
Abstract
Background:
The aim of this clinical study was to determine the prevalence of SHOX haploinsufficiency in a population of short stature patients and describe their anthropometric measurements.
Methods:
574 short statured patients were evaluated in a single center (1992–2015). SHOX copy number was detected by quantitative polymerase chain reaction (qPCR) in 574 subjects, followed by multiplex ligation-dependent probe amplification (MLPA) and DNA sequencing in subjects with SHOX haploinsufficiency. We evaluated anthropometric measurements at birth, and at first examination. Skeletal abnormalities were recorded for patients with SHOX haploinsufficiency.
Results:
Thirty-two patients were excluded due to Turner syndrome (n = 28), SRY-positive 46,XX male karyotype (n = 1), or lacked clinical follow-up information (n = 3). The prevalence of SHOX haploinsufficiency was 9 out of 542 (1.7%). The nine children had decreased height −2.85 (0.6) SD scores (SDS) (mean (SD)) and weight −2.15 (1.36) SDS, P < 0.001 and P = 0.001, respectively. The sitting height/height ratio was increased, P = 0.04. Madelung deformity was diagnosed in three patients. Mean height was −2.9 (0.4) SDS at baseline and increased by 0.25 (0.2) SDS, P = 0.046, after 1 y of growth hormone (GH) treatment.
Conclusion:
The prevalence of SHOX haploinsufficiency was 1.7%. The clinical findings indicating SHOX haploinsufficiency among the nine children were disproportionate short stature and forearm anomalies.
Similar content being viewed by others

Main
Short stature is defined by a height below −2 SD scores (SDS) from an age- and gender-related population mean. The etiologies of short stature are numerous including endocrine disease such as growth hormone deficiency and various chronic diseases (1).
Longitudinal growth is regulated by the growth hormone/insulin-like growth factor-I axis (GH-IGF-I axis). The free biological active form of IGF-I is inducing the chondrocytes of the epiphyseal plates in the tubular bones to proliferate (2). In addition, FGF receptors (3) and the short stature homeobox-containing (SHOX) gene are regulating linear growth of the skeleton (4).
In the absence of any known causes of short stature, the patient is diagnosed with idiopathic short stature, which is an exclusion diagnosis defined by a height below −2 SDS without any causative disorders (1). However, it has been estimated that 1.1–16.9% of the idiopathic short stature patients have a haploinsufficiency of the SHOX gene (4,5,6,7,8,9,10,11,12,13,14,15,16).
The SHOX gene resides in the pseudoautosomal region 1 (PAR1) of both sex chromosomes; it escapes X inactivation (17) and the gene is expressed by growth plate chondrocytes (18,19). SHOX haploinsufficiency disorders include Leri-Weill dyschondrosteosis (20), Langer mesomelic dysplasia (21) and is involved in short stature and skeletal abnormalities of Turner syndrome (22). It is important to identify a patient with SHOX haploinsufficiency in order for the patient to benefit from possible GH therapy, and to convey the necessary genetic counseling. The aim of this study was (i) to evaluate the prevalence of SHOX haploinsufficiency in a population of Danish short stature patients and (ii) to describe clinical and biochemical characteristics of the patients.
Methods
Study Population
This study was a retrospective clinical study of 574 patients with growth retardation refereed to our tertiary pediatric endocrine department during the period of May 1992 to April 2015. The patients were selected due to the following inclusion criteria: (i) A diagnosis of short stature (ICD10 DE34.3) (ii) SHOX gene evaluation by qPCR. (iii) Available record files with necessary follow-up information. Data on anthropometric measurements and hormone analyses were collected from the patients’ files.
Clinical Examination
Height and sitting height were measured on a wall-mounted stadio-meter to the nearest 0.1 cm by trained staff. Weight was measured using electronic scales to the nearest 0.1 kg and BMI was calculated (weight in kilogram/height in meters2). SDS were derived for sitting height, birth weight, birth length, height, weight, and BMI using normal reference populations (23,24,25). Target height (SDS) was calculated as the mean of the maternal and paternal height (SDS). Data on pubertal development (Tanner stage (26,27)), age at menarche, and phenotypic characteristics (Madelung deformities, sitting height, brachymetacarpy, hyperconvex nails, cubitus valgus, scoliosis, short neck, micrognathia, high arched palate, muscular hypertrophy, increased papillae mammae gap, thoracic hypertrophy, naevi, and short lower leg) were obtained from the patient record files if available.
X-ray Investigations
Bone age and bone health index (BHI) was determined using an automated evaluation system (BoneXpert, Visiana, Denmark) (28). BHI is a size-corrected measure of the cortical thickness of the three middle metacarpals and is used to assess the bone mass of the patient. The calculation of the BHI (SDS) was based upon a large normative dataset of individual sex- and bone age-dependent SDS (29).
Karyotyping
Karyotyping was performed on lymphocytes isolated from peripheral blood using routine G-banding and counting of at least 10 metaphases, three of which were fully analyzed.
SHOX Copy Number Analysis by qPCR
The copy numbers of the SHOX gene were analyzed by qPCR with a forward and reverse primer targeting exon 1. Forward primer: 5′-CTC-CTA-CCC-GCC-TGT-CCA-3′. Reverse primer: 5′-TCC-GCG-CGT-CTC-TTT-CTA-CT-3′. The GAPDH gene, which is located on an autosome (chromosome 12), was used as the internal control, using forward primer: 5′-CTC-CCC-ACA-CAC-ATG-CAC-TTA-3′, and the reverse primer: 5′-TTG-CCA-AGT-TGC-CTG-TCC-TT-3′. The qPCR was performed on a Mx3000P platform from Stratagene (Cedar Creek, TX) as previously described (30).
In cases where the gene copy number assessed by qPCR was incompatible with the patient’s karyotype or if the phenotype clearly indicated SHOX haploinsufficiency, the patient was further screened for deletions or mutations in other exons of the SHOX gene by multiplex ligation-dependent probe amplification (MLPA) and SHOX gene sequencing.
MLPA
SHOX deletions were detected by using the MRC-Holland MLPA kit (Salsa P018-E and F1; MRC-Holland, Netherlands) according to the instructions of the manufacturer. The P018-G1 SHOX probe-mix contains 48 MLPA probes. Seven of the probes were specific for each exon of the SHOX gene. In total 26 probes targeted sequences located in the SHOX gene and in regions corresponding to the regulatory areas of SHOX transcription. The probe-mix also included 13 probes located outside of the PAR regions and 9 autosomal probes. Data analysis was performed using either the GeneMarker v.2.0 (Softgenetics, Stage College, PA) or Coffalyser software v. 9.4 (software by MRC-Holland, Netherlands) (31).
DNA Sequencing
The coding and intron flanking sequences of SHOX (NM_000451.3 and NM_006883.2) were PCR amplified and the products were subjected to Sanger sequencing by a standard approach on an ABI3130XL sequencer. Primers and PCR conditions are available upon request.
Hormone Assays
All blood samples were analyzed at Department of Growth and Reproduction, Rigshospitalet, Copenhagen, Denmark. Serum IGF-I concentrations were determined by three different assays during the 23-y study period. Until 2008 IGF-I concentrations were determined by radioimmunoassay serum as previously described (32). Limit of detection for IGF-I was 20 ng/ml and the coefficients of variation for the inter- and intra-assay were 15 and 6%, respectively. IGF-I (SDS) was calculated from our reference data (32). From 2008 to 2013, serum IGF-I concentrations were determined by using a solid-phase enzyme-labeled chemiluminescent immunometric assay (Immulite 2000, Diagnostic Products Corporation, L.A., CA) (33). Limit of detection was 20 ng/ml and the inter- and intra-assay coefficients of variations were 5.9 and 2.0%, respectively. IGF-I (SDS) was calculated from our reference data (34). From 2013, IGF-I concentrations were measured by an iSYS assay on IDS-iSYS Multidiscipline Automated Analyser (Immunodiagnosticsystems, UK). Limit of detection for IGF-I was 8.8 μg/l with an inter-assay coefficients of variation of 4.7–7.2%.
Statistical Analysis
The frequency of the SHOX haploinsufficiency is presented in percentage. We performed descriptive statistics on all patients. Variables are shown as standard deviation scores. Statistical analyses on SDS were done by using a One-Sample T-test. A P value < 0.05 was considered significant. For the evaluation of height (SDS) and IGF-I (SDS) prior to GH therapy and after 1 y of treatment, we performed a Wilcoxon Signed Ranks test. We used nonparametric testing by Mann-Whitney U-test when comparing the clinical characteristics of the patients with SHOX haploinsufficiency to the patients with a normal copy number of the SHOX gene. All statistical analyses were performed by using IBM SPSS Statistics for Windows, version 22.0 Armonk, NY: IBM Corp.
Ethical Considerations
All blood samples and genetic analyses were taken as part of the clinical follow-up of the patients. The project was approved by Danish Data Protection Agency permission for biobank: RH-2016–177, I-Suite no: 04732.
Results
In the cohort of 574 patients, 41 patients had one copy number of the SHOX gene. Thirty-two of these had either Turner syndrome (n = 28), a SRY-positive 46,XX male karyotype (n = 1), or lacked clinical follow-up information (n = 3).
Nine children (three boys) out of 542 patients (318 boys) had one copy number of the SHOX gene, and 533 patients had 2 SHOX gene copy numbers, resulting in a prevalence of 1.7%. The age of the children with SHOX haploinsufficiency at referral ranged between 0.5 and 14 y. Two girls (ID no. 6 and 9) were adopted from China and India, respectively.
SHOX gene point mutations or deletions are shown for each child (n = 9) in Table 1 . Three children (two boys) had known affected relatives.
Anthropometric Measurements
Height, weight, BMI, and pubertal stage at referral for each child with SHOX haploinsufficiency are shown in Table 1 . Height at referral and first available sitting height/height ratio are illustrated in Figures 1 and 2 comparing the nine children with SHOX haploinsufficiency to the 533 patients with two SHOX copies. The sitting height/height ratio was above +2 SDS in 4 children (1 boy) with SHOX haploinsufficiency. Anthropometric measurements at birth and at time of first examination for the children with SHOX haploinsufficiency are presented as standard deviation scores in Figure 3 . The children with SHOX haploinsufficiency had significantly decreased height −2.85 (0.6) SDS (mean (SD)) and weight −2.15 (1.36) SDS, P < 0.001 and P = 0.001, respectively. The sitting height/height ratio was significantly increased 2.17 (1.28) SDS, P = 0.04, whereas BMI did not differ. We found a decreased BHI in five children (two boys) but this did not reach statistical significance.

Longitudinal growth in short statured children with SHOX haploinsufficiency compared to short statured children with two copies of SHOX. (a) Male. (b) Female. Longitudinal growth in short statured children with two SHOX copies (open circles, n = 533), short statured children with SHOX gene haploinsufficiency (black circles, n = 6) and children with Leri-Weill dyschondrosteosis, short stature and SHOX gene haploinsufficiency (black triangles, n = 3) compared to a Danish normal reference population (25). Black lines correspond to −2 SD, mean and +2 SD.

Sitting height/height ratio in short statured children with SHOX haploinsufficiency compared to short statured children with two copies of SHOX. (a) Male. (b) Female. Sitting height/height ratio in short statured children with two SHOX copies (open circles, n = 157), short statured children with SHOX gene haploinsufficiency (black circles, n = 4) and children with Leri-Weill dyschondrosteosis, short stature and SHOX gene haploinsufficiency (black triangles, n = 3) compared to a Danish normal reference population (23). Black lines correspond to −2 SD, mean and +2 SD.

Anthropometric measurements at first presentation for patients with SHOX gene haploinsufficiency (black circles, n = 6) and patients with Leri-Weill dyschondrosteosis and SHOX gene haploinsufficiency (black triangles, n = 3). Black stars represent the anthropometric measurements that differed significantly. Black lines represent mean values for each variable. BHI, bone health index; BW, birth weight; BL, birth length; SH/H, sitting height/height ratio. All data are expressed as SD scores.
Comparison of anthropometric measurements between the children with SHOX haploinsufficiency and patients with two SHOX copies are shown in Table 2 . Height (SDS) and the SH/H ratio were significantly different between the two groups, P = 0.002 and P = 0.048, respectively.
X-ray Investigations
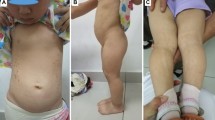
Madelung deformities were diagnosed in three children (one boy). One girl presented with Madelung deformity and bowing of corpus radii. One girl (ID no. 4) had a wrist deformity of the left wrist, but the deformity was not compatible with Madelung deformity. One girl with SHOX haploinsufficiency presented with scoliosis ( Table 1 ).
The bone age showed a mean delay of −1.04 (1.1) years (mean, (SD)) compared to chronological age ( Table 1 ).
Growth Hormone Therapy
Six children (one boy) of the nine patients with SHOX haploinsufficiency were treated with GH. One girl (ID no. 4) was treated with combined GH and gonadotropin-releasing hormone (GnRH) analog therapy. Changes in GH dose, height (SDS), and IGF-I (SDS) before and during the first year of GH treatment are shown in Figure 4 . Mean height (SDS) was −2.9 (0.4) SDS (mean (SD)) at baseline, and −2.67 (0.3) SDS, P = 0.046 after 1 y of treatment. Thus, average height gain was 0.25 (0.2) SDS after 12 mo of GH therapy.

Changes in GH dose, height (SDS) and IGF-I (SDS) before and during the first year of GH therapy. (a) Changes in GH dose (μg/kg/day) during the first 12 mo of GH therapy. (b) Changes in IGF-I (SDS) before and during the first 12 mo of GH therapy. (c) Changes in height (SDS) before and during the first 12 mo of GH therapy. Data are shown for six patients with SHOX gene haploinsufficiency. Black dashed lines represent the start of the GH therapy. Open square = ID no. 1. Open triangle = ID no. 4. Black square = ID no. 5. Black triangle = ID no. 6. Open circle = ID no. 7. Black circle = ID no. 8.
IGF-I (SDS) did increase from the low normal range to the high normal range during the first year of treatment in all patients, P = 0.028. Mean dose of GH was 28.8 (13.7) (μg/kg/day) ( Figure 4 ).
Discussion
In this large single-center study of 574 short statured patients, we found a prevalence of SHOX haploinsufficiency by qPCR of 1.7%. Children with SHOX haploinsufficiency showed moderate height gain following GH therapy.
Former studies have shown a prevalence of point mutations or deletions of the SHOX gene between 1.1–16.9% among patients with idiopatic short stature (4,5,6,7,8,9,10,11,12,13,14,15,16). Our clinical findings suggest that patients with forearm anomalies and short stature with an increased sitting height/height ratio are the most likely to have SHOX haploinsufficiency.
In our study population, four children were known with Madelung deformity or wrist deformity prior to SHOX gene analysis. Another study has shown that the prevalence of Madelung deformity is increasing substantially when radiographic evidence is included (35). It is likely that the frequency of Madelung deformity would be even higher if all patients in our study population were evaluated radiologically. Thus, we do not know if some of the 533 patients with two SHOX copies may have any undiagnosed skeletal lesions.
A phenotypic scoring system has been proposed by Rappold et al. as a way to select patients for SHOX screening, which included a variety of clinical characteristics (7). Our findings of short stature, low weight, and an increased sitting height/height ratio as a critical phenotypic characteristic are consistent with this scoring system. By contrast, BMI did not differ for the nine children with SHOX haploinsufficiency.
The sitting height/height ratio was significantly increased among the children with SHOX haploinsufficiency compared to patients with two SHOX copies. This confirms that an asymmetrical short stature is more frequently seen in our patients with SHOX haploinsufficiency.
Three children (two boys) included in this study had known affected relatives. A careful family history according to height (SDS), body proportions, and dysmorphic characteristics of relatives is necessary when selecting patients for SHOX screening, since SHOX is inherited in a pseudoautosomal dominant manner. Former studies have shown that it is not uncommon for patients with SHOX haploinsufficiency to have an affected parent (7,13).
GH therapy of children with SHOX haploinsufficiency resulted in increasing serum IGF-I levels during the first year of GH treatment. The children experienced a moderate height gain of 0.25 (0.2) SDS, which is less compared to previous studies of SHOX-deficient patients receiving GH therapy (11,35,36). This could be due to the use of lower GH doses in our study of 29 µg/kg/d compared to a previously randomized, controlled study that was using GH doses of 50 µg/kg/d (36).
In our study, one girl (ID no. 4) initiated GnRH analog therapy 1 y after her GH treatment was started due to precocious puberty. She was furthermore a part of the North European Small for Gestational Age (NESGAS) study and on this behalf she received a fixed high GH dose. She reached a height above −1 SDS with 3 y of combined treatment. Other case studies have shown an improvement of the height gain in SHOX-deficient patients treated with combined GH and GnRH analog therapy (37,38), but the long-term outcome of this combined treatment remains still to be elucidated in a large cohort of patients.
In addition, it is important to remember that the SHOX gene is expressed in the chondrocytes of the growth plates in the tubular bones (18,19). It has been estimated that SHOX haploinsufficiency is causing a more rapid fusion of the growth plates resulting in a downward shift of the growth velocity by the time the patients reach puberty (39). This could be an explanation of why some SHOX-deficient patients do not benefit from GH therapy. Furthermore, patients with deletions or mutations delimited to the SHOX gene’s enhancer regions might show a greater respond to GH therapy (40). For this reason, it may be advantageous to start GH therapy as early as possible and carefully examine which SHOX-deficient patients that are responding appropriate to the GH therapy. Whether the SHOX-deficient patients would benefit even more with a combined treatment of GH and GnRH analog therapy is still unclear.
In conclusion, we demonstrated SHOX haploinsufficiency in nine children corresponding to a prevalence of 1.7% among 542 patients with short stature. Due to the importance of early diagnosis and treatment, screening for SHOX haploinsufficiency among disproportionate short statured children must be considered.
Statement of Financial Support
No financial support was received in support of this study.
Disclosure
The authors have no conflict of interest to disclose.
References
Cohen P, Rogol AD, Deal CL, et al.; 2007 ISS Consensus Workshop participants. Consensus statement on the diagnosis and treatment of children with idiopathic short stature: a summary of the Growth Hormone Research Society, the Lawson Wilkins Pediatric Endocrine Society, and the European Society for Paediatric Endocrinology Workshop. J Clin Endocrinol Metab 2008;93:4210–7.
Chia DJ. Minireview: mechanisms of growth hormone-mediated gene regulation. Mol Endocrinol 2014;28:1012–25.
Decker E, Durand C, Bender S, et al. FGFR3 is a target of the homeobox transcription factor SHOX in limb development. Hum Mol Genet 2011;20:1524–35.
Rao E, Weiss B, Fukami M, et al. Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat Genet 1997;16:54–63.
Rappold GA, Fukami M, Niesler B, et al. Deletions of the homeobox gene SHOX (short stature homeobox) are an important cause of growth failure in children with short stature. J Clin Endocrinol Metab 2002;87:1402–6.
Stuppia L, Calabrese G, Gatta V, et al. SHOX mutations detected by FISH and direct sequencing in patients with short stature. J Med Genet 2003;40:E11.
Rappold G, Blum WF, Shavrikova EP, et al. Genotypes and phenotypes in children with short stature: clinical indicators of SHOX haploinsufficiency. J Med Genet 2007;44:306–13.
Huber C, Rosilio M, Munnich A, Cormier-Daire V ; French SHOX GeNeSIS Module. High incidence of SHOX anomalies in individuals with short stature. J Med Genet 2006;43:735–9.
Rosilio M, Huber-Lequesne C, Sapin H, Carel JC, Blum WF, Cormier-Daire V. Genotypes and phenotypes of children with SHOX deficiency in France. J Clin Endocrinol Metab 2012;97:E1257–65.
Binder G, Ranke MB, Martin DD. Auxology is a valuable instrument for the clinical diagnosis of SHOX haploinsufficiency in school-age children with unexplained short stature. J Clin Endocrinol Metab 2003;88:4891–6.
Binder G, Schwarze CP, Ranke MB. Identification of short stature caused by SHOX defects and therapeutic effect of recombinant human growth hormone. J Clin Endocrinol Metab 2000;85:245–9.
Morizio E, Stuppia L, Gatta V, et al. Deletion of the SHOX gene in patients with short stature of unknown cause. Am J Med Genet A 2003;119A:293–6.
Jorge AA, Souza SC, Nishi MY, et al. SHOX mutations in idiopathic short stature and Leri-Weill dyschondrosteosis: frequency and phenotypic variability. Clin Endocrinol (Oxf) 2007;66:130–5.
Caliebe J, Broekman S, Boogaard M, et al. IGF1, IGF1R and SHOX mutation analysis in short children born small for gestational age and short children with normal birth size (idiopathic short stature). Horm Res Paediatr 2012;77:250–60.
Hirschfeldova K, Solc R, Baxova A, et al. SHOX gene defects and selected dysmorphic signs in patients of idiopathic short stature and Léri-Weill dyschondrosteosis. Gene 2012;491:123–7.
Sandoval GT, Jaimes GC, Barrios MC, Cespedes C, Velasco HM. SHOX gene and conserved noncoding element deletions/duplications in Colombian patients with idiopathic short stature. Mol Genet Genomic Med 2014;2:95–102.
Ellison JW, Wardak Z, Young MF, Gehron Robey P, Laig-Webster M, Chiong W. PHOG, a candidate gene for involvement in the short stature of Turner syndrome. Hum Mol Genet 1997;6:1341–7.
Marchini A, Marttila T, Winter A, et al. The short stature homeodomain protein SHOX induces cellular growth arrest and apoptosis and is expressed in human growth plate chondrocytes. J Biol Chem 2004;279:37103–14.
Munns CJ, Haase HR, Crowther LM, et al. Expression of SHOX in human fetal and childhood growth plate. J Clin Endocrinol Metab 2004;89:4130–5.
Binder G, Renz A, Martinez A, et al. SHOX haploinsufficiency and Leri-Weill dyschondrosteosis: prevalence and growth failure in relation to mutation, sex, and degree of wrist deformity. J Clin Endocrinol Metab 2004;89:4403–8.
Zinn AR, Wei F, Zhang L, et al. Complete SHOX deficiency causes Langer mesomelic dysplasia. Am J Med Genet 2002;110:158–63.
Clement-Jones M, Schiller S, Rao E, et al. The short stature homeobox gene SHOX is involved in skeletal abnormalities in Turner syndrome. Hum Mol Genet 2000;9:695–702.
Hertel NT, Scheike T, Juul A, et al. [Body proportions of Danish children. Curves for sitting height ratio, subischial length and arm span]. Ugeskr Laeger 1995;157:6876–81.
Niklasson A, Albertsson-Wikland K. Continuous growth reference from 24th week of gestation to 24 months by gender. BMC Pediatr 2008;8:8.
Tinggaard J, Aksglaede L, Sørensen K, et al. The 2014 Danish references from birth to 20 years for height, weight and body mass index. Acta Paediatr 2014;103:214–24.
Marshall WA, Tanner JM. Variations in the pattern of pubertal changes in boys. Arch Dis Child 1970;45:13–23.
Marshall WA, Tanner JM. Variations in pattern of pubertal changes in girls. Arch Dis Child 1969;44:291–303.
Thodberg HH, Kreiborg S, Juul A, Pedersen KD. The BoneXpert method for automated determination of skeletal maturity. IEEE Trans Med Imaging 2009;28:52–66.
Martin DD, Heckmann C, Jenni OG, Ranke MB, Binder G, Thodberg HH. Metacarpal thickness, width, length and medullary diameter in children–reference curves from the First Zürich Longitudinal Study. Osteoporos Int 2011;22:1525–36.
Ottesen AM, Garn ID, Aksglaede L, Juul A, Rajpert-De Meyts E. A simple screening method for detection of Klinefelter syndrome and other X-chromosome aneuploidies based on copy number of the androgen receptor gene. Mol Hum Reprod 2007;13:745–50.
Gatta V, Palka C, Chiavaroli V, et al. Spectrum of phenotypic anomalies in four families with deletion of the SHOX enhancer region. BMC Med Genet 2014;15:87.
Juul A, Bang P, Hertel NT, et al. Serum insulin-like growth factor-I in 1030 healthy children, adolescents, and adults: relation to age, sex, stage of puberty, testicular size, and body mass index. J Clin Endocrinol Metab 1994;78:744–52.
Jensen RB, Thankamony A, O’Connell SM, et al. A randomised controlled trial evaluating IGF1 titration in contrast to current GH dosing strategies in children born small for gestational age: the North European Small-for-Gestational-Age Study. Eur J Endocrinol 2014;171:509–18.
Sørensen K, Aksglaede L, Petersen JH, Andersson AM, Juul A. Serum IGF1 and insulin levels in girls with normal and precocious puberty. Eur J Endocrinol 2012;166:903–10.
Salmon-Musial AS, Rosilio M, David M, et al. Clinical and radiological characteristics of 22 children with SHOX anomalies and familial short stature suggestive of Léri-Weill Dyschondrosteosis. Horm Res Paediatr 2011;76:178–85.
Blum WF, Crowe BJ, Quigley CA, et al.; SHOX Study Group. Growth hormone is effective in treatment of short stature associated with short stature homeobox-containing gene deficiency: Two-year results of a randomized, controlled, multicenter trial. J Clin Endocrinol Metab 2007;92:219–28.
Ogata T, Onigata K, Hotsubo T, Matsuo N, Rappold G. Growth hormone and gonadotropin-releasing hormone analog therapy in haploinsufficiency of SHOX. Endocr J 2001;48:317–22.
Scalco RC, Melo SS, Pugliese-Pires PN, et al. Effectiveness of the combined recombinant human growth hormone and gonadotropin-releasing hormone analog therapy in pubertal patients with short stature due to SHOX deficiency. J Clin Endocrinol Metab 2010;95:328–32.
Fukami M, Nishi Y, Hasegawa Y, et al. Statural growth in 31 Japanese patients with SHOX haploinsufficiency: support for a disadvantageous effect of gonadal estrogens. Endocr J 2004;51:197–200.
Donze SH, Meijer CR, Kant SG, et al. The growth response to GH treatment is greater in patients with SHOX enhancer deletions compared to SHOX defects. Eur J Endocrinol 2015;173:611–21.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Marstrand-Joergensen, M., Jensen, R., Aksglaede, L. et al. Prevalence of SHOX haploinsufficiency among short statured children. Pediatr Res 81, 335–341 (2017). https://doi.org/10.1038/pr.2016.233
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2016.233
- Springer Nature America, Inc.
This article is cited by
-
SHOX haploinsufficiency presenting with isolated short long bones in the second and third trimester
European Journal of Human Genetics (2018)