
Abstract
The “bedside-to-bench” Congenital Central Hypoventilation Syndrome (CCHS) research journey has led to increased phenotypic-genotypic knowledge regarding autonomic nervous system (ANS) regulation, and improved clinical outcomes. CCHS is a neurocristopathy characterized by hypoventilation and ANS dysregulation. Initially described in 1970, timely diagnosis and treatment remained problematic until the first large cohort report (1992), delineating clinical presentation and treatment options. A central role of ANS dysregulation (2001) emerged, precipitating evaluation of genes critical to ANS development, and subsequent 2003 identification of Paired-Like Homeobox 2B (PHOX2B) as the disease-defining gene for CCHS. This breakthrough engendered clinical genetic testing, making diagnosis exact and early tracheostomy/artificial ventilation feasible. PHOX2B genotype-CCHS phenotype relationships were elucidated, informing early recognition and timely treatment for phenotypic manifestations including Hirschsprung disease, prolonged sinus pauses, and neural crest tumors. Simultaneously, cellular models of CCHS-causing PHOX2B mutations were developed to delineate molecular mechanisms. In addition to new insights regarding genetics and neurobiology of autonomic control overall, new knowledge gained has enabled physicians to anticipate and delineate the full clinical CCHS phenotype and initiate timely effective management. In summary, from an initial guarantee of early mortality or severe neurologic morbidity in survivors, CCHS children can now be diagnosed early and managed effectively, achieving dramatically improved quality of life as adults.
Similar content being viewed by others

Main
Congenital Central Hypoventilation Syndrome (CCHS) patients characteristically present with diminutive tidal volumes and monotonous respiratory rates asleep and awake, with more profound alveolar hypoventilation typically occurring during sleep (1,2). These individuals thus become hypoxemic and hypercarbic, with diminished or absent ventilatory and arousal responses to these endogenous challenges. Nonetheless, they maintain the ability to consciously alter the rate and depth of breathing. Historically, CCHS was diagnosed in the newborn period, yet we now know that cases can be diagnosed later in infancy, childhood or adulthood (3,4,5,6,7,8,9,10,11). Hirschsprung disease (HSCR) and tumors of neural crest origin present in varied combinations with CCHS, in addition to a spectrum of symptoms compatible with autonomic nervous system dysregulation (ANSD) (1,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29). While the spectrum and variability of CCHS-associated symptoms have made diagnosis and treatment complex, these very complexities led to the discovery of its etiology and the consequent successes in diagnosis and treatment. Here we describe some of these discoveries and advances, which over the last five decades have transformed diagnosis, treatment, and quality of life for CCHS patients.
The Early Story of CCHS
The 1970 report of an infant with “failure of automatic control of ventilation” (30) was the first to describe the CCHS control of breathing deficit, including extraordinary details regarding the child’s autonomic dysregulation. Over the next few years, four additional CCHS patients were reported with clinical courses complicated by cor pulmonale, seizures, neurologic abnormalities and high mortality, but no identifiable neuropathology on autopsy (31,32).
Three cases reported by Hunt and colleagues in 1978 and 1979 (33,34) introduced two important strategies: (i) medical management to prevent or reverse cor pulmonale, and decrease risk for hypoxic encephalopathy, permanent disability or death, and (ii) successful introduction of diaphragm pacing. These infants proved unresponsive to respiratory stimulants, and one developed bilateral ganglioneuroblastomas, suggesting a phenotype not limited to the brainstem. Two of the three cases died before age two, in part due to the cor pulmonale and associated hypoxic encephalopathy, and the third died suddenly and unexpectedly as a young adult.
In 1978, Haddad and colleagues reported three additional CCHS infants (35). This report highlighted an association of CCHS with HSCR and tumors of neural crest origin. The report described an infant who exhibited postnatal cyanosis, improved ventilation in rapid eye movement (REM) sleep, attenuated ventilatory response to hypercarbia, lack of heart rate variability, HSCR and multiple ganglioneuroblastomas. Additionally, this report described CCHS in sisters, the first suggestion that CCHS is familial. While the pathophysiology remained quite unclear, linking CCHS to HSCR and neural crest tumors, and identifying familial recurrence were essential steps in understanding these entities.
In the following decades, additional CCHS cases were reported, though still totaling fewer than 100. These reports, combined with the first large cohort studies appearing in the early 1990s (13), resulted in a broad understanding of the phenotype and provided hints to the varying organ systems involved, the need for early diagnosis and referral, for vigilant ventilatory management, and for vigorous efforts to support an age-appropriate quality of life. These early cases collectively made it clear that CCHS is far more than a simple orphan disorder of respiratory control. While CCHS-related morbidity and mortality had decreased, major gaps in knowledge remained—in particular, the underlying cause of the disease.
Uncovering Clues to the Cause: Autonomic Dysfunction and Familial Recurrence
In response to increasing identification of CCHS, specialized centers were established to advance CCHS care. These centers would aggregate the rare disease patients needed for research analysis, allowing elucidation of CCHS-associated symptoms, uncovering of fundamental systems involved, and defining familial inheritance. Together, these advances would form the basis of future inquiry and discovery.
Dysfunction of the ANS
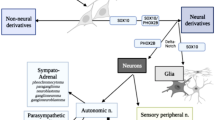
The effects of the ANS are expansive ( Figure 1 ), thus diagnosing and delineating symptoms associated with ANSD is complicated, particularly in a rare disease with variable expression. In 1999, the American Thoracic Society published the first official statement on CCHS (1). This statement reported 160–180 known living cases of CCHS, describing symptoms of and conditions associated with CCHS, and recognized critical diagnosis of abnormal respiratory control and related cardiorespiratory and autonomic symptoms. This report expanded the perception of CCHS beyond a control of breathing deficit with asleep hypoventilation, emphasizing that some patients also presented with awake hypoventilation, cardiovascular abnormalities, hypothermia, seizures, neuroblastomas, constipation, HSCR, and ophthalmologic abnormalities. The American Thoracic Society statement encouraged a more integrated view of CCHS symptoms, placing them in context of more general ANSD.

Organ systems affected by the autonomic nervous system. Solid and dashed lines represent pre- and postganglionic fibers, respectively. Reproduced with permission from ref. 99.
The Hunt for a Gene
Early CCHS reports identified families with disease recurrence, suggesting a hereditary phenomenon (13,35,36,37), and segregation analyses provided evidence for the inheritance pattern of CCHS that suggested a Mendelian codominant transmission (12,38). Thus, researchers began to search for CCHS-causing mutations, focusing primarily on genes implicated in HSCR. While these studies provided interesting candidates for CCHS etiology, results were inconclusive and no single gene emerged as a clear causative factor (39,40,41).
The Breakthrough: PHOX2B as the Disease-Defining Gene for CCHS
A New Suspect
The Paired-Like Homeobox 2B (PHOX2B) gene encodes a highly conserved homeodomain transcription factor. Early work indicated PHOX2B may play a key role in embryologic development of the ANS, including circuits thought to underlie chemosensory functions (42,43). PHOX2B was further implicated in intestinal innervation, linking this gene to the gastrointestinal phenotype as well. Recognizing the overlap in phenotypic manifestations of CCHS with PHOX2B developmental roles, work began to evaluate PHOX2B in CCHS cohorts.
A Link is Identified
In 2003, Amiel and colleagues reported mutations in PHOX2B in 20 of 29 (69%) mainly French CCHS cases (44). The majority of these mutations (18 of 20) were in-frame expansions of a 20-alanine repeat region found in exon 3 of PHOX2B between 25 and 29 alanines (now termed as polyalanine repeat expansion mutations, PARMs). The two remaining patients had frameshift mutations, part of a group of rarer mutations in PHOX2B now termed non-PARMs (NPARMs). Neither mutation type was found in any controls, nor in parents of the CCHS cases. Similarly, a 2003 report identified PHOX2B PARM and NPARM mutations in 5 of 10 Japanese CCHS cases (41). These studies implicated PHOX2B in CCHS pathogenesis, but the full story was not yet clear.
Mounting Evidence of PHOX2B as the Disease-Defining Gene for CCHS
Concurrently, Weese-Mayer and colleagues (39) evaluated candidate CCHS genes in a larger cohort of US cases and controls. Surprisingly, PHOX2B mutations were identified in 100% of 67 CCHS cases in this cohort. Sixty-six of these patients had heterozygous PHOX2B PARMs, with expansions ranging from 25–33 alanine repeats. The remaining patient had a nonsense NPARM. This not only confirmed the role of PHOX2B mutations in CCHS, but identified PHOX2B as the disease-defining gene. This study also identified a genotype-phenotype relationship, with longer PARM expansions showing more severe phenotypes. Finally, this study identified somatic mosaicism in parents of a subset of CCHS probands, indicating that PARM expansion length was stable and could be inherited in an autosomal dominant mode.
Confirmed: PHOX2B as the Disease-Defining Gene for CCHS
In subsequent years, evaluation of PHOX2B in CCHS cohorts expanded. In 2004, cases were reported from Italy, Germany, and the Netherlands (3) that identified PHOX2B PARMs of 25–33 repeats in 21 (88%) and NPARMs in 2 (8%) of 24 CCHS cases. This study confirmed the correlation between PARM length and disease severity. Importantly, this report also demonstrated a technical complication in standard PCR methods that could cause false negative results for PARMs. With this information, the Amiel group (re)analyzed their cohort and identified a PHOX2B mutation in 93% of 174 subjects with CCHS from multiple nations, including 7 of the 9 “mutation-negative” patients reported in 2003 (45). These, and subsequent studies, confirmed PHOX2B as the disease-defining gene in CCHS (39).
PHOX2B Mutations and Inheritance
Development of Diagnostic Testing
The 2003 Weese-Mayer report established the first clinically available assay for diagnosis of CCHS, a method for identifying PARM and most frameshift NPARM mutations, together causative in ~95% of CCHS patients (patent donated to Chicago Community Foundation; proceeds support CCHS research) (39). This test (www.genetests.org; Rush University PHOX2B Screening Test) can be used for prenatal diagnosis and family testing (including detection of low-level mosaicism). Subsequently, many labs began to offer diagnostic sequencing of PHOX2B (www.genetests.org). Sequencing PHOX2B in these assays enables diagnosis of all known NPARM and PARM mutations. The disadvantage is sequencing tests often fail to detect lower-level mosaicism (46), which has been identified both in asymptomatic parents of CCHS cases and rarely in cases with mild CCHS symptoms (unpublished results). Additionally, variations in copy number of PHOX2B (whole and partial gene deletions) have been recently identified in cases with variable, CCHS-like phenotypes (47). Accordingly, labs now offer PHOX2B deletion/duplication testing. While it is possible that mutations in genes with roles similar to PHOX2B could cause a phenotype similar to CCHS, this has not yet been reported to our knowledge. Consequently, a mutation in PHOX2B is considered requisite to a diagnosis of CCHS (2). Approximately 1,300 patients with CCHS have been diagnosed since 1970. However, as two studies indicate that the incidence of CCHS is ~1/150,000-1/200,000 live births, it is clear that CCHS remains grossly under-diagnosed (48,49). PHOX2B testing has drastically improved CCHS diagnosis, with >80% of known CCHS cases with PARMs now being diagnostically confirmed in the first 60 d of life (unpublished observation).
Delineating Mutation Types
Over 90% of CCHS patients will be heterozygous for an in-frame PARM mutation, with the addition of 4–13 alanines to the normal 20 alanine repeat. These mutations produce genotypes of 20/24 to 20/33 (normal genotype is 20/20). Over 100 different NPARM mutations have been identified in nearly 200 patients (2) (unpublished results) including missense, nonsense, frameshift, stop codon and likely splice-site mutations ( Figure 2 ). The majority of NPARMs cause very severe forms of CCHS, typically including extensive intestinal aganglionosis and tumors of neural crest origin. However, the protein and cellular effects are varied, with variable expressivity and incomplete penetrance. Indeed, a small subset of these NPARMs have been shown to cause only tumor in one patient, only HSCR in another, and CCHS with or without tumor or HSCR in others, with differences arising even within a single family. Currently, this variability in expression of phenotypes remains poorly defined.

CCHS-associated PHOX2B gene mutations. Previously reported polyalanine repeat expansion mutations (PARMs) and non-PARM (NPARMs). Adapted with permission from ref. 99.
Mosaicism
Parental somatic mosaicism has been confirmed in several large cohorts (11,50,51,52,53). The range of mosaicism is estimated to be 9–35% (51) with stable transmission of specific mutations to offspring. Though low level mosaic carriers are not expected to be symptomatic, it is possible that some may present with subclinical phenotypes, and these may worsen with challenges such as respiratory infection, use of respiratory depressants, and generally declining health (51) (personal communication). It is thus recommended that carriers of mosaic PHOX2B mutations undergo careful comprehensive evaluation and follow-up.
Inheritance
Parents of CCHS patients should be tested for PHOX2B mutations, to determine if mosaicism exists (both for family planning and to address possible health risks for mosaic carriers). As low-level mosaicism is difficult to detect using sequencing, a sizing/genotyping test such as the PHOX2B Screening Test is suggested (46). Germline mosaicism has been identified for PHOX2B mutations (54). Thus prenatal testing is suggested for individuals with CCHS and all parents of CCHS patients, including both somatic mosaic carriers of PHOX2B mutations, and those who tested negative for somatic mosaicism.
Advances at the Bench
Uncovering the Molecular Basis of Disease
Since the discovery that PHOX2B was disease-defining for CCHS, work has intensified to uncover both the normal function of PHOX2B and the molecular ramifications of abnormal function. Polyalanine tract expansions, particularly in transcription factors, are a poorly understood but emerging cause of human disease. It is clear that in other polyalanine diseases, as in CCHS, disease severity typically increases with increasing expansion length (55,56). Work in animal and cellular models has clarified aspects of the molecular mechanisms of PHOX2B mutations, and the resulting cellular and neurological dysfunction. These results have been augmented by imaging and postmortem autopsy studies in CCHS patients, which have helped to direct bench work in the cell and mouse and evaluate bench results in the human model.
Animal Models
The early knockout mouse model (homozygous null) revealed potential developmental roles for Phox2b that could account for some of the clinical picture seen in CCHS, with deficits throughout respiratory (and autonomic) reflex arcs and including the carotid bodies (57,58). A heterozygous null mutant reproduced some of the chemosensory deficit seen in CCHS, but effects were transient (58). More targeted mutant models did not reproduce the widespread anatomical lesions seen in the knockout mouse, but showed cell loss in a candidate site for central chemosensory function. Knock-in of an analog of the human 20/27 PARM reduced the chemoreflex and produced potentially lethal hypoventilation in neonatal mice (59). However, restricting the mutation to a pontine cellular lineage reproduced the chemosensory deficit without resulting in lethal hypoventilation (60). Both manipulations resulted in cell loss in a neural population thought to be crucial to a central chemosensory response, the medullary retrotrapezoid nucleus (RTN) (59,60).
The RTN, which likely overlaps functionally with a center identified in neonatal mice called the parafacial respiratory group (61,62), has become one of the most studied candidates for central chemosensory drive and integration. Evidence for this functional role of the RTN is quite robust in rodent models (63). First, a subset of Phox2b expressing cells in this region is intrinsically sensitive to extracellular pH (64). Second, anatomical evidence indicates that the RTN is well-integrated into respiratory neural circuits in a way that would allow integration of afferent input from peripheral chemosensors, efferent connections to respiratory rhythm and pattern circuits, as well as input from neuromodulatory centers (65,66). Finally, targeted manipulation of Phox2b-positive cells in the RTN can alter baseline ventilation as well as ventilatory response to hypercapnia in a state-dependent manner (67,68,69). Specifically, the influence of the RTN on respiratory frequency seems to be limited to behavioral states in which medullary respiratory rhythm generators are running relatively autonomously (quiet wakefulness, and non-REM sleep), possibly explaining the more frequent hypoventilation often seen in CCHS patients during these states (70).
Efforts to translate these results to infants have had mixed results. Two studies that attempted to identify the human homolog of the rodent RTN pointed to ponto-medullary regions that were not completely consistent with each other (71,72). Autopsy studies in CCHS have indicated damage in other medullary areas as well (13,73,74,75). Nonetheless, a working hypothesis is that a human homolog of the RTN does exist and plays a crucial role in CCHS pathophysiology. Still, many features of the phenotype may be a result of functional loss of other cell populations and fiber tracts. Imaging studies suggest relatively widespread brain injury in CCHS (76), and it seems likely that in a subset of cases, the damage may be progressive (though not necessarily due to the disease itself) (77).
Cellular Models
Cellular models have also been developed to evaluate pathogenic mechanisms of PHOX2B mutations. In these models, both PARM and NPARM PHOX2B constructs have demonstrated an impaired ability to transactivate some downstream targets. Mutant PHOX2B PARM constructs have demonstrated an inverse correlation between trans-activating ability and length of the polyalanine expanded tract (78,79,80). In frameshift NPARMs, the impairment was correlated with length of the disrupted C-terminal sequence (78,79,81). These transactivation studies give insight into possible mechanisms of PHOX2B mutations, and offer insight into the basis of the genotype-phenotype relationship.
Localization of both the wild type and expanded PHOX2B proteins, including the possible role of protein aggregation on localization, has also been evaluated. While cell models suggest almost exclusive nuclear localization of the wild type PHOX2B protein, PARMs induced partial or complete cytoplasmic mislocalization and protein aggregation (78,79). Longer PARMs were associated with larger protein aggregates in increasing proportions of cells (79). This finding implicates mutant protein mislocalization as a possible pathogenetic mechanism. In these models, PARMs have also demonstrated a partial dominant negative effect by interfering with normal function of the wild type protein, though this interaction is weak (82).
Finally, cell models demonstrate DNA binding is reduced for PHOX2B PARM proteins with expansions of 29 alanines and above, with the spontaneous formation of oligomers making them unavailable for DNA binding (78). Similarly, NPARMs have predominately shown complete loss of DNA binding, despite correct nuclear localization (78,79). PHOX2B NPARM constructs retain their ability to suppress cellular proliferation, without promoting differentiation, suggesting a mechanism which might promote development of neural crest tumors (83) and explain the high prevalence of neuroblastoma in NPARM patients.
The ability to replicate PARM and NPARM pathogenic mechanisms in cell models holds great potential for developing therapeutic interventions. Indeed, experiments in these models have allowed in vitro analysis to screen several molecules for effects on promotion of nuclear localization of PHOX2B and recovery of the protein’s transactivation potential (81,84).
Brain Imaging
Brains imaging studies and fMRI responses have complemented the relatively limited available data from autopsy and animal models. Additionally, these studies have identified structural deficiencies in CCHS cases which contribute to the observed respiratory and autonomic phenotypes (76,77,85,86). These findings may be primarily due to PHOX2B mutation-induced failure of neurogenesis in the human embryo, but a significant contribution from postnatal hypoxic, hypercarbic, or perfusion damage cannot be excluded.
Investigating the Locus Coeruleus in CCHS Utilizing Human, Animal, and Cellular Systems
A recent study of two postmortem CCHS cases (one PARM and one NPARM) (87) reported neuronal loss within the locus coeruleus, the major noradrenergic nucleus of the brain, connected to systemic arousal and sympathetic activation (88). The same study used a transgenic mouse model to evaluate timing of developmental defects and to confirm that expression of this NPARM leads to deleterious effects in the locus coeruleus (though a previous study did not find deleterious effects on the locus coeruleus with the 20/27 PARM mutation (59)). Since the PHOX2A gene is required for development of the locus coeruleus and interacts with PHOX2B, it has been suggested that PHOX2B mutations may cause a dominant-negative effect on PHOX2A. The results from cellular models have indicated that PHOX2B mutants do not interfere with the localization and transcriptional activity of PHOX2A, but did identify an unexpected synergistic effect through PHOX2A and PHOX2B mutation heterodimer formation (82). These studies highlight the collective advances made possible by studying PHOX2B mutations in human, animal, and cellular models.
Advances at the Bedside
With PHOX2B established as the disease-defining gene for CCHS and early analysis indicating a relationship between specific PHOX2B mutations and CCHS phenotypes, the opportunity for better understanding of CCHS and anticipatory management emerged.
Genotype-Based Anticipatory Management
Among individuals with PARMs, a relationship has been identified for physiologic ANSD that includes artificial ventilatory needs, risk of cardiac sinus pauses, heart rate variability, and a number of symptoms of ANSD (2,3,39,50). Knowledge of genotype provides a benchmark for the medical team in providing optimal life support and evaluation from diagnosis onward, with the aim of optimizing outcome ( Table 1 ).
Ventilatory needs. Patients with short PARM mutations (20/24–20/25) will typically require night-time only ventilation. With rare exception, the patients with longer PARM or NPARM mutations have more severe hypoventilation, often necessitating 24 h/d ventilatory support. Knowledge of genotype allows for more clear recommendations for ventilatory management, especially since children with CCHS lack overt behavioral effects of hypoventilation. Comprehensive physiologic testing awake and asleep in activities of daily living enable health care personnel to determine the severity of the hypoventilation and be aggressive about placement of tracheostomy and introducing continuous mechanical ventilation.
Sinus pause. Children with the 20/27 genotype are very likely to have ≥3 second pauses (>80%), those with 20/25 genotype are very unlikely to have such pauses in childhood (0%) (25), and the children with the 20/26 genotype often have an intermediate sinus pause frequency (~20%). Introduction of 72-hour Holter recordings every 12 mo at a minimum has allowed for early identification of these sinus pauses permitting timely cardiac pacemaker placement. Heart rate variability is characteristically decreased in CCHS (27) due to a decrease in cardiac baroreflex sensitivity and blunted sympathetic response (8,23). A recent report demonstrated a genotype-phenotype relationship for heart rate variability during exogenous ventilatory response testing, enabling assessment of risk for sinus pauses (29).
Anatomic ANSD. Amongst PARMs, evaluation for HSCR is based on clinical symptoms in recognition of overall risk of 20% across the 20/26–20/33 genotypes. PHOX2B mutations in mouse models exhibit sympathetic agangliogenesis, displaying decreased proliferation of neural crest-derived cells and development of gastrointestinal glia cells (89). Even in cases without HSCR diagnosis, cases display symptoms of lower esophageal dysfunction and intestinal dysmotility that result in symptoms such as severe constipation and abnormal esophageal motility (15). Risk of a tumor of neural crest origin is <2% overall, and even then only among the longer PARMs. However, overall risk for HSCR and/or neuroblastoma in NPARMS is >50%. Screening for neural crest tumors is advised in children with 20/28–20/33 genotypes (in addition to all children with NPARMs). Though the rare tumors are expected to be ganglioneuromas or ganglioneuroblastomas in the PARMs, a neuroblastoma was reported in a child with the 20/33 genotype (90).
Identifying Late-onset CCHS
Late-onset CCHS patients typically have a PARM (20/24 or 20/25) and present after 1 mo of age. A few children with an NPARM will present late as well. This later presentation reflects the variable penetrance of these mutations and potential need for an environmental cofactor. Late-onset CCHS should be considered among individuals with unexplained centrally mediated hypoventilation and/or cyanosis or seizures with sedation or anesthesia, recurrent severe pulmonic infection with difficulty weaning from ventilatory support, obstructive sleep apnea refractory to intervention, or unexplained neurocognitive impairment. With a heightened level of suspicion, increasing numbers of older children and adults have been diagnosed with late-onset CCHS and properly treated (2,7,51).
Optimizing Neurocognitive Performance
Impaired oxygen delivery to the brain, whether acute or chronic, can have detrimental effects on neurocognitive development. Individuals with CCHS require assisted ventilation, but the optimum method can include positive pressure ventilation via tracheostomy, bi-level positive airway pressure via mask, negative pressure ventilation and diaphragm pacing. The American Thoracic Society statement recommends positive pressure ventilation via tracheostomy in the first several years of life to ensure optimal oxygenation and ventilation (1,2). The method of choice in later years will depend on a variety of factors including severity of disease, patient age, level of patient and family cooperation, availability and quality of home health care, amongst other factors. The level of oxygen stability obtained with each varies. Thus, the method of respiratory assistance is likely to play a factor in neurocognitive outcome. Past literature has indicated deficiencies of mental abilities in school-age children with CCHS (91,92,93,94).
Even in preschool years, children with CCHS demonstrate reduced neurocognitive performance (95). In these cases, PHOX2B genotype was clearly associated with both mental and motor outcome ( Table 2 ). This association was also found with CCHS-related features such as severe cyanotic breath holding spells, sinus pauses, seizures, and severity of hypoventilation ( Table 3 ). It is unclear if this association is intrinsic to the specific mutation, the phenotypes associated with the mutation, or most likely both. Despite the delays observed, 29% of subjects had mid-average mental development scores, and 13% performed above that level. These findings suggest the potential for excellent neurocognitive outcome. This potential appears greatest in individuals with a 20/25 genotype in this cohort where average Bayley mental scores were over the population mean of 100. In sharp contrast, a 2015 report identified remarkably low IQs in a cohort of 19 Japanese children with the 20/25 genotype, with 42% of these cases reported as having displayed “mental retardation” (48). Many of these 20/25 genotype Japanese patients were diagnosed after the first month of life and some were managed with minimal support including home oxygen only, despite clear recommendations against such support (1,2). While it is not possible to directly compare the cohorts and determine if the dramatic differences are due to standard of care, age at diagnosis, genetic background, testing methods employed, or other, it seems clear given the disparity in outcome within the same genotype, that early recognition and conservative management are essential to insuring optimized neurocognitive outcome. Recognizing this, neurodevelopmental monitoring would be most beneficial beginning in early infancy.
Applying the Lessons Learned and Future Directions
While great progress has been made in CCHS, many potential avenues remain for improving the lives of these patients. One potentially important approach is interfering in the basic pathogenic process of CCHS, or compensating for the hallmark deficit with a well-targeted pharmacological or technological intervention (96,97). While the seeming neurodevelopmental primacy of PHOX2B has discouraged speculation about fundamental interventions, hope can be found in anatomical evidence that a brainstem nucleus (area postrema) may be affected by PHOX2B mutations but may also undergo significant postnatal development (98). Together with recent evidence that the respiratory control phenotype in CCHS may be preserved in some patients early in life (29) ( Figure 3 ), these results suggest there may be a substrate for intervention in the fundamental pathological processes of CCHS even after birth.

Chemosensory response maturation in CCHS cases and controls. Response was measured by change in weight-normalized ▵VE/unit change in EtCO2 during 3-minute challenge of inspired 7% FiCO2/14% FiO2 with minimal ventilator support in 32 CCHS cases (64 trials) and 15 controls (15 trials). Vertical bars: 5% CIs for the regression model (lighter shades indicate CIs spanning 0, i.e., a nonsignificant difference from 0 at P = 0.05). Gender: females shown as squares, males as circles. Genotypes: 20/25 (orange), 20/26 (yellow), and 20/27 (purple); NPARMs (aqua), PHOX2B whole gene deletion (blue), and controls (black). Line segments represent repeated trials. Reproduced with permission from ref. (29).
Before PHOX2B was identified as the disease-defining gene in CCHS, diagnosis was often delayed, introduction of artificial ventilation was variable and often inadequate, leading to high risk for neurocognitive impairment, prolonged hospitalization, and early death. In sharp contrast, knowledge of the role of PHOX2B in the disease has enabled today’s physician to confirm CCHS with diagnostic testing in the first weeks of life, initiate early and aggressive artificial ventilation, and anticipate management pertinent to sinus pauses, HSCR, risk for neural crest tumors, ANSD and severity of hypoventilation. While life expectancy for patients identified since the discovery of PHOX2B as the disease-defining gene and development of CCHS diagnostic testing in 2003 remains unclear, the majority of CCHS patients are now living well into adulthood with drastically improved neurocognitive outcome.
In summary, the “bedside-to-bench” CCHS research journey has taken this rare neurocristopathy to the forefront of genetics and neurobiology of autonomic control overall. Study of CCHS and the discovery of the role of PHOX2B have enabled new insights into development and control of central respiratory drive, chemosensation, and autonomic function, and may yield new insights into more common disorders such as sudden infant death syndrome and ventilatory instability in premature infants. This remarkable journey to understand and effectively treat this rare disease has fostered an entire scientific discipline studying control of breathing, autonomic regulation, and neurocristopathies. The vision for the next leg of this journey is continued integration of new knowledge, building on the discoveries of the past 50 y to further advance CCHS clinical care.
Disclosure
The authors do not have any financial disclosures or conflicts of interest to report.
References
Weese-Mayer DE, Shannon DC, Keens TG, Silvestri JM. Idiopathic congenital central hypoventilation syndrome: diagnosis and management. American Thoracic Society. Am J Respir Crit Care Med 1999;160:368–73.
Weese-Mayer DE, Berry-Kravis EM, Ceccherini I, Keens TG, Loghmanee DA, Trang H ; ATS Congenital Central Hypoventilation Syndrome Subcommittee. An official ATS clinical policy statement: Congenital central hypoventilation syndrome: genetic basis, diagnosis, and management. Am J Respir Crit Care Med 2010;181:626–44.
Matera I, Bachetti T, Puppo F, et al. PHOX2B mutations and polyalanine expansions correlate with the severity of the respiratory phenotype and associated symptoms in both congenital and late onset Central Hypoventilation syndrome. J Med Genet 2004;41:373–80.
Repetto GM, Corrales RJ, Abara SG, et al. Later-onset congenital central hypoventilation syndrome due to a heterozygous 24-polyalanine repeat expansion mutation in the PHOX2B gene. Acta Pediatr 2009;98:192–5.
Trang H, Laudier B, Trochet D, et al. PHOX2B gene mutation in a patient with late-onset central hypoventilation. Pediatr Pulmonol 2004;38:349–51.
Weese-Mayer DE, Berry-Kravis EM, Zhou L. Adult identified with congenital central hypoventilation syndrome–mutation in PHOX2b gene and late-onset CHS. Am J Respir Crit Care Med 2005;171:88.
Antic NA, Malow BA, Lange N, et al. PHOX2B mutation-confirmed congenital central hypoventilation syndrome: presentation in adulthood. Am J Respir Crit Care Med 2006;174:923–7.
Diedrich A, Malow BA, Antic NA, et al. Vagal and sympathetic heart rate and blood pressure control in adult onset PHOX2B mutation-confirmed congenital central hypoventilation syndrome. Clin Auton Res 2007;17:177–85.
Doherty LS, Kiely JL, Deegan PC, et al. Late-onset central hypoventilation syndrome: a family genetic study. Eur Respir J 2007;29:312–6.
Barratt S, Kendrick AH, Buchanan F, Whittle AT. Central hypoventilation with PHOX2B expansion mutation presenting in adulthood. Thorax 2007;62:919–20.
Parodi S, Bachetti T, Lantieri F, et al. Parental origin and somatic mosaicism of PHOX2B mutations in Congenital Central Hypoventilation Syndrome. Hum Mutat 2008;29:206.
Weese-Mayer DE, Silvestri JM, Huffman AD, et al. Case/control family study of autonomic nervous system dysfunction in idiopathic congenital central hypoventilation syndrome. Am J Med Genet 2001;100:237–45.
Weese-Mayer DE, Silvestri JM, Menzies LJ, Morrow-Kenny AS, Hunt CE, Hauptman SA. Congenital central hypoventilation syndrome: diagnosis, management, and long-term outcome in thirty-two children. J Pediatr 1992;120:381–7.
Goldberg DS, Ludwig IH. Congenital central hypoventilation syndrome: ocular findings in 37 children. J Pediatr Ophthalmol Strabismus 1996;33:175–80.
Faure C, Viarme F, Cargill G, Navarro J, Gaultier C, Trang H. Abnormal esophageal motility in children with congenital central hypoventilation syndrome. Gastroenterol 2002;122:1258–63.
Pine DS, Weese-Mayer DE, Silvestri JM, Davies M, Whitaker AH, Klein DF. Anxiety and congenital central hypoventilation syndrome. Am J Psychiatry 1994;151:864–70.
Silvestri JM, Weese-Mayer DE, Flanagan EA. Congenital central hypoventilation syndrome: cardiorespiratory responses to moderate exercise, simulating daily activity. Pediatr Pulmonol 1995;20:89–93.
Paton JY, Swaminathan S, Sargent CW, Hawksworth A, Keens TG. Ventilatory response to exercise in children with congenital central hypoventilation syndrome. Am Rev Respir Dis 1993;147:1185–91.
Shea SA, Andres LP, Paydarfar D, Banzett RB, Shannon DC. Effect of mental activity on breathing in congenital central hypoventilation syndrome. Respir Physiol 1993;94:251–63.
Spengler CM, Banzett RB, Systrom DM, Shannon DC, Shea SA. Respiratory sensations during heavy exercise in subjects without respiratory chemosensitivity. Respir Physiol 1998;114:65–74.
Shea SA, Andres LP, Shannon DC, Banzett RB. Ventilatory responses to exercise in humans lacking ventilatory chemosensitivity. J Physiol 1993;468:623–40.
Shea SA, Andres LP, Shannon DC, Guz A, Banzett RB. Respiratory sensations in subjects who lack a ventilatory response to CO2 . Respir Physiol 1993;93:203–19.
Trang H, Girard A, Laude D, Elghozi JL. Short-term blood pressure and heart rate variability in congenital central hypoventilation syndrome (Ondine’s curse). Clin Sci (Lond) 2005;108:225–30.
O’Brien LM, Holbrook CR, Vanderlaan M, Amiel J, Gozal D. Autonomic function in children with congenital central hypoventilation syndrome and their families. Chest 2005;128:2478–84.
Gronli JO, Santucci BA, Leurgans SE, Berry-Kravis EM, Weese-Mayer DE. Congenital central hypoventilation syndrome: PHOX2B genotype determines risk for sudden death. Pediatr Pulmonol 2008;43:77–86.
Silvestri JM, Hanna BD, Volgman AS, Jones PJ, Barnes SD, Weese-Mayer DE. Cardiac rhythm disturbances among children with idiopathic congenital central hypoventilation syndrome. Pediatr Pulmonol 2000;29:351–8.
Woo MS, Woo MA, Gozal D, Jansen MT, Keens TG, Harper RM. Heart rate variability in congenital central hypoventilation syndrome. Pediatr Res 1992;31:291–6.
Saiyed R, Rand CM, Carroll MS, et al. Congenital central hypoventilation syndrome (CCHS): Circadian temperature variation. Pediatr Pulmonol 2016;51:300–7.
Carroll MS, Patwari PP, Kenny AS, Brogadir CD, Stewart TM, Weese-Mayer DE. Residual chemosensitivity to ventilatory challenges in genotyped congenital central hypoventilation syndrome. J Appl Physiol (1985) 2014;116:439–50.
Mellins RB, Balfour HH Jr, Turino GM, Winters RW. Failure of automatic control of ventilation (Ondine’s curse). Report of an infant born with this syndrome and review of the literature. Medicine (Baltimore) 1970;49:487–504.
Deonna T, Arczynska W, Torrado A. Congenital failure of automatic ventilation (Ondine’s curse). A case report. J Pediatr 1974;84:710–4.
Shannon DC, Marsland DW, Gould JB, Callahan B, Todres ID, Dennis J. Central hypoventilation during quiet sleep in two infants. Pediatrics 1976;57:342–6.
Hunt CE, Matalon SV, Thompson TR, et al. Central hypoventilation syndrome: experience with bilateral phrenic nerve pacing in 3 neonates. Am Rev Respir Dis 1978;118:23–8.
Hunt CE, Inwood RJ, Shannon DC. Respiratory and nonrespiratory effects of doxapram in congenital central hypoventilation syndrome. Am Rev Respir Dis 1979;119:263–9.
Haddad GG, Mazza NM, Defendini R, et al. Congenital failure of automatic control of ventilation, gastrointestinal motility and heart rate. Medicine (Baltimore) 1978;57:517–26.
Khalifa MM, Flavin MA, Wherrett BA. Congenital central hypoventilation syndrome in monozygotic twins. J Pediatr 1988;113:853–5.
Hamilton J, Bodurtha JN. Congenital central hypoventilation syndrome and Hirschsprung’s disease in half sibs. J Med Genet 1989;26:272–4.
Marazita ML, Maher BS, Cooper ME, et al. Genetic segregation analysis of autonomic nervous system dysfunction in families of probands with idiopathic congenital central hypoventilation syndrome. Am J Med Genet 2001;100:229–36.
Weese-Mayer DE, Berry-Kravis EM, Zhou L, et al. Idiopathic congenital central hypoventilation syndrome: analysis of genes pertinent to early autonomic nervous system embryologic development and identification of mutations in PHOX2b. Am J Med Genet A 2003;123A:267–78.
Amiel J, Salomon R, Attié T, et al. Mutations of the RET-GDNF signaling pathway in Ondine’s curse. Am J Hum Genet 1998;62:715–7.
Sasaki A, Kanai M, Kijima K, et al. Molecular analysis of congenital central hypoventilation syndrome. Hum Genet 2003;114:22–6.
Pattyn A, Morin X, Cremer H, Goridis C, Brunet JF. Expression and interactions of the two closely related homeobox genes Phox2a and Phox2b during neurogenesis. Development 1997;124:4065–75.
Young HM, Ciampoli D, Hsuan J, Canty AJ. Expression of Ret-, p75(NTR)-, Phox2a-, Phox2b-, and tyrosine hydroxylase-immunoreactivity by undifferentiated neural crest-derived cells and different classes of enteric neurons in the embryonic mouse gut. Dev Dyn 1999;216:137–52.
Amiel J, Laudier B, Attié-Bitach T, et al. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat Genet 2003;33:459–61.
Trochet D, O’Brien LM, Gozal D, et al. PHOX2B genotype allows for prediction of tumor risk in congenital central hypoventilation syndrome. Am J Hum Genet 2005;76:421–6.
Jennings LJ, Yu M, Zhou L, Rand CM, Berry-Kravis EM, Weese-Mayer DE. Comparison of PHOX2B testing methods in the diagnosis of congenital central hypoventilation syndrome and mosaic carriers. Diagn Mol Pathol 2010;19:224–31.
Jennings LJ, Yu M, Rand CM, et al. Variable human phenotype associated with novel deletions of the PHOX2B gene. Pediatr Pulmonol 2012;47:153–61.
Shimokaze T, Sasaki A, Meguro T, et al. Genotype-phenotype relationship in Japanese patients with congenital central hypoventilation syndrome. J Hum Genet 2015;60:473–7.
Trang H, Dehan M, Beaufils F, Zaccaria I, Amiel J, Gaultier C ; French CCHS Working Group. The French Congenital Central Hypoventilation Syndrome Registry: general data, phenotype, and genotype. Chest 2005;127:72–9.
Berry-Kravis EM, Zhou L, Rand CM, Weese-Mayer DE. Congenital central hypoventilation syndrome: PHOX2B mutations and phenotype. Am J Respir Crit Care Med 2006;174:1139–44.
Trochet D, de Pontual L, Straus C, et al. PHOX2B germline and somatic mutations in late-onset central hypoventilation syndrome. Am J Respir Crit Care Med 2008;177:906–11.
Meguro T, Yoshida Y, Hayashi M, et al. Inheritance of polyalanine expansion mutation of PHOX2B in congenital central hypoventilation syndrome. J Hum Genet 2012;57:335–7.
Bachetti T, Parodi S, Di Duca M, Santamaria G, Ravazzolo R, Ceccherini I. Low amounts of PHOX2B expanded alleles in asymptomatic parents suggest unsuspected recurrence risk in congenital central hypoventilation syndrome. J Mol Med (Berl) 2011;89:505–13.
Rand CM, Yu M, Jennings LJ, et al. Germline mosaicism of PHOX2B mutation accounts for familial recurrence of congenital central hypoventilation syndrome (CCHS). Am J Med Genet A 2012;158A:2297–301.
Brown LY, Brown SA. Alanine tracts: the expanding story of human illness and trinucleotide repeats. Trends Genet 2004;20:51–8.
Lavoie H, Debeane F, Trinh QD, et al. Polymorphism, shared functions and convergent evolution of genes with sequences coding for polyalanine domains. Hum Mol Genet 2003;12:2967–79.
Pattyn A, Morin X, Cremer H, Goridis C, Brunet JF. The homeobox gene Phox2b is essential for the development of autonomic neural crest derivatives. Nature 1999;399:366–70.
Dauger S, Pattyn A, Lofaso F, et al. Phox2b controls the development of peripheral chemoreceptors and afferent visceral pathways. Development 2003;130:6635–42.
Dubreuil V, Ramanantsoa N, Trochet D, et al. A human mutation in Phox2b causes lack of CO2 chemosensitivity, fatal central apnea, and specific loss of parafacial neurons. Proc Natl Acad Sci USA 2008;105:1067–72.
Ramanantsoa N, Hirsch MR, Thoby-Brisson M, et al. Breathing without CO2 chemosensitivity in conditional Phox2b mutants. J Neurosci 2011;31:12880–8.
Onimaru H, Ikeda K, Kawakami K. Phox2b, RTN/pFRG neurons and respiratory rhythmogenesis. Respir Physiol Neurobiol 2009;168:13–8.
Amiel J, Dubreuil V, Ramanantsoa N, et al. PHOX2B in respiratory control: lessons from congenital central hypoventilation syndrome and its mouse models. Respir Physiol Neurobiol 2009;168:125–32.
Guyenet PG, Bayliss DA. Neural control of breathing and CO2 homeostasis. Neuron 2015;87:946–61.
Bayliss DA, Barhanin J, Gestreau C, Guyenet PG. The role of pH-sensitive TASK channels in central respiratory chemoreception. Pflugers Arch 2015;467:917–29.
Barna BF, Takakura AC, Moreira TS. Acute exercise-induced activation of Phox2b-expressing neurons of the retrotrapezoid nucleus in rats may involve the hypothalamus. Neuroscience 2014;258:355–63.
Rosin DL, Chang DA, Guyenet PG. Afferent and efferent connections of the rat retrotrapezoid nucleus. J Comp Neurol 2006;499:64–89.
Takakura AC, Barna BF, Cruz JC, Colombari E, Moreira TS. Phox2b-expressing retrotrapezoid neurons and the integration of central and peripheral chemosensory control of breathing in conscious rats. Exp Physiol 2014;99:571–85.
Abbott SB, Stornetta RL, Coates MB, Guyenet PG. Phox2b-expressing neurons of the parafacial region regulate breathing rate, inspiration, and expiration in conscious rats. J Neurosci 2011;31:16410–22.
Abbott SB, Stornetta RL, Fortuna MG, et al. Photostimulation of retrotrapezoid nucleus phox2b-expressing neurons in vivo produces long-lasting activation of breathing in rats. J Neurosci 2009;29:5806–19.
Burke PG, Kanbar R, Basting TM, et al. State-dependent control of breathing by the retrotrapezoid nucleus. J Physiol 2015;593:2909–26.
Rudzinski E, Kapur RP. Phox2b immunolocalization of the candidate human retrotrapezoid nucleus. Pediatr Dev Pathol 2010;13:291–9.
Lavezzi AM, Weese-Mayer DE, Yu MY, et al. Developmental alterations of the respiratory human retrotrapezoid nucleus in sudden unexplained fetal and infant death. Auton Neurosci 2012;170:12–9.
Tomycz ND, Haynes RL, Schmidt EF, Ackerson K, Kinney HC. Novel neuropathologic findings in the Haddad syndrome. Acta Neuropathol 2010;119:261–9.
Cutz E, Ma TK, Perrin DG, Moore AM, Becker LE. Peripheral chemoreceptors in congenital central hypoventilation syndrome. Am J Respir Crit Care Med 1997;155:358–63.
Ozawa Y, Okado N. Alteration of serotonergic receptors in the brain stems of human patients with respiratory disorders. Neuropediatrics 2002;33:142–9.
Harper RM, Kumar R, Macey PM, Harper RK, Ogren JA. Impaired neural structure and function contributing to autonomic symptoms in congenital central hypoventilation syndrome. Front Neurosci 2015;9:415.
Kumar R, Woo MS, Macey PM, Woo MA, Harper RM. Progressive gray matter changes in patients with congenital central hypoventilation syndrome. Pediatr Res 2012;71:701–6.
Trochet D, Hong SJ, Lim JK, et al. Molecular consequences of PHOX2B missense, frameshift and alanine expansion mutations leading to autonomic dysfunction. Hum Mol Genet 2005;14:3697–708.
Bachetti T, Matera I, Borghini S, Di Duca M, Ravazzolo R, Ceccherini I. Distinct pathogenetic mechanisms for PHOX2B associated polyalanine expansions and frameshift mutations in congenital central hypoventilation syndrome. Hum Mol Genet 2005;14:1815–24.
Borghini S, Bachetti T, Fava M, et al. The TLX2 homeobox gene is a transcriptional target of PHOX2B in neural-crest-derived cells. Biochem J 2006;395:355–61.
Bachetti T, Bocca P, Borghini S, et al. Geldanamycin promotes nuclear localisation and clearance of PHOX2B misfolded proteins containing polyalanine expansions. Int J Biochem Cell Biol 2007;39:327–39.
Di Lascio S, Belperio D, Benfante R, Fornasari D. Alanine expansions associated with congenital central hypoventilation syndrome impair PHOX2B homeodomain-mediated dimerization and nuclear import. J Biol Chem 2016;291:13375–93.
Raabe EH, Laudenslager M, Winter C, et al. Prevalence and functional consequence of PHOX2B mutations in neuroblastoma. Oncogene 2008;27:469–76.
Di Zanni E, Bachetti T, Parodi S, et al. In vitro drug treatments reduce the deleterious effects of aggregates containing polyAla expanded PHOX2B proteins. Neurobiol Dis 2012;45:508–18.
Macey PM, Moiyadi AS, Kumar R, Woo MA, Harper RM. Decreased cortical thickness in central hypoventilation syndrome. Cereb Cortex 2012;22:1728–37.
Patwari PP, Carroll MS, Rand CM, Kumar R, Harper R, Weese-Mayer DE. Congenital central hypoventilation syndrome and the PHOX2B gene: a model of respiratory and autonomic dysregulation. Respir Physiol Neurobiol 2010;173:322–35.
Nobuta H, Cilio MR, Danhaive O, et al. Dysregulation of locus coeruleus development in congenital central hypoventilation syndrome. Acta Neuropathol 2015;130:171–83.
Samuels ER, Szabadi E. Functional neuroanatomy of the noradrenergic locus coeruleus: its roles in the regulation of arousal and autonomic function part II: physiological and pharmacological manipulations and pathological alterations of locus coeruleus activity in humans. Curr Neuropharmacol 2008;6:254–85.
Nagashimada M, Ohta H, Li C, et al. Autonomic neurocristopathy-associated mutations in PHOX2B dysregulate Sox10 expression. J Clin Invest 2012;122:3145–58.
Armstrong AE, Weese-Mayer DE, Mian A, et al. Treatment of neuroblastoma in congenital central hypoventilation syndrome with a PHOX2B polyalanine repeat expansion mutation: New twist on a neurocristopathy syndrome. Pediatr Blood Cancer 2015;62:2007–10.
Zelko FA, Nelson MN, Leurgans SE, Berry-Kravis EM, Weese-Mayer DE. Congenital central hypoventilation syndrome: neurocognitive functioning in school age children. Pediatr Pulmonol 2010;45:92–8.
Marcus CL, Jansen MT, Poulsen MK, et al. Medical and psychosocial outcome of children with congenital central hypoventilation syndrome. J Pediatr 1991;119:888–95.
Oren J, Kelly DH, Shannon DC. Long-term follow-up of children with congenital central hypoventilation syndrome. Pediatrics 1987;80:375–80.
Silvestri JM, Weese-Mayer DE, Nelson MN. Neuropsychologic abnormalities in children with congenital central hypoventilation syndrome. J Pediatr 1992;120:388–93.
Charnay AJ, Antisdel-Lomaglio JE, Zelko FA, et al. Congenital central hypoventilation syndrome: neurocognition already reduced in preschool-aged children. Chest 2016;149:809–15.
Nattie E. Ondine Undone. N Engl J Med 2015;373:573–5.
Kanbar R, Stornetta RL, Cash DR, Lewis SJ, Guyenet PG. Photostimulation of Phox2b medullary neurons activates cardiorespiratory function in conscious rats. Am J Respir Crit Care Med 2010;182:1184–94.
Gokozan HN, Baig F, Corcoran S, et al. Area postrema undergoes dynamic postnatal changes in mice and humans. J Comp Neurol 2016;524:1259–69.
Weese-Mayer DE, Rand CM, Berry-Kravis EM, et al. Congenital central hypoventilation syndrome from past to future: model for translational and transitional autonomic medicine. Pediatr Pulmonol 2009;44:521–35.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Weese-Mayer, D., Rand, C., Zhou, A. et al. Congenital central hypoventilation syndrome: a bedside-to-bench success story for advancing early diagnosis and treatment and improved survival and quality of life. Pediatr Res 81, 192–201 (2017). https://doi.org/10.1038/pr.2016.196
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2016.196
- Springer Nature America, Inc.