an important modifier of glutamatergic neurotransmission. J Neurochem. 2015;135:1062–79.
Williams LE, Featherstone DE. Regulation of hippocampal synaptic strength by glial xCT. J Neurosci. 2014;34:16093–102.
Mosienko V, Teschemacher AG, Kasparov S. Is L-lactate a novel signaling molecule in the brain? J Cereb Blood Flow Metab. 2015;35:1069–75.
Bergersen LH, Gjedde A. Is lactate a volume transmitter of metabolic states of the brain? Front Neuroenergetics. 2012;4:5.
Lauritzen KH, Morland C, Puchades M, Holm-Hansen S, Hagelin EM, Lauritzen F, et al. Lactate receptor sites link neurotransmission, neurovascular coupling, and brain energy metabolism. Cereb Cortex. 2014;24:2784–95.
Bozzo L, Puyal J, Chatton JY. Lactate modulates the activity of primary cortical neurons through a receptor-mediated pathway. PLoS ONE. 2013;8:e71721.
Sala N, Suys T, Zerlauth JB, Bouzat P, Messerer M, Bloch J, et al. Cerebral extracellular lactate increase is predominantly nonischemic in patients with severe traumatic brain injury. J Cereb Blood Flow Metab. 2013;33:1815–22.
Carpenter KL, Jalloh I, Hutchinson PJ. Glycolysis and the significance of lactate in traumatic brain injury. Front Neurosci. 2015;9:112.
Ichai C, Payen JF, Orban JC, Quintard H, Roth H, Legrand R, et al. Half-molar sodium lactate infusion to prevent intracranial hypertensive episodes in severe traumatic brain injured patients: a randomized controlled trial. Intensive Care Med. 2013;39:1413–22.
Lin AP, Ramadan S, Stern RA, Box HC, Nowinski CJ, Ross BD, et al. Changes in the neurochemistry of athletes with repetitive brain trauma: preliminary results using localized correlated spectroscopy. Alzheimer’s Res Ther. 2015;7:13.
Kierans AS, Kirov II, Gonen O, Haemer G, Nisenbaum E, Babb JS, et al. Myoinositol and glutamate complex neurometabolite abnormality after mild traumatic brain injury. Neurology. 2014;82:521–8.
Gasparovic C, Yeo R, Mannell M, Ling J, Elgie R, Phillips J, et al. Neurometabolite concentrations in gray and white matter in mild traumatic brain injury: an 1H-magnetic resonance spectroscopy study. J Neurotrauma. 2009;26:1635–43.
Yeo RA, Gasparovic C, Merideth F, Ruhl D, Doezema D, Mayer AR. A longitudinal proton magnetic resonance spectroscopy study of mild traumatic brain injury. J Neurotrauma. 2011;28:1–11.
Kawai N, Maeda Y, Kudomi N, Yamamoto Y, Nishiyama Y, Tamiya T. Focal neuronal damage in patients with neuropsychological impairment after diffuse traumatic brain injury: evaluation using (1)(1)C-flumazenil positron emission tomography with statistical image analysis. J Neurotrauma. 2010;27:2131–8.
Kraus MF, Smith GS, Butters M, Donnell AJ, Dixon E, Yilong C, et al. Effects of the dopaminergic agent and NMDA receptor antagonist amantadine on cognitive function, cerebral glucose metabolism and D2 receptor availability in chronic traumatic brain injury: a study using positron emission tomography (PET). Brain Inj. 2005;19:471–9.
Green RE. Editorial: brain injury as a neurodegenerative disorder. Front Hum Neurosci. 2015;9:615.
Hellewell S, Semple BD, Morganti-Kossmann MC. Therapies negating neuroinflammation after brain trauma. Brain Res. 2016;1640(Pt A):36–56.
Schurman LD, Lichtman AH. Endocannabinoids: a promising impact for traumatic brain injury. Front Pharmacol. 2017;8:69.
McGinn MJ, Povlishock JT. Cellular and molecular mechanisms of injury and spontaneous recovery. Handb Clin Neurol. 2015;127:67–87.
Lu J, Gary KW, Copolillo A, Ward J, Niemeier JP, Lapane KL. Randomized controlled trials in adult traumatic brain injury: a review of compliance to CONSORT statement. Arch Phys Med Rehabil. 2015;96:702–14.
Maas AI, Menon DK, Lingsma HF, Pineda JA, Sandel ME, Manley GT. Re-orientation of clinical research in traumatic brain injury: report of an international workshop on comparative effectiveness research. J Neurotrauma. 2012;29:32–46.
Tosetti P, Hicks RR, Theriault E, Phillips A, Koroshetz W, Draghia-Akli R, et al. Toward an international initiative for traumatic brain injury research. J Neurotrauma. 2013;30:1211–22.
Manley GT, Maas AI. Traumatic brain injury: an international knowledge-based approach. JAMA: J Am Med Assoc. 2013;310:473–4.
Yue JK, Vassar MJ, Lingsma HF, Cooper SR, Okonkwo DO, Valadka AB, et al. Transforming research and clinical knowledge in traumatic brain injury pilot: multicenter implementation of the common data elements for traumatic brain injury. J Neurotrauma. 2013;30:1831–44.
Maas AI, Menon DK, Steyerberg EW, Citerio G, Lecky F, Manley GT, et al. Collaborative European NeuroTrauma Effectiveness Research in Traumatic Brain Injury (CENTER-TBI): a prospective longitudinal observational study. Neurosurgery. 2015;76:67–80.
Sorani MD, Yue JK, Sharma S, Manley GT, Ferguson AR, Investigators TT, et al. Genetic data sharing and privacy. Neuroinformatics. 2015;13:1–6.
FITBIR. Federal Interagency Traumatic Brain Injury Research Informatics System. https://fitbir.nih.gov/.
Acknowledgements
The authors would like to thank Martin Ngwenya for assistance with preparation of the figures.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
McGuire, J.L., Ngwenya, L.B. & McCullumsmith, R.E. Neurotransmitter changes after traumatic brain injury: an update for new treatment strategies. Mol Psychiatry 24, 995–1012 (2019). https://doi.org/10.1038/s41380-018-0239-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-018-0239-6
- Springer Nature Limited
This article is cited by
-
Preclinical Studies on Mechanisms Underlying the Protective Effects of Propranolol in Traumatic Brain Injury: A Systematic Review
Journal of Neuroimmune Pharmacology (2024)
-
Exploring the molecular mechanisms of PPARγ agonists in modulating memory impairment in neurodegenerative disorders
Molecular Biology Reports (2024)
-
Psychiatric sequelae of traumatic brain injury — future directions in research
Nature Reviews Neurology (2023)