
Abstract
Autosomal recessive (AR) gene defects are the leading genetic cause of intellectual disability (ID) in countries with frequent parental consanguinity, which account for about 1/7th of the world population. Yet, compared to autosomal dominant de novo mutations, which are the predominant cause of ID in Western countries, the identification of AR-ID genes has lagged behind. Here, we report on whole exome and whole genome sequencing in 404 consanguineous predominantly Iranian families with two or more affected offspring. In 219 of these, we found likely causative variants, involving 77 known and 77 novel AR-ID (candidate) genes, 21 X-linked genes, as well as 9 genes previously implicated in diseases other than ID. This study, the largest of its kind published to date, illustrates that high-throughput DNA sequencing in consanguineous families is a superior strategy for elucidating the thousands of hitherto unknown gene defects underlying AR-ID, and it sheds light on their prevalence.
Similar content being viewed by others

Introduction
Intellectual disability (ID) or delayed psychomotor development are by far the most common reasons for referral to genetic services, and most severe forms are caused by single genetic defects. In Western populations where parental consanguinity is rare and families are usually small, most affected individuals are of sporadic cases. As shown by array CGH [1] and more recently, whole exome (WES) or whole genome sequencing (WGS) of patients and their parents [2, 3], dominant de novo copy number variants (CNVs) and mutations in single genes account for ID in the majority of these individuals [4, 5]. Autosomal recessive inheritance was rarely observed, at least in the first larger NGS-based trio studies of this kind [6, 7], where low sequencing depth may have hampered the identification of compound heterozygotes (discussed in ref. [5]). In a recent comprehensive study of children with severe developmental disorders, autosomal recessive defects accounted for 11.7% of all cases with a clear molecular diagnosis, whereas apparently disease-causing autosomal dominant de novo mutations were seen almost 5 times as often [8]. Not long ago, a meta-analysis of 2104 trios identified 10 novel genes for ID [9].
These observations, as well as the availability of conceptually simple ‘Trio sequencing’ strategies for their identification, explain why in recent years, genetic research into ID and related disorders has been dominated by the de novo mutation paradigm [2, 8, 10,11,12]. Most of the dominant de novo mutations identified are inactivate or one copy of a deleted haploinsufficient gene, whereas gain-of-function or dominant negative mutations seem to be much rarer. Since only a minority of the ~20,000 protein-coding human genes are dosage-sensitive [13], it is not surprising that according to recent estimates, there may be less than 500 genes in the human genome where functional loss of one copy is associated with ‘autism spectrum disorder’ (ASD) [14, 15], a collective term that includes ID and autism. ‘Trio sequencing’ of individuals with ID/ASD and their parents has already identified more than 400 of these genes [9, 16, 17].
In contrast, the molecular elucidation of recessive forms of ID is still in its infancy. By performing large-scale autozygosity mapping in unrelated consanguineous families, we were the first to show that autosomal recessive ID (ARID) is extremely heterogeneous [18, 19]. Extrapolation from the hitherto identified ~120 genes for recessive ID on the human X-chromosome [20] suggests that mutations in more than 3000 human genes may be associated with autosomal recessive ID (ARID) [4]. To date, less than 600 of these genes have been identified [16, 17].
Only recently, ARID has gained popularity as a promising target for research into the development and function of the human brain (e.g., see ref. [21]). Yet, the elucidation of ARID is also of considerable importance for global health care, which is still widely disregarded. Recent population studies have shown that the prevalence of ID is highly correlated with the frequency and the degree of parental consanguinity. In the offspring of double cousin or uncle-niece unions, ID is 3 to 4 times more common than in children of unrelated parents [22,23,24,25], and there is compelling evidence for the involvement of recessive gene defects (e.g., see refs. [5, 25,26,27]). Over 1 billion people live in countries where consanguineous marriage is common [28], and it has been estimated that couples related as second cousins or closer and their offspring account for 10.4% of the global population [29].
In outbred Western populations, the search for recessive causes of ID has turned out to be tedious (e.g., see refs. [30, 31]). Even trio sequencing in several thousand affected families yielded only a modest number of novel ARID genes [8, 32]. In 2011, we performed large-scale high-throughput sequencing in cohorts of consanguineous families to speed up the search for novel ARID (candidate) genes [33] but it took several years before other groups joined in [34,35,36,37,38,39].
Here we report on WES and WGS studies in 404 consanguineous families with ID, about three times as many as analyzed 5 years ago by our group [33]. The vast majority of these are from Iran, where almost 40% of all children have related parents [40,41,42,43]. These investigations shed more light on the genetic causes of ARID. Moreover, they significantly broadened the basis for the diagnosis and prevention of cognitive disorders in Iran and beyond.
Materials and methods
This study comprised 404 out of 757 families with ≥ 2 affected individuals recruited during the past 10 years. Initial screening revealed Fragile X syndrome in 46 (6.1%) of these families which were excluded. In other families, different monogenic causes of ID have been identified prior to the NGS era (reviewed by refs. [5] and [44]), and many families could not be reached or were excluded because of poor parental cooperation, borderline ID or genetic heterogeneity.
From each family of our cohort, one affected patient was selected for sequencing.
Genomic DNA (gDNA) was extracted from peripheral blood, the quality of which was controlled by Nanodrop2000 (Thermo Scientific), and approximately 2 µg gDNA was used for constructing deep sequencing libraries. In the course of this study, four different deep sequencing protocols were employed, i.e., target enrichment sequencing (TES) using the Illumina GAII sequencer, whole exome sequencing (WES) on the Illumina HiSeq2000 sequencer, and whole genome sequencing (WGS) on the Illumina HiSeq X Ten sequencer (Macrogen) followed by sequence alignment and variant calling using the DRAGEN infrastructure and pipeline (www.edicogenome.com). For some of the families, WGS was (also) performed by Complete Genomics, following the service provider’s standard procedure, using short-read paired sequencing and the CGATM tools for data analysis (www.completegenomics.com/). Details about the performance of these protocols, including the average coverage depth (non-redundant reads only) per sample and the percentage of the targeted coding regions covered by 10, 20, or 30 reads, are shown in the Table S6 and discussed below.
We analyzed TES and WES data by using our previously published Medical Resequencing Analysis Pipeline (MERAP) [45] (for details, see Materials and Methods Supplement; MERAP procedure). A particular strength of this pipeline is the detection of small, medium-sized, and large copy number variants. Therefore, and because homozygous CNVs turned out to be very rare in the families studied, array CGH was soon discontinued.
All potentially ID-causing variants detected by NGS were validated by Sanger sequencing, and for those identified by WES and WGS, co-segregation studies were performed, including all available and informative family members. A novel algorithm was developed to detect runs of homozygous markers (ROHs) encompassing possibly disease-causing mutations in the exome and genome of ARID patients, and comparison of ROHs spanning identical mutations in apparently unrelated families revealed shared haplotypes for all of them (for details, see Fig. 2, Supplement and Table S7). To further enrich them for pathogenic mutations, variants were also filtered in several other ways (for details, see Table S1), including pathogenicity prediction for missense variants using four established prediction tools, and selected for absence or very low allele frequencies in the ExAC database and our own in-house cohort which largely consists of Iranian families. Moreover, we have shown that novel ID genes are functionally related to known ID genes, either by protein-protein or through regulatory interaction (for details, see Materials and Methods, Supplement). Variants in known genes were scored using the ACMG variant interpretation guidelines [46]. Of these, 83.3% were classified as pathogenic or likely pathogenic. A few variants of uncertain significance (VUS) were retained as likely relevant based on a variety of criteria (e.g., low allele frequency or not even listed in ExAC; confined to a single family in our in-house database; called as pathogenic by at least 2 out of 4 relevant algorithms [PolyPhen2, SIFT, MutationTaster, CADD]; distinctive clinical phenotype; location within ROHs; supporting evidence from functional studies and expression data). The same criteria were also employed to classify mutations in novel candidate genes. Nevertheless, rigorous confirmation of the pathogenicity of these variants will have to include future functional studies and/or the identification of identical variants in other affected families.

Protein-protein interaction network linking 39 known (green nodes) and 48 novel genes (orange nodes) for recessive forms of ID that were identified during this study. Five genes have been identified as well by other studies during the course of this work (light yellow nodes). Interactions were retrieved from the ConsensusPathDB resource (Kamburov U, Stelzl H, Lehrach R Herwig. Nucleic Acids Res. 2013). Known ARID genes refer to recent publications [17, 33,34,35] and are labeled according to the number of supporting references (dark green = high number of references). Enriched protein complexes and pathways are shown as colored clouds
To validate previously reported ARID candidate genes [33], we have also generated fly models and performed behavioral tests [47, 48]. Other previously identified ARID candidate genes could be confirmed by identifying allelic mutations in unrelated families (for details, see Materials and Methods Supplement; Fly Models, Drosophila behavior testing and Table S3).
Results
In 219 out of 404 families investigated (54.2%) we identified likely disease-causing DNA variants in novel candidate genes and in known genes, all co-segregating with ID. As expected for affected offspring of healthy consanguineous parents, the vast majority of these turned out to be autozygous for autosomal recessive defects. Compound heterozygosity was confined to a single family (M135, see Table S1) with a frameshift and a missense change in the MADD gene, which has a role in synaptic vesicle transport.
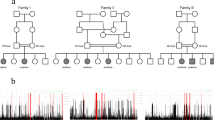
Likely disease-relevant variants in known or novel X-chromosomal (candidate) genes were found in 26 (23 genes) out of 219 consanguineous families (11.9%). For all of these, inheritance patterns were compatible with X-linkage. Pedigrees of all families with mutations in novel candidate genes are shown in Fig. S1. Five of the novel candidate genes had not been implicated in ID before (see Tables S1), two variants were identified in known non-ID disease genes (DIAPH2 and XPNPEP2), and two (KIF4A [49] and WDR13 [50]) had been linked to ID in a single family and are confirmed as X-linked intellectual disability (XLID) genes by this study. These data suggest that including Fragile X syndrome, X-linked gene defects may account for almost 18% of the consanguineous families with ID in Iran (see also refs. [51, 52]). Pathogenic mutations in DDX3X and PHF8 were observed in two affected brother pairs (families M030 and M9100013, respectively, see Table S1) but not in blood of their mothers, suggesting maternal germ cell mosaicism. A similar frequency of de novo mutations shared by siblings had been reported before [14].
For 26 autosomal and X-chromosomal genes we found allelic mutations in two or more families (Table 1). Most of these are known ID genes, and several had already been described in our previous study [33]. Four genes (IPP, ITGAV, RNFT2, and TTC5) had not yet been linked to any disease and PIDD1 only very recently [39]. For two known disease genes (AK1 and ALS2), an association with ID had not been reported before.
Two hundred and fifteen different, likely disease-causing variants were identified, 127 in known and 88 in novel (candidate) genes (Table 2). Of note, 11 out of 127 likely causative variants were found in genes that had been previously implicated in diseases other than ID. Of the 127 variants observed in known genes, 57 (44.9%) were loss of function (LOF) mutations including large deletions, stop-gain, frameshift, extension, and splice site variants, while 70 (55.1%) were missense variants. With 42%, the proportion of LOF variants was slightly lower in families with novel (candidate) genes and the proportion of missense mutations or small in-frame deletions was a little higher (58%). While these differences are not statistically significant, they might indicate that our criteria for selecting missense mutations in novel genes were slightly too permissive. On the other hand, it is noteworthy that in a recent study of de novo mutations causing ‘autism spectrum disorder‘ (ASD) [14], the inferred relative contribution of gene-disrupting (43%) and missense mutations (57%) was very similar to our findings.
There was almost no overlap between the genes implicated in ARID by our present study and 847 genes thought to be functionally redundant for which homozygous mutations have been recently identified in healthy adults with related parents [53]. Only 4 of the genes mutated in our families (CLN3, CLIP1, POMGNT1, and SASS6) were listed as potentially redundant, but there is solid evidence linking all four to recessive cognitive disorders. CLN3 (OMIM #204200) and POMGNT1 (OMIM #613151) are known genes for neuronal ceroid lipofuscinosis and congenital dystroglycanopathy with mental retardation, respectively, whereas for CLIP1 and SASS6, homozygous deleterious mutations have been identified in at least two unrelated ARID families (this study and ref. [54]).
Further, albeit indirect support for the reliability of our findings had been obtained from the confirmation of previous results. Of the 50 novel ARID candidate genes presented previously [33], > 30 have been firmly implicated in ARID through identification of additional families with allelic mutations, our investigation of fly models (Table S2 and Fig. S2) or in other ways (for a comprehensive overview, see Table S3). For many of the remaining candidate genes, mouse models with behavioral abnormalities have been reported (e.g., see http://www.informatics.jax.org). Moreover, the introduction of MERAP, a comprehensive Medical Resequencing Analysis Pipeline with its integrated Logit pathogenicity score [45] has greatly improved the identification and ranking of likely disease-causing sequence variants. Therefore, we believe that eventually, most of the candidate genes presented here will be confirmed. Indeed, this expectation has already been met for a variety of (former) candidate genes identified in the course of this study, including FMN2 [55], CLIP1 [56], CAPN10 [57], MFSD2A [58, 59], SLC6A17 [60], HNMT [61], DDX3X [62], TAF6 [34], and TAF1 [20, 63], which have been recently published by us and/or other groups.
ARID is often associated with microcephaly
In our previous study [33], novel forms of ARID had been considered as non-syndromic if index patients showed no obvious clinical symptoms other than ID. In many of these families, however, re-examination including affected siblings revealed additional clinical signs that had been overlooked before. In the present study, thorough clinical examination of all affected family members and comparison with families carrying allelic mutations allowed us to classify as syndromic 200 out of 219 families with a defined disease-causing variant. In 86 of the 219 ‘identified’ ARID families, the average occipito-frontal diameter (OFC) of affected individuals was at least 2 SD lower than the mean, and mutations in 30 novel candidate genes for ID were found to be associated with microcephaly (see Table S1, column G, and clinical description, Text File S1). Very severe microcephaly, with OFC < −7 standard deviations (SD) [64] was observed in families with homozygous, likely damaging mutations in the genes PPP1R35, GUF1, METTL5, PUS7, and TBC1D23 (see more detailed information in Supplementary Text).
ID with moderate microcephaly (OFC: −5 SD to −3 SD) was observed for 11 novel genes. For three of these, a second affected family with an allelic mutation has established their involvement in ID and microcephaly.
Defects in three genes, i.e., SP2, CLPTM1, and MADD, were found to be associated with enlarged head size. The most striking head enlargement (OFC: + 4 and + 5 SD, respectively) was observed in two children with mild to moderate ID (see family M135 in Supplementary Text S1) and compound heterozygosity for two allelic MADD mutations.
Of note, in two of three genes previously linked to non-ID disorders, PLIN1 (OFC: −3.5 SD) and YARS (OFC: −9.5 SD), we observed likely causative variants associated with microcephaly, and for AK1 mutation, an association with enlarged head size has been observed.
Epilepsy is also common, but autism is rare
Epilepsy was observed in 62 families of our cohort (28%), involving 33 known and 29 novel candidate genes (see Table S1, column H, and Supplementary Text). Of note, three out of 33 known genes (ALS2, FDPS, and XPNPEP2) had not been linked to ID before. Thus, after microcephaly, epilepsy was the most common additional finding in families with ARID. As judged from MRI results, which were only available for a minority of the ARID families, structural brain abnormalities and/or leukoencephalopathy are also fairly common (Table S1).
Prominent signs of autism were only present in 8 of the 219 families, involving 5 known and 3 hitherto unknown ID genes. These findings corroborate our earlier observation that compared to sporadic forms of ID seen in outbred populations, autism is rare in patients with recessive forms of ID [33]. Among the relevant known ID genes, four genes (ADSL, SHANK3, GRM1, and CNTNAP2) have been implicated in autism before. Novel ID- and autism-associated (candidate) genes included SP2, TRIM47, and EZH1. The association of SP2 mutations with autism and large head size is noteworthy because it has been reported that autistic children tend to have large brains [65]. SP2 is a cell cycle regulator gene, in which, deletion leads to the interruption of neurogenesis in embryonic and postnatal brain [66]. TRIM47 is expressed in fetal astrocytes and may be involved in brain development [67]. The homozygous truncating mutation observed in the EZH1 gene is of particular interest. Heterozygous de novo mutations in the paralogous EZH2 gene are associated with Weaver syndrome [68], characterized by developmental delay, overgrowth and dysmorphic signs. Both EZH1 and EZH2 catalyze mono-methylation, di-methylation, and tri-methylation of histone H3 at lysine 27 (H3K27me2/3) [69], but EZH1 is less abundant in embryonic stem cells and has weaker methyltransferase activity. We show here that homozygous loss of EZH1 function is also associated with overgrowth (see clinical description of family M8800071 in Supplementary Text and Fig. S3), but otherwise there was little phenotypic overlap with Weaver syndrome. In another family with ID and autism, we found a likely disease-causing variant in the XPNPEP2 gene. XPNPEP2 may be involved in cleavage of neuropeptide Y [70], a neuromodulator implicated in controlling the energy balance and behavior.
Allelic mutations causing recessive or dominant ID
In six previously described genes for autosomal dominant ID (ADID) or related disorders (CACNA1C, SCN8A, SETBP1, SHANK3, ATP1A3, PRRT2), we have identified recessive sequence variants that co-segregated with ID in consanguineous ARID families (Table S1). Four of these are likely mild missense mutations, as evidenced by moderate Logit pathogenicity scores (see Table S1). This may explain why heterozygous carriers in the respective families are healthy and only homozygotes have ID, as shown for a Pro→His mutation in CACNA1C, an Arg→His mutation in SCN8A, a Glu→Gly mutation in SETBP1 and a Val→Ala mutation in SHANK3 (Table S1). Dominant ATP1A3 mutations (see OMIM *182350) have been identified in dystonia, alternating hemiplegia of childhood and in the severe CAPOS syndrome. The Arg476Cys variant found in family M204 has high pathogenicity scores, but has not been linked to disease before. It is listed 65 times in the ExAC database, but exclusively in heterozygotes. In family M204, the phenotype of two homozygous females born to healthy second cousin parents overlapped with CAPOS syndrome, including severe ID, cerebellar ataxia with quadrupedal gait, short stature (< 3% ile) and in one, seizures since infancy. Reduced body size has also been noted in a mouse model for this disorder (see OMIM *182350).
For PRRT2 (see OMIM *614386), heterozygous loss-of function (LOF) mutations have been described in families with three different dominant disorders, i.e., infantile convulsions with paroxysmal choreoathetosis, episodic dyskinesia and infantile benign familial seizures. A recurrent c.649dupC frameshift mutation in the PRRT2 gene has been identified as the most common cause of all three conditions, which have been observed in different members of the respective families [71]. In family M003 reported here, homozygosity of c.649dupC is associated with moderate ID, strabism, seizures, and spasticity; their parents are healthy and do not have a history of infantile seizures. To our knowledge, homozygous truncating PRRT2 mutations with ID have not been described before.
For TBC1D23 and HIVEP3, presented here as novel ARID (candidate) genes (families M268 and M8700057 in Table S1), heterozygous de novo mutations have been described in individuals with autism and schizophrenia, respectively [17, 72].
Discussion
Many of the ~570 genes hitherto implicated in ARID [5, 16, 17, 44] code for metabolic enzymes, and their defects often cause severe or even lethal inborn errors of metabolism. In our study, we have focused on familial ID, and many affected individuals were recruited as adolescents or even adults. This may explain why in our cohort, the proportion of families with metabolic defects is much lower. Detailed information about the function of all novel ARID (candidate) genes is provided in Table S1 (see column U). Grouping them into functional classes is somewhat arbitrary because many genes have pleiotropic functions. As previously reported [33], ARID is often caused by mutations in genes with essential ‘housekeeping functions’ such as DNA transcription and translation, cell division, protein degradation, or energy metabolism. Here we show that the spectrum of recessive gene defects leading to ID is much wider. In particular, novel ARID genes are tightly connected with known ARID genes at the level of protein–protein interactions often functioning in fundamental biological processes such as TFIID, elongation and 7SK RNP complexes (Fig. 1). A detailed functional characterization of the novel ARID (candidate) genes is summarized in Tables S4.1–4.7 and different aspects are shown in Figs. S4–S6.

Regional clustering of recurrent ARID mutations suggest short half-life and rapid replacement of serious recessive disease-causing gene defects in Iran
Apart from the functional classes mentioned above, defects involving synaptic function, cell migration, cell signaling and remarkably, innate immunity form visible clusters (see Table S1, column T). In total, 7 of the novel ARID (candidate) genes presented here have a role in innate immunity, and at least three (HIVEP3, PIDD1, and TMED7-TICAM2) are upstream regulators of NF-kappa B (see Table S1). Two ARID genes, CC2D1A [73] and TRAPPC9 [74] have been linked to innate immunity before; inactivation of these genes leads to up-regulation of NF-kappa B signaling. Recently, excessive NF-kappa B signaling has also been implicated in the pathogenesis of Rett syndrome and presented as possible new route for alleviating the course of this severe X-linked neurodevelopmental disorder [75]. Using homozygosity mapping, Gamsiz ED et al., (2015) identified novel rare, recessive loci, which include a protein truncating mutation in CC2D1A in consanguineous families with syndromes such as autism symptoms [76].
Functions of ARID and ADID genes
Dominant de novo mutations in fragile X mental retardation protein (FMRP)-interactors and chromatin-remodeling genes are common in sporadic forms of ID and autism, and the same is true for genes coding for post-synaptic density proteins (e.g., see refs. [14, 15, 72, 77]). In contrast, only two of the novel ARID genes identified by our present study qualify as chromatin-remodeling genes (SMYD5 and EZH1, with ATF7IP as possible third). Three of the novel ARID (candidate) genes (CTNNA2, FSCN1, and ITSN1), and a known disease gene reported with non-ID phenotype in OMIM (AK1) code for postsynaptic density proteins (http://www.genes2cognition.org/db/GeneList) [78]. ALS2, another known non-ID disease gene, is the only ARID gene among the top 40 FRMP targets that have been linked to ID or autism [79]. Most FMRP targets and genes implicated in sporadic forms of ID and autism code for exceptionally long, highly brain-expressed proteins [80] whereas ARID proteins tend to be shorter, and as shown here, they are less often involved in multi-protein complexes. These differences may explain why protein interaction and regulatory networks for recessive forms of ID show little overlap with published ones (e.g., see refs. [81, 82]), (see Fig. S7 and Tables S5.1–5.13).
Geographical clustering of recurrent mutations
The scarcity of compound heterozygosity in Iran may reflect the tradition to marry within families or large clans with ‘private’ recessive defects (e.g., see refs. [52, 83]). This is supported by regional clustering of apparently unrelated families with identical mutations. Six likely disease-causing variants were observed in two different families of this cohort. One of these (AP4M1 p.E193K) had already been described in our previous study (family M004 in ref. [33]), and a mutation in another, seventh gene (PRRT2 p.Arg217Profs*8, family M003, Table S1) had also been observed before (family M010 [33]). Most of the families carrying matching mutations turned out to be from the same or neighboring provinces or even the same town (see Fig.1). Haplotype analyses confirmed their identity by descent, even for three families with the recurrent AP4M1 p.E193K mutation living in different regions of Iran, thereby ruling out the possibility of a mutational hotspot (see Table S7). To our knowledge, none of these recurrent mutations has been described outside Iran so far. On the other hand, the relatively small size of the shared haplotypes argues against the possibility that these mutations are evolutionarily young. While in consanguineous demes, genetic drift will rapidly lead to loss of internal diversity at a given locus, there is evidence that the overall gene diversity in the population as a whole will remain remarkably stable [84], which may explain these observations.
Spectrum of ARID genes in neighboring populations
Recently, several groups have looked for genetic defects causing neurodevelopmental disorders in neighboring countries where parental consanguinity is also common. We have compared the outcome of our previous [33] and the current investigations with combined data from Arab countries [34, 37, 38, 85], and with the results of studies conducted in Turkey [35] and Pakistan [36, 39]. Of the 228 known and novel ARID (candidate) genes carrying mutations in our (mainly Persian) cohort, only 28, 11, and 25, respectively, were found to be mutated in the cohorts from Arab countries, from Turkey and from Pakistan (Fig. 3, Table S8).

Genes mutated in autosomal recessive ID and related neurodevelopmental disorders in predominantly Iranian (cohort A, 228 genes [5, 33, 44], and this study), Arabs (cohort B, 252 genes [34, 37, 38, 85]), Turkish families (cohort C, 67 genes [35]), Pakistani Families (cohort D, 100 genes [36, 39]) (see Table S8)
Of note, no single ARID gene was found to be mutated in all 4 cohorts, which corroborates the conclusion that in highly consanguineous populations, most severe recessive disease-causing mutations are confined to clans and do not spread much farther, and that in ARID, the locus heterogeneity is extremely high [19]. Thus, compared to the estimated 500 genes involved in ADID [14, 15] the number of ARID genes must be large and is likely to run into the thousands [4].
Marrying within families or clans should also favor the regional clustering of genetic risk factors for related complex diseases. Thus, genetic factors predisposing to other neuro-psychiatric disorders are not likely to spread in these countries either, and common associated markers are expected to be rare, not only in conditions such as autism and schizophrenia where GWAS cannot work due to reduced fecundity of affected individuals and rapid turnover of the predisposing genetic factors [21, 86].
The quest for ID genes: an unaccomplished mission
In view of the rapidly growing capacity for whole exome or whole genome sequencing, the detection of genetic variants is no longer a problem, but assessing the clinical relevance of genetic variants is still a bottleneck. Algorithms predicting the pathogenicity of missense variants in known disease-associated genes have improved, but their reliability is still limited. Gene- or pathway-specific functional tests have been employed to study mutations implicated in immunodeficiencies [87] and defects in the blood coagulation pathway [88], but devising analogous functional tests for neuropsychiatric disorders [21] is a much greater challenge. For ARID, this approach is no realistic option given the plethora of functionally different ARID genes and the high proportion of families with likely causative mutations in novel genes, indicating that most ARID genes have not been identified yet (see also Fig. 3).
Therefore, the search for gene defects that cause or predispose to ID and/or related disorders has to remain a priority of research into neuropsychiatric disorders until most of the underlying gene defects are known. In line with previous considerations [89], our results suggest that there are more recessive than dominant forms of ID, and their overdue systematic elucidation will generate a wealth of new data on the development and function of the central nervous system. Searching for multiple allelic mutations in cohorts of consanguineous families with two or more affected children is the strategy of choice for identifying hitherto unknown ARID genes. The success of this approach is primarily dependent on the number of families studied, and at least in principle, it does not rely on functional clues which are often scant or absent. The identification of most or all single-gene defects causing ID and related neurodevelopmental disorders will be a major step towards understanding the function of the human brain in health and disease.
Where are the missing mutations?
We detected likely disease-causing variants in 219 out of 404 consanguineous ID families (54.2%). This finding was comparable to that of our previous, smaller study (57% [33]), but lower than in two other recent investigations [34, 35]. The lower mutation yield of our present study may reflect the inclusion of families which had been unsuccessfully screened by targeted exon sequencing before. Of note, 11 families harbored variants in genes known to be associated with diseases other than ID.
These studies and the unexpected paucity of compound heterozygosity highlight the importance of linkage information and suggest that combining autozygosity mapping with WES is a superior strategy for identifying disease-causing mutations in consanguineous families. Autozygous genomic segments harboring most of the recessive mutations can also be identified by sequencing several individuals per family, which is a time-saving, albeit more expensive alternative to prior linkage studies [35].
Not unexpectedly, the additional diagnostic yield of WGS was limited suggesting that the vast difference between WES and WGS reported by others may in the first place reflect technical differences rather than indicating that WGS is fundamentally superior to WES. In principle, of course, WGS should allow to detect all kinds of mutations everywhere in the genome, not only in coding regions and exon-flanking splice sites. In practice, however, this advantage is rather theoretical as long as we cannot reliably identify functionally relevant sequence variants in the non-coding portion of the genome, including deep intronic or even exonic mutations affecting splicing, but also enhancer, repressor or insulator mutations in intergenic regions. Recent studies suggest that exonic variants enhancing or silencing splicing [90] or generating novel splice sites [91] are important ‘sinks’ of disease-causing mutations, and novel algorithms have been developed that promise to facilitate their identification.
Other mechanisms that may cause ID in sporadic patients such as dominant de novo mutations, polygenic inheritance or epigenetic changes that are not directly related to changes in the DNA sequence are unlikely to cause ID in multiple children of healthy consanguineous parents, and recurrent parental germline mutations are also rare.
Many of the mutations missed by the afore-mentioned studies may involve non-coding regulatory sequences. Therefore, their identification and functional characterization is of central fundamental and diagnostic importance. For defining regulatory sequences in the genome ([92] and references therein) it is advantageous to study defects with highly specific, recognizable phenotypes; thus, ID does not qualify. It is also unlikely that algorithms for assessing the functional relevance of non-coding sequence variants will be available soon. However, searching for regulatory mutations in autozygous genome intervals of large ARID families is a viable option, as previously documented for X-linked ID [93]. Large ARID families from our cohort where even WGS failed to identify a likely causative mutation should be particularly suitable for this purpose.
The spectrum of ARID gene defects identified in predominantly Iranian, Arabian, Turkish, and Pakistani families shows little overlap, as illustrated above. In countries where parental consanguinity has been practiced for many generations, deleterious recessive mutations are expected to differ between demes or clans, and the gene defects present in the entire population are a more or less stochastic sample from the large pool of gene defects that can give rise to ARID. Secondly, in samples of limited size, the frequency of specific gene defects causing ARID will differ from their prevalence in the population. Given the relatively small cohorts of ARID families studied to date and the very high number of potential ARID genes, this sampling error is presumably large. Indeed, this is supported by the limited number of overlapping gene defects identified in two separate cohorts of families sampled from the Iranian population [33] (this study, see Fig. S8). Therefore, it may be possible to consider these cohorts and those from Arab countries, Turkey and Pakistan as independent samples from the same gene pool, and the combined outcome of these studies may provide a sufficiently broad basis for estimating the total number of ARID genes [94].
In conclusion, this study has identified numerous novel ARID genes, as well as likely ID-causing mutations in a large number of genes that had not been implicated in ARID before. It revealed that most forms of ARID are syndromic, with microcephaly being present in almost half of the families, while autism is rare; and that genomic sequencing and autozygosity mapping in consanguineous families is the strategy of choice for identifying novel ARID genes. Our study showed that the implementation of WES or WGS might be an efficient diagnostic strategy for countries where parental consanguinity is common and recessive disorders are a central problem of health care. In outbred Western populations, large consanguineous families are rare, and even the largest pilot studies may be too small for elucidating recessive disorders in a systematic fashion.
References
Shaw-Smith C, Redon R, Rickman L, Rio M, Willatt L, Fiegler H, et al. Microarray based comparative genomic hybridisation (array-CGH) detects submicroscopic chromosomal deletions and duplications in patients with learning disability/mental retardation and dysmorphic features. J Med Genet. 2004;41:241–8.
Gilissen C, Hehir-Kwa JY, Thung DT, van de Vorst M, van Bon BW, Willemsen MH, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014;511:344–7.
Vissers LE, de Ligt J, Gilissen C, Janssen I, Steehouwer M, de Vries P, et al. A de novo paradigm for mental retardation. Nat Genet. 2010;42:1109–12.
Ropers HH. Genetics of early onset cognitive impairment. Annu Rev Genom Hum Genet. 2010;11:161–87.
Musante L, Ropers HH. Genetics of recessive cognitive disorders. Trends Genet. 2014;30:32–39.
de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367:1921–9.
Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–82.
Fitzgerald TW, Gerety SS, Jones WD, van Kogelenberg M, King DA, McRae J, et al. Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015;519:223–8.
Lelieveld SH, Reijnders MR, Pfundt R, Yntema HG, Kamsteeg EJ, de Vries P, et al. Meta-analysis of 2104 trios provides support for 10 new genes for intellectual disability. Nat Neurosci. 2016;19:1194–6.
Hamdan FF, Srour M, Capo-Chichi JM, Daoud H, Nassif C, Patry L, et al. De novo mutations in moderate or severe intellectual disability. PLoS Genet. 2014;10:e1004772.
Krumm N, O’Roak BJ, Shendure J, Eichler EE. A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 2014;37:95–105.
Yuen RK, Thiruvahindrapuram B, Merico D, Walker S, Tammimies K, Hoang N, et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat Med. 2015;21:185–91.
MacArthur DG, Balasubramanian S, Frankish A, Huang N, Morris J, Walter K, et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–8.
Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–21.
Ronemus M, Iossifov I, Levy D, Wigler M. The role of de novo mutations in the genetics of autism spectrum disorders. Nat Rev Genet. 2014;15:133–41.
Kochinke K, Zweier C, Nijhof B, Fenckova M, Cizek P, Honti F, et al. Systematic phenomics analysis deconvolutes genes mutated in intellectual disability into biologically coherent modules. Am J Hum Genet. 2016;98:149–64.
Vissers LE, Gilissen C, Veltman JA. Genetic studies in intellectual disability and related disorders. Nat Rev Genet. 2016;17:9–18.
Kuss AW, Garshasbi M, Kahrizi K, Tzschach A, Behjati F, Darvish H, et al. Autosomal recessive mental retardation: homozygosity mapping identifies 27 single linkage intervals, at least 14 novel loci and several mutation hotspots. Hum Genet. 2011;129:141–8.
Najmabadi H, Motazacker MM, Garshasbi M, Kahrizi K, Tzschach A, Chen W, et al. Homozygosity mapping in consanguineous families reveals extreme heterogeneity of non-syndromic autosomal recessive mental retardation and identifies 8 novel gene loci. Hum Genet. 2007;121:43–8.
Hu H, Haas SA, Chelly J, Van Esch H, Raynaud M, de Brouwer AP, et al. X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes. Mol Psychiatry. 2016;21:133–48.
Heinzen EL, Neale BM, Traynelis SF, Allen AS, Goldstein DB. The genetics of neuropsychiatric diseases: looking in and beyond the exome. Annu Rev Neurosci. 2015;38:47–68.
Durkin MS, Hasan ZM, Hasan KZ. Prevalence and correlates of mental retardation among children in Karachi, Pakistan. Am J Epidemiol. 1998;147:281–8.
Kaufman L, Ayub M, Vincent JB. The genetic basis of non-syndromic intellectual disability: a review. J Neurodev Disord. 2010;2:182–209.
Saad HA, Elbedour S, Hallaq E, Merrick J, Tenenbaum A. Consanguineous marriage and intellectual and developmental disabilities among Arab Bedouins Children of the Negev Region in Southern Israel: a pilot study. Front Public Health. 2014;2:3.
Weller M, Tanieri M, Pereira JC, Almeida Edos S, Kok F, Santos S. Consanguineous unions and the burden of disability: a population-based study in communities of Northeastern Brazil. Am J Hum Biol. 2012;24:835–40.
Monies D, Abouelhoda M, AlSayed M, Alhassnan Z, Alotaibi M, Kayyali H, et al. The landscape of genetic diseases in Saudi Arabia based on the first 1000 diagnostic panels and exomes. Hum Genet. 2017;136:921–39.
Fareed M, Afzal M. Estimating the inbreeding depression on cognitive behavior: a population based study of child cohort. PLoS ONE. 2014;9:e109585.
Hamamy H. Consanguineous marriages: preconception consultation in primary health care settings. J Community Genet. 2012;3:185–92.
Bittles AH, Black ML. Evolution in health and medicine Sackler colloquium: consanguinity, human evolution, and complex diseases. Proc Natl Acad Sci USA. 2010;107 Suppl 1:1779–86.
Schuurs-Hoeijmakers JHM, Hehir-Kwa JY, Pfundt R, van Bon BWM, de Leeuw N, Kleefstra T, et al. Homozygosity mapping in outbred families with mental retardation. Eur J Human Genet. 2011;19:597–601.
Schuurs-Hoeijmakers JHM, Vulto-van Silfhout AT, Vissers LELM, van de Vondervoort IIGM, van Bon BWM, de Ligt J, et al. Identification of pathogenic gene variants in small families with intellectually disabled siblings by exome sequencing. J Med Genet. 2013;50:802–11.
Akawi NA, Al-Jasmi F, Al-Shamsi AM, Ali BR, Al-Gazali L. LINS, a modulator of the WNT signaling pathway, is involved in human cognition. Orphanet J Rare Dis. 2013;8:87.
Najmabadi H, Hu H, Garshasbi M, Zemojtel T, Abedini SS, Chen W, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478:57–63.
Alazami AM, Patel N, Shamseldin HE, Anazi S, Al-Dosari MS, Alzahrani F, et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 2015;10:148–61.
Karaca E, Harel T, Pehlivan D, Jhangiani SN, Gambin T, Coban Akdemir Z, et al. Genes that affect brain structure and function identified by rare variant analyses of mendelian neurologic disease. Neuron. 2015;88:499–513.
Riazuddin S, Hussain M, Razzaq A, Iqbal Z, Shahzad M, Polla DL, et al. Exome sequencing of Pakistani consanguineous families identifies 30 novel candidate genes for recessive intellectual disability. Mol Psychiatry. 2017;22:1604–14.
Reuter MS, Tawamie H, Buchert R, Hosny Gebril O, Froukh T, Thiel C, et al. Diagnostic yield and novel candidate genes by exome sequencing in 152 consanguineous families with neurodevelopmental disorders JAMA Psychiatry. 2017;74:293–9.
Anazi S, Maddirevula S, Faqeih E, Alsedairy H, Alzahrani F, Shamseldin HE, et al. Clinical genomics expands the morbid genome of intellectual disability and offers a high diagnostic yield Mol Psychiatry. 2017;22:615–24.
Harripaul R, Vasli N, Mikhailov A, Rafiq MA, Mittal K, Windpassinger C, et al. Mapping autosomal recessive intellectual disability: combined microarray and exome sequencing identifies 26 novel candidate genes in 192 consanguineous families. Mol Psychiatry. 2017 Apr 11. https://doi.org/10.1038/mp.2017.60. [Epub ahead of print]
Saadat M, Ansari-Lari M, Farhud DD. Consanguineous marriage in Iran. Ann Hum Biol. 2004;31:263–9.
Jalal Abbasi-Shavazi M, McDonald P, Hosseini-Chavoshi M. Modernization or cultural maintenance: the practice of consanguineous marriage in Iran. J Biosoc Sci. 2008;40:911–33.
Najmabadi H, Ghamari A, Sahebjam F, Kariminejad R, Hadavi V, Khatibi T, et al. Fourteen-year experience of prenatal diagnosis of thalassemia in Iran. Community Genet. 2006;9:93–97.
Sloan-Heggen CM, Babanejad M, Beheshtian M, Simpson AC, Booth KT, Ardalani F, et al. Characterising the spectrum of autosomal recessive hereditary hearing loss in Iran. J Med Genet. 2015;52:823–9.
Kahrizi, K. and Najmabadi, H. 2015. Genetics of Recessive Cognitive Disorders. eLS. 1–21. Wiley Online Library.
Hu H, Wienker TF, Musante L, Kalscheuer VM, Kahrizi K, Najmabadi H, et al. Integrated sequence analysis pipeline provides one-stop solution for identifying disease-causing mutations. Hum Mutat. 2014;35:1427–35.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Keleman K, Kruttner S, Alenius M, Dickson BJ. Function of the Drosophila CPEB protein Orb2 in long-term courtship memory. Nat Neurosci. 2007;10:1587–93.
Siwicki KK, Ladewski L. Associative learning and memory in Drosophila: beyond olfactory conditioning. Behav Process. 2003;64:225–38.
Willemsen MH, Ba W, Wissink-Lindhout WM, de Brouwer AP, Haas SA, Bienek M, et al. Involvement of the kinesin family members KIF4A and KIF5C in intellectual disability and synaptic function. J Med Genet. 2014;51:487–94.
Whibley AC, Plagnol V, Tarpey PS, Abidi F, Fullston T, Choma MK, et al. Fine-scale survey of X chromosome copy number variants and indels underlying intellectual disability. Am J Hum Genet. 2010;87:173–88.
Pouya AR, Abedini SS, Mansoorian N, Behjati F, Nikzat N, Mohseni M, et al. Fragile X syndrome screening of families with consanguineous and non-consanguineous parents in the Iranian population. Eur J Med Genet. 2009;52:170–3.
Hosseini-Chavoshi M, Abbasi-Shavazi MJ, Bittles AH. Consanguineous marriage, reproductive behaviour and postnatal mortality in contemporary Iran. Hum Hered. 2014;77:16–25.
Narasimhan VM, Hunt KA, Mason D, Baker CL, Karczewski KJ, Barnes MR, et al. Health and population effects of rare gene knockouts in adult humans with related parents. Science. 2016;352:474–7.
Khan MA, Rupp VM, Orpinell M, Hussain MS, Altmuller J, Steinmetz MO, et al. A missense mutation in the PISA domain of HsSAS-6 causes autosomal recessive primary microcephaly in a large consanguineous Pakistani family. Hum Mol Genet. 2014;23:5940–9.
Law R, Dixon-Salazar T, Jerber J, Cai N, Abbasi AA, Zaki MS, et al. Biallelic truncating mutations in FMN2, encoding the actin-regulatory protein Formin 2, cause nonsyndromic autosomal-recessive intellectual disability. Am J Hum Genet. 2014;95:721–8.
Larti F, Kahrizi K, Musante L, Hu H, Papari E, Fattahi Z, et al. A defect in the CLIP1 gene (CLIP-170) can cause autosomal recessive intellectual disability. Eur J Hum Genet. 2015;23:331–6.
Oladnabi M, Musante L, Larti F, Hu H, Abedini SS, Wienker T, et al. New evidence for the role of calpain 10 in autosomal recessive intellectual disability: identification of two novel nonsense variants by exome sequencing in Iranian families. Arch Iran Med. 2015;18:179–84.
Alakbarzade V, Hameed A, Quek DQ, Chioza BA, Baple EL, Cazenave-Gassiot A, et al. A partially inactivating mutation in the sodium-dependent lysophosphatidylcholine transporter MFSD2A causes a non-lethal microcephaly syndrome. Nat Genet. 2015;47:814–7.
Guemez-Gamboa A, Nguyen LN, Yang H, Zaki MS, Kara M, Ben-Omran T, et al. Inactivating mutations in MFSD2A, required for omega-3 fatty acid transport in brain, cause a lethal microcephaly syndrome. Nat Genet. 2015;47:809–13.
Iqbal Z, Willemsen MH, Papon MA, Musante L, Benevento M, Hu H, et al. Homozygous SLC6A17 mutations cause autosomal-recessive intellectual disability with progressive tremor, speech impairment, and behavioral problems. Am J Hum Genet. 2015;96:386–96.
Heidari A, Tongsook C, Najafipour R, Musante L, Vasli N, Garshasbi M, et al. Mutations in the histamine N-methyltransferase gene, HNMT, are associated with nonsyndromic autosomal recessive intellectual disability. Hum Mol Genet. 2015;24:5697–710.
Snijders Blok L, Madsen E, Juusola J, Gilissen C, Baralle D, Reijnders MR, et al. Mutations in DDX3X are a common cause of unexplained intellectual disability with gender-specific effects on Wnt signaling. Am J Hum Genet. 2015;97:343–52.
O’Rawe JA, Wu Y, Dorfel MJ, Rope AF, Au PY, Parboosingh JS, et al. TAF1 variants are associated with dysmorphic features, intellectual disability, and neurological manifestations. Am J Hum Genet. 2015;97:922–32.
Nellhaus G. Head circumference from birth to eighteen years. Practical composite international and interracial graphs. Pediatrics. 1968;41:106–14.
Davis JM, Keeney JG, Sikela JM, Hepburn S. Mode of genetic inheritance modifies the association of head circumference and autism-related symptoms: a cross-sectional study. PLoS One. 2013;8:e74940.
Liang H, Xiao G, Yin H, Hippenmeyer S, Horowitz JM, Ghashghaei HT. Neural development is dependent on the function of specificity protein 2 in cell cycle progression. Development. 2013;140:552–61.
Vandeputte DA, Meije CB, van Dartel M, Leenstra S, IJ-K H, Das PK, et al. GOA, a novel gene encoding a ring finger B-box coiled-coil protein, is overexpressed in astrocytoma. Biochem Biophys Res Commun. 2001;286:574–9.
Parkel S, Lopez-Atalaya JP, Barco A. Histone H3 lysine methylation in cognition and intellectual disability disorders. Learn Mem. 2013;20:570–9.
Conway E, Healy E, Bracken AP. PRC2 mediated H3K27 methylations in cellular identity and cancer. Curr Opin Cell Biol. 2015;37:42–48.
Frerker N, Wagner L, Wolf R, Heiser U, Hoffmann T, Rahfeld JU, et al. Neuropeptide Y (NPY) cleaving enzymes: structural and functional homologues of dipeptidyl peptidase 4. Peptides. 2007;28:257–68.
Ono S, Yoshiura K, Kinoshita A, Kikuchi T, Nakane Y, Kato N, et al. Mutations in PRRT2 responsible for paroxysmal kinesigenic dyskinesias also cause benign familial infantile convulsions. J Hum Genet. 2012;57:338–41.
Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–99.
Basel-Vanagaite L, Attia R, Yahav M, Ferland RJ, Anteki L, Walsh CA, et al. The CC2D1A, a member of a new gene family with C2 domains, is involved in autosomal recessive non-syndromic mental retardation. J Med Genet. 2006;43:203–10.
Mir A, Kaufman L, Noor A, Motazacker MM, Jamil T, Azam M, et al. Identification of mutations in TRAPPC9, which encodes the NIK- and IKK-beta-binding protein, in nonsyndromic autosomal-recessive mental retardation. Am J Hum Genet. 2009;85:909–15.
Kishi N, MacDonald JL, Ye J, Molyneaux BJ, Azim E, Macklis JD. Reduction of aberrant NF-kappaB signalling ameliorates Rett syndrome phenotypes in Mecp2-null mice. Nat Commun. 2016;7:10520.
Gamsiz ED, Sciarra LN, Maguire AM, Pescosolido MF, van Dyck LI, Morrow EM. Discovery of rare mutations in autism: elucidating neurodevelopmental mechanisms. Neurotherapeutics. 2015;12:553–71.
Coba MP, Komiyama NH, Nithianantharajah J, Kopanitsa MV, Indersmitten T, Skene NG, et al. TNiK is required for postsynaptic and nuclear signaling pathways and cognitive function. J Neurosci. 2012;32:13987–99.
Bayes A, van de Lagemaat LN, Collins MO, Croning MD, Whittle IR, Choudhary JS, et al. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat Neurosci. 2011;14:19–21.
Suhl JA, Chopra P, Anderson BR, Bassell GJ, Warren ST. Analysis of FMRP mRNA target datasets reveals highly associated mRNAs mediated by G-quadruplex structures formed via clustered WGGA sequences. Hum Mol Genet. 2014;23:5479–91.
Ouwenga RL, Dougherty J. Fmrp targets or not: long, highly brain-expressed genes tend to be implicated in autism and brain disorders. Mol Autism. 2015;6:16.
Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, Vitkup D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron. 2011;70:898–907.
O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43:585–9.
Basel-Vanagaite L, Taub E, Halpern GJ, Drasinover V, Magal N, Davidov B, et al. Genetic screening for autosomal recessive nonsyndromic mental retardation in an isolated population in Israel. Eur J Hum Genet. 2007;15:250–3.
Krawczak M, Barnes R. How obedience of marriage rules may counteract genetic drift. J Community Genet. 2010;1:23–28.
Shaheen R, Patel N, Shamseldin H, Alzahrani F, Al-Yamany R, AL A, et al. Accelerating matchmaking of novel dysmorphology syndromes through clinical and genomic characterization of a large cohort. Genet Med. 2016;18:686–95.
Power RA, Kyaga S, Uher R, MacCabe JH, Langstrom N, Landen M, et al. Fecundity of patients with schizophrenia, autism, bipolar disorder, depression, anorexia nervosa, or substance abuse vs their unaffected siblings. JAMA Psychiatry. 2013;70:22–30.
Casanova JL, Conley ME, Seligman SJ, Abel L, Notarangelo LD. Guidelines for genetic studies in single patients: lessons from primary immunodeficiencies. J Exp Med. 2014;211:2137–49.
Zhang B, Spreafico M, Zheng C, Yang A, Platzer P, Callaghan MU, et al. Genotype-phenotype correlation in combined deficiency of factor V and factor VIII. Blood. 2008;111:5592–5600.
Ropers HH. New perspectives for the elucidation of genetic disorders. Am J Hum Genet. 2007;81:199–207.
Soukarieh O, Gaildrat P, Hamieh M, Drouet A, Baert-Desurmont S, Frebourg T, et al. Exonic splicing mutations are more prevalent than currently estimated and can be predicted by using in silico tools. PLoS Genet. 2016;12:e1005756.
Lee M, Roos P, Sharma N, Atalar M, Evans TA, Pellicore MJ, et al. Systematic computational identification of variants that activate exonic and intronic cryptic splice sites. Am J Hum Genet. 2017;100:751–65.
Lupianez DG, Spielmann M, Mundlos S. Breaking TADs: how alterations of chromatin domains result in disease. Trends Genet. 2016;32:225–37.
Huang L, Jolly LA, Willis-Owen S, Gardner A, Kumar R, Douglas E, et al. A noncoding, regulatory mutation implicates HCFC1 in nonsyndromic intellectual disability. Am J Hum Genet. 2012;91:694–702.
Bohning D, Rocchetti I, Alfo M, Holling H. A flexible ratio regression approach for zero-truncated capture-recapture counts. Biometrics. 2016;72:697–706.
Acknowledgements
We are grateful to all patients and families for their participation in this study, S. Banihashemi and Kh. Jalalvand for technical support and Gabriele Eder for assisting us with the preparation of the manuscript. We would also like to thank the NHLBI GO Exome Sequencing Project and its ongoing studies, as well as the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about. This research was supported by the European Union through FP7 project GENCODYS, grant no 241995 (organizer: Hans van Bokhoven), the Max Planck Innovation Funds and the Ministry of Health and Medical Education, Islamic Republic Iran. Additional support was received from the Iranian National Science Foundation (grant no.s 92038458 and 92035782), the Iranian National Elite Foundation and the Iranian Science Elite Federation.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Hu, H., Kahrizi, K., Musante, L. et al. Genetics of intellectual disability in consanguineous families. Mol Psychiatry 24, 1027–1039 (2019). https://doi.org/10.1038/s41380-017-0012-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-017-0012-2
- Springer Nature Limited
This article is cited by
-
Identification of novel likely pathogenic variant in CDH23 causing non-syndromic hearing loss, and a novel variant in OTOGL in an extended Iranian family
Egyptian Journal of Medical Human Genetics (2024)
-
In silico analysis of mutation spectrum of Ehlers–Danlos, osteogenesis imperfecta, and cutis laxa overlapping phenotypes in Iranian population
Egyptian Journal of Medical Human Genetics (2024)
-
Bi-allelic truncating variants in CASP2 underlie a neurodevelopmental disorder with lissencephaly
European Journal of Human Genetics (2024)
-
New insights into the clinical and molecular spectrum of the MADD-related neurodevelopmental disorder
Journal of Human Genetics (2024)
-
Novel compound heterozygous ABCA2 variants cause IDPOGSA, a variable phenotypic syndrome with intellectual disability
Journal of Human Genetics (2024)