
Abstract
The visualization of polymer structures at the molecular level is one of the most important issues in polymer science; however, even using scanning probe microscopy techniques such as atomic force microscopy (AFM), it is still challenging to observe soft polymer materials at the molecular level. In this review, we show that using two-dimensional (2D) samples suitable for AFM observation, especially Langmuir–Blodgett films, and by optimizing the scan conditions, it is possible to obtain molecular images of various polymer structures by tapping-mode AFM. The molecular-level observations included isolated polymer chains and their movements on substrates, 2D folded-chain crystals and their melting behavior, crystallization behavior of single isolated chains, supramolecular multistranded stereocomplexes and chain packing in monolayers. The molecular-level information obtained by this type of direct observation is expected to greatly improve our understanding of physical properties of polymers.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Introduction
Visualization of polymer structures at the molecular level is one of the most important issues in polymer science. The invention of scanning probe microscopy (SPM), including scanning tunneling microscopy1 and atomic force microscopy (AFM),2 has enabled us to observe materials at the molecular or even atomic level. However, observation of soft materials such as polymers at the molecular level is still a difficult task. Because SPM visualizes materials by scanning surfaces, the best resolution with SPM is attained by using flat samples—in particular, molecularly flat samples. This is because a flat sample allows us to minimize the feedback during scanning without the risk of crashing the probe onto the sample surface. As a result, we can reduce the force acting on the sample to avoid damage to the sample in observations at high magnification. Hence, we might expect that the most suitable sample for polymers is polymer Langmuir–Blodgett (LB) films.3, 4 Polymers with an appropriate hydrophilic group spread on a water surface with each repeating unit absorbed on the water surface as a monolayer. By compressing the monolayer on the water surface with a moving barrier, it transforms from the isolated chains to an amorphous film, and further to a crystalline film depending on the nature of the polymer. The resulting single monolayer can be deposited on a substrate for observation.
Using polymer LB films, we have successfully observed various polymer structures at the molecular level by conventional tapping-mode (intermittent contact-mode) AFM. There are several imaging modes used in AFM. Tapping mode is the most popular, in which a cantilever (a plate spring with a sharp probe tip attached at the end of the plate) is forced to oscillate to gently tap a sample; the amplitude of the oscillation of the cantilever is used as a feedback signal. The force on the sample in tapping-mode AFM is significantly smaller compared with that of contact-mode AFM, which directly scans the sample with the cantilever. Thus, soft materials such as polymers can be observed without significant damage. In this tapping mode, if a viscoelastic interaction exists between the cantilever and the sample, a phase retardation of the cantilever oscillation occurs from which a phase image can be constructed. In reality, the phase image contains not only the viscoelastic interaction but also other effects such as feedback error; therefore, physical interpretation of the phase image is not straightforward. However, the phase images usually exhibit a higher contrast compared with the height images, and they are especially useful in high-magnification observation. The resolution of tapping-mode AFM is generally believed to be inferior to that of contact-mode AFM, but we have found that a resolution close to or slightly better than 1 nm can be attained by tapping-mode AFM on polymer two-dimensional (2D) films.
Our observations include (1) an isolated chain5, 6 and its movement in humid air,7, 8 (2) a folded-chain crystal9 and its melting behavior,10 (3) a single-chain crystal,11 (4) a multistranded helix of a stereocomplex,12, 13 (5) chain packing in an LB film14 and (6) molecular images of chains solubilized in miscible-blend monolayers.15 These molecular images were obtained from 2D samples but were found to closely resemble those expected in 3D states; thus, they provide new molecular insights in understanding polymer structures in both 2D and 3D states. In this review, we focus on our attempts to conduct AFM observations of polymer structures at the molecular level by tapping-mode AFM using polymer 2D films, especially those prepared by the LB technique. Readers who are interested in other current topics concerning AFM observation of polymers are referred to recent reviews and books.16, 17, 18, 19, 20, 21, 22, 23, 24
Isolated chains
The conformation of isolated polymer chains is one of the most fundamental physical factors contributing to our understanding of the properties of polymers. However, even in 1996, almost 10 years after the invention of AFM,2 obtaining a clear AFM observation of a synthetic polymer chain remained a challenge. Evidence of string-like structures could be found in the literature, but no valid identification of the structures was possible. To identify a polymer chain, it is necessary at a minimum to evaluate the string length by visualizing the chain ends and to estimate the molecular weight. Large biomolecules such as DNA have been studied extensively by AFM.25, 26 The thickness of a DNA strand is almost one order of magnitude larger compared with that of conventional synthetic polymers composed of simple carbon–carbon bonds; as a result, DNA can be observed even by transmission electron microscopy (TEM) using a metal shadowing technique; it has also been visualized extensively by AFM. However, synthetic polymers cannot be visualized by TEM, and observation by AFM has been challenging.
To observe an isolated chain by AFM, we must immobilize it on a 2D substrate. The LB technique is one of the best ways to do this; in fact, immediately after the invention of SPM, poly(methyl methacrylate) (PMMA) LB films were imaged, but unfortunately the process did not reveal meaningful molecular information.27 To circumvent the difficulties of observation, we used polystyrene-b-poly(methyl methacrylate) (PS-b-PMMA) block copolymer instead of a PMMA homopolymer.5, 6 PS contains no hydrophilic groups and therefore does not spread on a water surface but collapses to form a particle much thicker than a monolayer; by contrast, PMMA, which contains a hydrophilic ester group, spreads as a monolayer. Therefore, the PS particle can be used as a probe for observation of a PMMA chain that emanates from it. Previously, we reported that PS monomolecular particles, each of which contains a single PS molecule, can be obtained by spreading an extremely dilute PS solution (~10−6g ml−1) in benzene on a water surface.28, 29, 30 The concentration was 2–3 orders of magnitude more dilute compared with the overlapping concentration of the polymer.
PS-b-PMMA was spread under similar conditions to form a PS single-block particle, a particle that contains a single PS block chain, from which a PMMA block chain emanated. Figure 1 shows an AFM height image of PS-b-PMMA on mica prepared in this way. From a central PS aggregate, three PMMA chains emanated. This AFM image was observed after exposure a deposited PS-b-PMMA sample in humid air: 100% relative humidity (RH) for 1 h and 79.3% RH for an additional 26 h. Immediately after being deposited on mica, the PMMA chains formed a condensed monolayer around the PS particles, preventing visualization of the conformations of the PMMA chains. After exposure to the humid air, however, the conformation of the PMMA chains markedly expanded such that the conformations of individual chains were isolated and could be clearly observed. A thin adsorbed water layer formed on the mica in humid air (~0.5 nm thick at 79.3% RH31), which enabled movement and rearrangement of the PMMA conformation on mica. The size of the PS single-block particle could be estimated from the molecular weight of the PS block and the measured thickness (2.5 nm) assuming an oblate ellipsoidal shape and a density equal to that of bulk PS. As shown in the schematic representation in Figure 1b, the central core contained three PS single-block particles from which three strands (A, B and C) emanated. The curvilinear lengths of the strands were ~30–60% of the fully stretched PMMA block chain for this molecular weight. This is reasonable because AFM ignores small loops and waviness less than the resolution of AFM. From both the agreement between the number of the PS particles and strands and the reasonable chain lengths obtained from the molecular weight, the strands were identified as PMMA chains. In this study, a block copolymer was used to identify and visualize the polymer chains; however, such a restriction is no longer necessary. Additionally, the LB technique that is used to deposit a polymer in a well-controlled manner is not necessary because we now know that simple spin casting of a polymer solution on a substrate is sufficient for observation of isolated chains. Followed by the observation of PMMA chains, various structures such as polymer brushes,32, 33 dendron polymers,32, 34 polyelectrolytes35 and star polymers36 were observed. Direct observations of isolated polymer chains are, at present, one of the essential components of polymer research.

(a) Atomic force microscopy (AFM) height image of polystyrene-b-poly(methyl methacrylate) (PS-b-PMMA) deposited on mica from a water surface, and then exposed to 100% relative humidity (RH) for 1 h and 79.3% RH for an additional 26 h. The number average molecular weight (Mn) was 1.55 × 105 for PS and 3.92 × 105 for PMMA, respectively. (b) Schematic representation of chain conformation. Reproduced with permission from Kumaki et al.5 Copyright (1996) American Chemical Society.
Movement of isolated chains
In addition to the static structures discussed above, the dynamic movements of isolated chains in various environments were studied. Gallyamov and co-workers22, 37 observed conformational switching of a linear polymer and polymer brushes upon environmental changes between humid air and organic vapors. Sheiko and co-workers38 successfully observed the movements of individual polymer brush molecules in a droplet spreading on a substrate.
We observed reptational movements of isotactic (it) PMMA chains on a substrate in humid air.7 Figure 2 shows movements of it-PMMA chains on mica over 82.5 min in 71% RH and a further 54 min in 54% RH. The AFM height image at 0 min and the trajectories of the center of mass of the chains (black lines: 71% RH; white lines: 54% RH) are shown in Figure 2a. The movements were accelerated at high humidity. Chains labeled by underlined characters (e.g., g, j and h) moved freely without being permanently fixed on the substrate. They moved along their length as shown in Figure 2b. Chains labeled by characters without underlines are fixed on the substrate over at least a portion of the chain during observation. For example, as shown in Figure 2b, n was fixed at the middle of the chain (arrow); the residual chains moved laterally around this fixed point. These movements bear a striking resemblance to a real snake, which moves along its length if we let it move freely; if we trap a part of the body, however, the rest of the body moves laterally around the fixed point. The origin of the reptational movement of it-PMMA can be explained as shown schematically in Figure 2c. In humid air, the segments repeatedly attach and detach with a help of an adsorbed water layer. If some loops happen to form, they move along their length and, as a result, the chain moves along its length. At the lower humidity of 34% RH shown in Figure 2d, the translational movements were restricted, but detailed movements inside a chain could be observed. A small loop (indicated by an arrow) moved along the chain, in a similar manner to the schematic representation in Figure 2c.

Movements of isotactic-poly(methyl methacrylate) (it-PMMA) (Mn=1.76 × 105) chains on mica under high humidity. (a) Atomic force microscopy (AFM) height image of it-PMMA chains on mica. The trajectories of the center of mass of the chains from 0 to 82.5 min at 71% relative humidity (RH) (black line) and from 82.5 to 136.5 min in 54% RH (white line) are shown. (b) Time lapse of the chain shapes for freely moving chains (g, h and j) and an anchored chain (n). (c) Schematic representation of reptational chain movements on mica. (d) Time-lapse AFM height images of an it-PMMA chain at 34% RH. Reproduced with permission from Kumaki et al.7 Copyright (2006) American Chemical Society.
Such dynamic observations are useful, providing a wealth of information that static observations do not, including chain movements, the rigidity or flexibility of chains and adhesion of the chain to the substrate at the molecular level.
Polymer crystals
During the initial studies using contact-mode AFM, many high-resolution images of polymer crystals were reported.39 However, at present, these are believed to reflect periodic responses of the crystal lattices and do not provide true molecular images of the polymers in the crystals. Important non-periodic structures, such as those associated with chain folding, tie chains and defects, were difficult to observe.
We have shown that true molecular images of crystals can be visualized by tapping-mode AFM using a crystalline LB film. Figure 3a shows an AFM height image of an it-PMMA monolayer, which was spread on a water surface, compressed to crystallize at a surface pressure of 10 mN m−1 and deposited on mica.9 Lamella crystals with chain packing perpendicular to the long axis of the lamella were clearly visualized. A 2D fast Fourier transform of the image indicated that the chain-to-chain distance was ~1.29 nm. Kusanagi et al.40 studied the structure of uniaxially stretched it-PMMA filaments by X-ray diffraction and reported the material to be a crystal composed of double-stranded helices of it-PMMA chains (Figure 3c). The chain-to-chain distance observed by AFM was in good agreement with their model (1.22 nm), indicating that our results support the double-stranded helix model. The average lamella width was much shorter compared with the length of the double-stranded helix composed of the it-PMMA, and the double-stranded helix was estimated to be folded 23 times on average in the lamella, if we assume that the double-stranded helix is composed of two it-PMMA chains. The folding number is halved if we assume that a double-stranded helix consists of an intertwined single it-PMMA chain. On close inspection of Figure 3a, a crystalline defect (1), regular chain folding (2) and tight tie chains (3) were also recognizable; schematic representations of possible chain packing are shown on the right. A vague structure between two lamellae (4) cannot be identified in this image, but if we treat the data as a movie constructed of a sequence of time-lapse AFM images, we are able to recognize it as a loose tie chain exhibiting considerable movement (See Supplementary Figure S1 and Supplementary Movie S1 in the Supplementary Information. The movie is very short, readers are recommended to view it in the repeated replay mode in their viewer.). We note that the use of 2D crystals allowed these important structures to be observed clearly for the first time. Figure 3b shows an AFM phase image of the crystal after being annealed at 50 °C for 12 h. The regular chain folding is more clearly visible.

(a) Atomic force microscopy (AFM) height image of a crystallized isotactic-poly(methyl methacrylate) (it-PMMA) (Mn=1.76 × 105) monolayer deposited on mica at 10 mN m−1. Schematic representation of possible chain conformations are shown on the right. (b) High-magnification AFM phase image of a crystallized it-PMMA monolayer on mica after being annealed at 50 °C for 12 h. Schematic representation of a possible chain conformation is shown on the right. (c) Molecular model of it-PMMA double-stranded helix, which is the basic unit of it-PMMA crystals. Reproduced with permission from Kumaki et al.9 Copyright (2005) American Chemical Society.
In this study, we found that a resolution close to 1 nm could be attained by tapping-mode AFM using regular 2D films deposited on the substrates. Similarly, using regular monolayers prepared by spin casting polymer solutions on a substrate followed by annealing under a solvent vapor to prepare a well-ordered monolayer on the substrate, we successfully observed molecular images of various helical polymers with a helical pitch and helical handedness for poly(phenyl acetylene)s, poly(phenyl isocyanide)s and an artificial double-stranded helix built up through interstrand amidinium-carboxylate salt bridges.19, 41, 42, 43, 44, 45, 46, 47
Recently, Mullin and Hobbs48 succeeded in observing a molecular image of a polyethylene crystal by torsional tapping-mode AFM using a T-shaped cantilever with an ultrasharp carbon whisker tip. They successfully resolved the minimum chain-to-chain packing down to 0.368 nm. Further improvements in the AFM technique will no doubt enable us to study polymer crystals in more detail.
Melting behavior of polymer crystals
The it-PMMA folded-chain crystal visible in Figure 3 is a true 2D crystal with a thickness equal to that of a single crystalline plane. Therefore, this is an ideal material to study the physical properties of the 2D state, and we attempted to study the melting behavior of the crystals. Figure 4a shows AFM phase images of an it-PMMA crystal with an Mn of 1.76 × 105 (it-PMMA(176k)) observed in situ at the temperatures indicated in the images, using a high-temperature AFM with a heated stage.10 With rising temperature, the lamellae gradually melted and at 100 °C most of them had melted. We note that the chain packing was clearly visible even after a significant degree of melting at 100 °C. Figure 4b shows the temperature dependence of the crystalline area. The crystalline area is the area ratio occupied by lamellae in AFM images; thus, it is an index of the 2D crystalline ratio. As the temperature rose, the area remained constant or slightly increased, possibly as a result of annealing of the crystals, and then rapidly decreased. From the inflection point, the 2D melting temperature (Tm) of the it-PMMA(176 k) was determined to be 80 °C, which is 54 °C lower compared with the bulk Tm (134 °C) of the it-PMMA(176 k); hence, a significant Tm depression was detected for the 2D crystal. In Figure 4b, the results for it-PMMA with a lower Mn of 1.2 × 104 (it-PMMA(12 k)) is also shown. It melted at an even lower temperature, and its Tm was determined to be 45 °C. Because the molecular weight dependence of the bulk Tm for this molecular weight regime was small (bulk Tm(12 k): 131 °C), an even larger Tm depression (86 °C) was observed for the it-PMMA(12 k). The widths of the 2D lamella of both polymers were not significantly different, as a result, the folding number of the chain for both polymers was detectably different (1.8 for it-PMMA(12 k), 23 for it-PMMA(176 k)); this may explain the significant molecular weight dependence of the 2D Tm. We confirmed that the melting was reversible; that is, once a crystal melted with increasing temperature, subsequent cooling resulted in full recovery of the crystal. This result confirms that the 2D Tm values determined here were not caused by an irreversible destruction of the 2D crystal by the scanning tip of the tapping-mode AFM, but are true thermodynamic transitions. At present, only a limited number of reports are available on the Tm depression of ultrathin polymer films, and those investigations used spin-cast films with inhomogeneous and poorly defined crystals, and the range of the film thickness was limited (thicker than 15 nm).49, 50 Our results provide clear evidence of Tm depression with a well-defined crystal down to the thickness of a single crystalline plane.

(a) Atomic force microscopy (AFM) phase images of two-dimensional (2D) folded-chain crystals of isotactic-poly(methyl methacrylate)(176k) (it-PMMA(176 k)) observed in situ at 30 °C, 80 °C and 100 °C by high-temperature AFM. (b) Crystalline area of 2D folded-chain crystals of it-PMMA(175 k) and it-PMMA(12 k) as a function of temperature. Melting temperatures of bulk crystals measured by differential scanning calorimetry are also shown by arrows. Reproduced with permission from Takanashi et al.10 Copyright (2013) American Chemical Society.
Single-chain crystals
Crystallization is an important process in polymer physics; however, the crystallization mechanism, especially at the molecular level, is still not well understood. If we could observe the crystallization behavior of a single isolated chain at the molecular level, we might gain new insights in our understanding of the crystallization mechanism. Figure 5d shows our approach to observe crystallization of a single isolated chain using a mixed monolayer.11 A small amount of high-molecular-weight it-PMMA chains are solubilized in an it-PMMA oligomer monolayer, the molecular weight of which is too small to allow it to crystallize. Then, if the mixed monolayer is compressed to a surface pressure higher compared with the crystalline transition, the high-molecular-weight it-PMMA is expected to crystallize, whereas the matrix monolayer stays amorphous. Figures 5a and b shows AFM images of a mixed monolayer, it-PMMA(758 k)/it-PMMA(590)=1/200 (wt wt−1), deposited below (Figure 5a, 1 mN m−1) and beyond (Figure 5b, 18 mN m−1) the crystallization transition. At 1 mN m−1, amorphous string-like it-PMMA(758 k) chains solubilized in an it-PMMA(590) monolayer are observed. The contrast comes from the different glass transition temperatures (Tg), as explained later; it-PMMA(758 k) (40 °C) was observed as being higher compared with the oligomer monolayer, which is liquid at room temperature. After compression at 18 mN m−1, the amorphous chain is converted to crystallites. A specific chain in Figure 5b converted to three small crystallites (1, 2 and 3) linked by an amorphous chain, as schematically shown in Figure 5d (right). In the magnified AFM image (c) of the crystallite 1 in Figure 5b, the crystallite is apparently an it-PMMA folded-chain crystal consisting of a double-stranded chain similar to those shown in Figures 3 and 4. Because this crystal formed from a single chain, the it-PMMA was assumed to intertwine to form a double-stranded helix at both ends (1, 3) and the middle (2) of the chain, which then immediately folded to form folded-chain crystals. In our method, we cannot directly observe the crystallization process in real time, but the crystallized monolayer is expected to contain crystals at various crystallization stages. If we carefully analyze them, we may be able to deduce how they crystallized. Figure 6 shows various single-chain crystals of it-PMMA(758 k). Single-chain crystals crystallized at one end (upper), at both ends (middle) and at both ends, as well as in the three points at the middle of the chain in a manner resembling a necklace (lower). In the table, 43 single-chain crystals are categorized by the number and position of the crystallites formed. Close inspection indicates that all single-chain crystals crystallized at least at one end (first line), then the other end crystallized (second line) and after both ends had crystallized, the middle of the chain crystallized (third to fifth lines). These results indicate that the chain end is more susceptible to crystallization compared with the middle of the chain in this system. We also studied single-chain crystals from various molecular weight it-PMMAs from 1.76 × 105 to 1.40 × 107, and found that although the molecular weight varied by almost a factor of 10, the size of the small crystallites formed in the single-chain crystals were almost the same and were estimated to be composed of a 1.14 × 105–2.41 × 105 molecular weight chain. The small crystallites may not be crystalline nuclei themselves, but may reflect them. Therefore, we presume that the size of the crystal nuclei is independent of the molecular weight of the polymers. The structure of single-chain crystals observed here, especially those small crystallites linked by an amorphous chain similar to a necklace, bears a close resemblance to a simulation of a single-chain crystal by Langevin dynamics by Muthukmar and co-workers.51

(a and b) Atomic force microscopy (AFM) height images of a mixed monolayer of isotactic-poly(methyl methacrylate)(758k) (it-PMMA(758 k))/it-PMMA(590)=1/200 (wt wt−1) deposited at (a) 1 mN m−1 and (b) 18 mN m−1, respectively. (c) Magnified AFM phase image corresponding to the square in (b). (d) Schematic representation of crystallization upon compression of a single isolated it-PMMA chain solubilized in its oligomer monolayer. Reproduced with permission from Anzai et al.11 Copyright (2015) American Chemical Society.

(Upper) Atomic force microscopy (AFM) height images of various single-chain crystals of isotactic-poly(methyl methacrylate)(758k) (it-PMMA(758 k)). (Lower) Forty-three single-chain crystals were classified by the number and the position of small crystallites in the chain. Reproduced with permission from Anzai et al.11 Copyright (2015) American Chemical Society.
Multistranded helical structure of PMMA stereocomplex
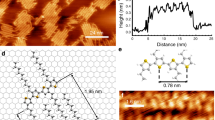
Stereoregular PMMAs, it- and syndiotactic (st) PMMAs, form a crystalline stereocomplex with a stoichiometry of it/st =1/2 in solutions and solids (for a review see Spěváček and Schneider52). For example, both it- and st-PMMA dissolve in acetonitrile to form a clear solution, and if one mixes the two clear solutions, each of which contains it- or st-PMMA, a crystalline precipitate immediately forms that consists of the PMMA stereocomplex. Schomaker and Challa53 proposed a double-stranded helical structure for the stereocomplex as shown in Figure 7a, left based on X-ray diffraction of a uniaxially stretched strand. A 9/1 (9 monomers per turn) it-PMMA helix with a helical pitch of 1.84 nm is loosely wound with a 18/1 (18 monomers per turn) st-PMMA helix with the same helical pitch. Thus, the PMMA stereocomplex is a unique multistranded helical structure composed of complementary it- and st-PMMA helices; such a structure has attracted the attention of researchers. However, the limited number of X-ray diffraction peaks obtained from the stereocomplex prevented definite identification of the structure for long time. Because a resolution close to or slightly better than 1 nm can be attained by AFM using LB films, we tried to observe directly a molecular image of the stereocomplex. Fortunately, the mixed monolayer of it- and st-PMMAs spread on a water surface was known to form a stereocomplex upon compression based on the surface pressure–area isotherms and infrared measurement of the resultant LB films.54

(a) Double-stranded and triple-stranded helix models for isotactic- and syndiotactic-poly(methyl methacrylate) (it- and st-PMMA) stereocomplex. (b) Atomic force microscopy (AFM) phase image of an it- and st-PMMA (it/st=1/2) mixed monolayer deposited on mica at 10 mN m−1. (c) Magnified AFM phase image corresponding to the square (c) in (b) with the triple-stranded helix model for a PMMA stereocomplex. Right- (A and B) and left- (C) handed helices are clearly shown. Reproduced with permission from Kumaki et al.12 Copyright (2007) Wiley-VCH.
Figures 7b and c show high-resolution AFM phase images of the PMMA stereocomplex prepared by the LB technique.12 Many spring-like structures are discernable, each of which is a stereocomplex molecule. As shown in the magnified image (Figure 7c), mixtures of right-handed (A and B) and left-handed (C) helices are present. From the AFM image, the width of the stereocomplex (2.4 nm) is close to that expected from the double-stranded helix model, but the helical pitch of the outer st-PMMA helix (0.92 nm) is only half that of the model (1.84 nm), indicating that the model should be reconsidered. If an st-PMMA is wound around an it-PMMA helix with a half-pitch, as shown in the AFM image, the it-PMMA helix should also wind with a double density to maintain the experimental stoichiometry (it/st=1/2). Thus, the previous model needs to be totally reconstructed. In Figure 7a, right, we propose a new triple-stranded helix model that agrees with both the AFM image and the stoichiometry. The outer st-PMMA winds with a half-pitch of 0.92 nm, a configuration that resembles the structure of the st-PMMA helix in the crystalline inclusion complex of st-PMMA/solvent molecules.55 Pure st-PMMA is difficult to crystallize, but it does cocrystallize with a small amount of specific solvent molecules, such as cyclohexanone and benzene, included in the cavity of the generated st-PMMA helices. The st-PMMA helix can be analyzed as a 74/4 (=18.5/1) helix with a helical pitch of 0.885 nm, very close to the st-PMMA helix used in the triple-stranded stereocomplex model. For the inner it-PMMA helix with double density, it is probably reasonable to select a thermodynamically stable helical structure of it-PMMA, namely, the double-stranded helix structure that forms in its crystal. The helical structure of the it-PMMA double-stranded helix in the triple-stranded helix (9/1 helix with a helical pitch of 1.84 nm) was slightly modified from the it-PMMA homocrystal (10/1 helix with a helical pitch of 2.1 nm; Kusanagi et al.40) to fit the X-ray diffraction and the stoichiometry. The exact helical structures of it- and st-PMMA in the triple-stranded helix need to be optimized based on experimental data, such as X-ray diffraction of a stretched strand. Recently, Yarovsky and co-workers56 optimized the triple-stranded helix model using a molecular dynamics simulation to fit X-ray powder diffraction data of a stereocomplex. Their optimized model suggests that a 9/1 it-PMMA double-stranded helix with 1.8 nm helical pitch was surrounded with a 20/1 st-PMMA single helix with an average helical pitch of 0.9 nm.
We note that AFM is a powerful new tool that can provide key information to understand the stereocomplex structure, which was too complicated to be analyzed using the conventional X-ray diffraction for over half a century.
Mechanism of PMMA stereocomplex formation
The PMMA stereocomplex is a rare complementary multistranded helix composed of synthetic polymers and is thus an ideal model to study how a multistranded helix forms. In this context, multistranded helical structures were investigated in detail by AFM as a function of the molecular weight of the component it- and st-PMMAs.13 To avoid uncertainty arising from the molecular weight distribution of the component polymers, we used uniform polymers obtained by fractionation of a specific molecular weight component from a polymer.57 Figure 8a shows an AFM phase image of a stereocomplex composed of it-PMMA 22 mers (it22) and st-PMMA 44 mers (st44) with the stoichiometric composition of it/st=1/2. A stereocomplex formed with a uniform length (lines) that was arranged in parallel to form lamellae that formed further stacked and regular aggregations. Note that there was no irregular large aggregation resulting from intermolecular connections, indicating that the stereocomplex was formed as a thermodynamically stable supramolecule with a definite size corresponding to the molecular weight of the component polymers.

(a) Atomic force microscopy (AFM) phase images of the stereocomplex composed of uniform isotactic-poly(methyl methacrylate) 22 mers (it-PMMA 22 mers) and syndiotactic-poly(methyl methacrylate) 44 mers (st-PMMA 44 mers) (it/st=1/2 in unit-molar base) deposited on mica at 10 mN m−1. (b) Molecular length distributions of the stereocomplexes composed of it22/st44 (1/2 in weight), it43/st44 (1/2 in weight) and it22/it43/st44 (0.34/0.66/2 in weight, corresponding to it/st=1/2 in weight and it22/it43=50/50 on polymer-molar basis) mixtures. (c) Schematic representation of possible chain arrangements in a stereocomplex composed of an it22/it43/st44 mixture. Reproduced with permission from Kumaki et al.13 Copyright (2010) American Chemical Society.
A detailed AFM study of the stereocomplexes as a function of the molecular weight of the component it- and st-PMMAs showed that (1) the size of the stereocomplex depends precisely on the molecular weights of the component PMMAs, (2) the it- or st-PMMA with the longer molecular length determines the total length of the stereocomplex and molecules of the shorter component associate until they fill up or cover the longer component and (3) the observed lengths of the stereocomplexes support the supramolecular triple-stranded helical structure composed of a double-stranded helix of two intertwined it-PMMA chains included in a single helix of st-PMMA. The stereocomplex shown in Figure 8a was therefore concluded to be composed of an it-PMMA double-stranded helix with two it22 molecules and a st-PMMA single-stranded helix with two st44 molecules, as shown in the schematic representation.
From the result (2) above, the size of an it-PMMA double-stranded helix inside the stereocomplex can be evaluated, if we use a st-PMMA helix sufficiently shorter than the it-PMMA helix. As schematically shown in Figure 8c, we examined the stereocomplex formed from a mixture of it22, it-PMMA 43 mer (it43) and st-PMMA 44 mer (st44). The length of the st44 helix (2.2 nm) is appropriately shorter than that of it22 (4.5 nm) and it43 (8.8 nm); therefore, we could evaluate the length of the it-PMMA double-stranded helix from the length of the resultant stereocomplex. As shown in Figure 8c, the it-PMMA double-stranded helix can be formed by selecting the same molecular weight as it22/it/22 and it43/it43 or mixing the different molecular weights as two it22 and one it43. Figure 8b shows that the length distribution of the resultant stereocomplex prepared from an it22 and it43 equimolar mixture on a polymer-molar basis with st44 was bimodal. Each peak was in good agreement with the distribution of a mixture of it22/st44 and it43/st44, respectively, and the number of stereocomplexes in each peak was identical, corresponding exactly to the mixing ratio of it22 and it43. If the it-PMMA double-stranded helix had formed from a mixture of different molecular weights, it22 and it44, the length distribution of the resultant stereocomplex would have differed from the mixing ratio of it22 and it43. Therefore, the result indicated that the it-PMMA double-stranded helix formed by selecting the same molecular weights, which we term molecular sorting (Figure 8c, right-top and -bottom), and no heterogeneous mixture formed from different molecular weights (Figure 8c, right-middle).
As mentioned above, the it- or st-PMMA with the longer length determined the total length of the resultant stereocomplex. It is of interest to ask why it-PMMA forms a double-stranded helix selecting the same molecular weight. We believe that this behavior originated from the molecular structures of the stereocomplex. The triple-stranded helix model is an inclusion complex of an it-PMMA double-stranded helix in a single-stranded st-PMMA helix, and not a simple triple-stranded helix in which three helices are intertwined. In the inclusion complex between the it-PMMA double helix and the st-PMMA helix, the longer component may determine the total length, on the other hand, in an intertwined helix of it-PMMA, chains may select the molecular weight of the other component. The triple-stranded helix model of the stereocomplex rationally explains the molecular weight dependence of the stereocomplex.
Selective associations, recognizing the structure and molecular weight of the pairing molecules, that is, molecular sorting, have so far been limited to systems containing strong intermolecular monomer–monomer interactions, such as helicates with metal–coordinate bonds58, 59 and peptide nucleic acids (PNAs) with hydrogen bonds.60 The present PMMA stereocomplex system is believed to form by van der Waals forces without specific monomer–monomer interactions, thus indicating that molecular weight recognition between complementary helices can be achieved without specific monomer–monomer interactions. This finding suggests that the multiple-stranded helical structure itself may have the ability to recognize molecular weight through a topological interaction.
Chain packing in a polymer monolayer
Although polymer Langmuir monolayers have been extensively studied as highly functional ultrathin films, the most fundamental issue in understanding a 2D film, the polymer chain packing in the film, is still not well understood. de Gennes61 first indicated that because a 2D ideal chain can be built up to 2D bulk density without interpenetrating into other chains, the polymer chains in a 2D state may exist as perfectly segregated chains (Figure 9a, right). However, there are contradictory results that support either segregated chain packing62, 63 or interpenetrating chain packing.14, 64, 65, 66, 67 Direct observation of polymer chain packing by AFM is expected to be the most direct way to understand this, but the limited resolution means that direct observation of the chain packing in the monolayer of a conventional polymer is still difficult.

(a) Schematic representation of possible chain packing in two-dimensional (2D) states. (b) Atomic force microscopy (AFM) phase image of a monolayer of polyisocyanide deposited on mica at 1 mN m−1. (c) Schematic representation of the polymer chain packing in (a). Some chains are highlighted for clarity. (d) Chemical structure of the polyisocyanide used. Reproduced with permission from Kumaki et al.14 Copyright (2010) American Chemical Society.
To overcome this difficulty, we used a rigid rod helical polymer that contains a long side group in the repeating unit.44 Figure 9a shows an AFM phase image of a monolayer of poly(phenyl isocyanide) bearing l-alanine residues with a long n-decyl chain as a pendant group (Figure 9d) deposited on mica at a surface pressure of 1 mN m−1.14 As a result of the long side chain, the chain-to-chain distance was sufficiently large (2.6 nm) to visualize the chain packing by AFM. As schematically shown in Figure 9b, the chain packing, including the chain ends, are satisfactorily visualized: some chains are highlighted for clarity. Note that the chains are packed into a 2D film without any stacking and are strongly interpenetrated. Unexpectedly, in some areas chains are folded into hairpin-like conformations and also some bundles of chains are folded similar to deformed rock strata. These highly forced conformations may be formed spontaneously when the polymer solution is spread on the water surface because of the condensed nature of the monolayer. Further systematic studies using more flexible polymers will make the chain packing in monolayers more clear.
Isolated chains solubilized in a polymer monolayer
Chain packing in miscible polymer blends cannot be visualized for 3D samples, but in 2D systems, chains solubilized in miscible polymer blend monolayer are expected to be observable. We found that a PMMA/poly(n-nonyl acrylate) (PNA) blend monolayer was miscible,68 and if a small amount of PMMA was solubilized in a PNA monolayer, the isolated PMMA chains solubilized in the PNA monolayer were clearly visible by AFM with clear contrast.15 Figure 10b shows an AFM height image of a PMMA/PNA (1/100 (wt wt−1)) monolayer deposited on mica at 1 mN m−1. Chain-like structures that are ~0.3 nm higher compared with the matrix monolayer were clearly observed. The average number of the chain-like structures in the AFM images (22 μm−2) was in good agreement with the number of PMMA chains in the monolayer expected from the LB deposition conditions (21 μm−2). The origin of the contrast is assumed to be the different rigidities of the polymers because of their different Tg (105 °C (PMMA), −89 °C (PNA)). If the polymers are tapped by a cantilever with the same force, the more rigid PMMA chains should be detected as being physically higher compared with the flexible PNA chains. Figure 10c shows a double-logarithmic plot of the radius of gyration (Rg) of the PMMA chains solubilized in PNA(13 k) monolayers as a function of the molecular weight of the PMMA. The parameter Rg scales with the molecular weight as Rg(PMMA)∝MPMMA~0.63. The scaling of Rg(PMMA) was larger compared with that of a 2D ideal chain (0.5) but smaller compared with that of a 2D chain in a good solvent (0.75). The absolute value of Rg(PMMA) was four to six times larger compared with that of the 2D ideal chain (perfectly segregated chain), indicating that the PMMA chain markedly expanded and interpenetrated into the PNA monolayer. We also evaluated the Rg of PMMA(210 k) solubilized in monolayers of different molecular weight PNA from 7 to 550 k, and found that the Rg(PMMA, 210 k) was independent of the molecular weights of the matrix PNA over a wide range. In a 3D homopolymer system, a polymer chain solubilized in a lower molecular weight matrix swells; however, a chain solubilized in a higher molecular weight matrix should shrink. Therefore, the Rg dependence of the PMMA on the Mn of the PNA matrix is markedly different from the case in 3D systems. One possible explanation is that the PMMA/PNA monolayer is a strongly miscible system and that the Rg of the PMMA is not strongly affected by the Mn of the matrix, although the scaling of Rg(PMMA) on Mn(PNA) (0.63) was slightly smaller compared with that in a good solvent (0.75). The other is that in the 2D system, a polymer chain has a somewhat larger Rg compared with the 2D ideal chain (segregated chain) but is not truly solubilized with the matrix polymer; as a result, the Rg is unaffected by the Mn of the PNA matrix. At present, it is not clear how the PMMA/PNA monolayer mixes; further systematic studies changing the interactions between components with various blend systems should clarify the nature of monolayers of 2D miscible-blend systems.

(a and b) Atomic force microscopy (AFM) height image of a mixed monolayer of poly(methyl methacrylate)(292k)/poly(n-nonyl acrylate) (PMMA(292k))/PNA(13k)) (=1/100 (wt wt−1)) deposited on mica at 1 mN m−1, with a schematic representation of the blend monolayer. (c) Rg of PMMA chains solubilized in PNA(13k) monolayers as a function of Mn of PMMA. Reproduced with permission from Sugihara et al.15 Copyright (2012) American Chemical Society.
Concluding remarks
In this review, we report that various polymer chain structures can be observed using 2D films prepared by the LB technique; the observations include isolated chains, chains in amorphous and crystalline states, miscible polymer blends and a supramolecular multistranded helix. The LB technique is superior in preparing well-ordered 2D films with homogeneous structures throughout a film, and produces samples suitable for high-resolution imaging. However, observation should be expanded to 3D samples. Further improvements of AFM instrumentation and cantilevers will provide a more robust method for observation of polymer structures at the molecular level. Recent more sophisticated AFM instruments, such as non-contact frequency modulation AFM,69 quartz turning fork cantilevers69 and torsional tapping-mode AFM,48 are still applied to polymer observations to only a limited degree, but are expected to improve our ability to observe polymers at the molecular level in the future.
References
Binning, G., Rohrer, H., Gerber, Ch. & Weibel, E. Surface studies by scanning tunneling microscopy. Phys. Rev. Lett. 49, 57 (1982).
Binnig, G., Quate, C. F. & Gerber, Ch. Atomic force microscope. Phys. Rev. Lett. 56, 930–933 (1986).
Gaines, G. L. Jr. Insoluble Monolayers at Liquid–Gas Interface, (Interscience, New York, NY, USA, 1966).
Ulman, A. An Introduction to Ultrathin Organic Films from Langmuir-Blodgett to Self-Assembly, (Academic Press, New York, NY, USA, 1991).
Kumaki, J., Nishikawa, Y. & Hashimoto, T. Visualization of single chain conformations of a synthetic polymer with atomic force microscopy. J. Am. Chem. Soc. 118, 3321–3322 (1996).
Kumaki, J. & Hashimoto, T. Conformational change in isolated single synthetic polymer chain on mica surface observed by atomic force microscopy. J. Am. Chem. Soc. 125, 4907–4917 (2003).
Kumaki, J., Kawauchi, T. & Yashima, E. 'Reptational' movements of single synthetic polymer chains on substrate observed by in-situ atomic force microscopy. Macromolecules 39, 1209–1215 (2006).
Kumaki, J., Kawauchi, T. & Yashima, E. Peculiar ‘reptational’ movements of single synthetic polymer chains on substrate observed by AFM. Macomol. Rapid Commun. 29, 406–411 (2008).
Kumaki, J., Kawauchi, T. & Yashima, E. Two-dimensional folded chain crystals of a synthetic polymer in a Langmuir-Blodgett film. J. Am. Chem. Soc. 127, 5788–5789 (2005).
Takanashi, Y. & Kumaki, J. Significant melting point depression of two-dimensional folded-chain crystals of isotactic poly(methyl methacrylate)s observed by high-resolution in-situ atomic force microscopy. J. Phys. Chem. B 117, 5594–2605 (2013).
Anzai, T., Kawauchi, M., Kawauchi, T. & Kumaki, J. Crystallization behavior of single isotactic poly(methyl methacrylate) chains visualizaaed by atomic force microscopy. J. Phys. Chem. B 119, 338–347 (2015).
Kumaki, J., Kawauchi, T., Okoshi, K., Kusanagi, H. & Yashima, E. Supramolecular helical structure of the stereocomplex composed of complementary isotactic and syndiotactic poly(methyl methacrylate)s as revealed by atomic force microscopy. Angew. Chem. Int. Ed. 46, 5348–5351 (2007).
Kumaki, J., Kawauchi, T., Ute, K., Kitayama, T. & Yashima, E. Molecular weight recognition in the multiple-stranded helix of a synthetic polymer without specific monomer-monomer interaction. J. Am. Chem. Soc. 130, 6373–6380 (2008).
Kumaki, J., Kajitani, T., Nagai, K., Okoshi, K. & Yashima, E. Visualization of polymer chain conformations in amorphous polyisocyanide Langmuir–Blodgett films by atomic force microscopy. J. Am. Chem. Soc. 132, 5604–5606 (2010).
Sugihara, K. & Kumaki, J. Visualization of two-dimensional single chain conformations solubilized in miscible polymer blend monolayer by atomic force microscopy. J. Phys. Chem. B 116, 6561–6568 (2012).
Samorí, P., Surin, M., Palermo, V., Lazzaroni, R. & Leclère, P. Functional polymers; scanning force microscopy insights. Phys. Chem. Chem. Phys. 8, 3927–3938 (2006).
Rabe, J. P. Molecular Workbench for imaging and manipulation of single macromolecules and their complexes with the scanning force microscope. Top. Curr. Chem. 285, 77–102 (2008).
Hobbs, J. K., Farrance, O. E. & Kailas, L. How atomic force microscopy has contributed to our understanding of polymer crystallization. Polymer 50, 4281–4292 (2009).
Kumaki, J., Sakurai, S.-i. & Yashima, E. Visualization of synthetic helical polymers by high-resolution atomic force microscopy. Chem. Soc. Rev. 38, 737–746 (2009).
McConney, M. E., Singamaneni, S. & Tsukruk, V. V. Probing soft matter with the atomic force microscopies: Imaging and force spectroscopy. Polym. Rev. 50, 235–286 (2010).
Shönherr, H. & Vancso, G. J. Scanning Force Microscopy of Polymers, (Springer, Heidelberg, Germany, 2010).
Gallyamov, M. O. Scanning force microscopy as applied to conformational studies in macromolecular research. Macromol. Rapid. Commun. 32, 1210–1246 (2011).
Magonov, S. High-resolution visualization and compositional analysis of polymers with atomic force microscopy. Int. J. Polym. Anal. Charact. 16, 505–518 (2011).
Tsukruk, V. V. & Singamaneni, S. Fundamentals and Practices, (Wiley-VCH, Weinheim,Germany, 2012).
Hansma, H. G., Vesenka, J., Siegerist, C., Kelderman, G., Morrett, H., Sinsheimer, R. L., Elings, V., Bustamante, C. & Hansma, P. K. Reproducible imaging and dissection of plasmid DNA under liquid with the atomic force microscope. Science 256, 1180–1184 (1992).
Hansma, H. G. & Hoh, J. H. Biomolecular imaging with the atomic force microscope. Annu. Rev. Biophys. Biomol. Struct. 23, 115–139 (1994).
Albrecht, T. R., Dovek, M. M., Lang, C. A., Grütter, P., Quate, C. F., Kuan, S. W. J., Frank, C. W. & Pease, R. F. W. Imaging and modification of polymers by scanning tunneling and atomic force microscopy. J. Appl. Phys. 64, 1178–1184 (1988).
Kumaki, J. Polystyrene monomolecular particles obtained by spreading dilute solutions on the water surface. Macromolecules 19, 2258–2263 (1986).
Kumaki, J. Monolayer of polystyrene monomolecular particles on a water surface studied by Langmuir-type film balance and transmission electron microscopy. Macromolecules 21, 749–755 (1988).
Kumaki, J. Accumulation of monomolecular polystyrene particles from a water surface onto a substrate. J. Polym. Sci. Part B 28, 105–111 (1990).
Beaglehole, D., Radlinska, E. Z., Ninham, B. W. & Christenson, H. K. Inadequacy of Lifshitz Theory for thin liquid films. Phys. Rev. Lett. 66, 2084–2087 (1991).
Sheiko, S. S. & Möller, M. Visualization of molecules—a first step to manipulation and controlled response. Chem. Rev. 101, 4099–4123 (2001).
Sheiko, S.S., Gerle, M., Fischer, M., Schmidt, M. & Möller, M. Worm-like polystyrene brushes in thin films. Langmuir 13, 5368–5772 (1997).
Percec, V., Ahn, C. -H., Ungar, G., Yeardley, D. J. P., Möller, M. & Sheiko, S. S. Spherical and cylindrical supermolecules from polymer backbones jacketed with quasi-equivalent dendritic coats. Nature 391, 161–164 (1998).
Minko, S., Kiriy, A., Gorodyska, G. & Stamm, M. Single flexible hydrophobic polyelectrolyte molecules adsorbed on solid substrate: transition between a stretched chain, necklace-like conformation and a globule. J. Am. Chem. Soc. 124, 3218–3219 (2002).
Kiriy, A., Gorodyska, G., Minko, S., Stamm, M. & Tsitsilianis, C. Single molecules and associates of heteroarm star copolymer visualized by atomic force microscopy. Macromolecules 36, 8704–8711 (2003).
Gallyamov, M. O., Tartsch, B., Khokhlov, A. R., Sheiko, S. S., Börner, H. G., Matyjaszewski, K. & Möller, M. Reversible collapse of brushlike macromolecules in ethanol and water vapours as revealed by real-time scanning force microscopy. Chem. Eur. J. 10, 4599–4605 (2004).
Xu, H., Shirvanyants, D., Beers, K., Matyjaszewski, K., Rubinstein, M. & Sheiko, S. S. Molecular motion in a spreading precursor film. Phys. Rev. Lett. 93, 206103 (2004).
Magonov, S. N. & Whangbo, M. -H. Surface Analysis with STM and AFM, Experimental and Theoretical Aspects of Image Analysis, (VCH, Weinheim, Germany, 1996).
Kusanagi, H., Tadokoro, H. & Chatani, Y. Double strand helix of isotactic poly(methyl methacrylate). Macromolecules 9, 531–532 (1976).
Sakurai, S.-i., Okoshi, K., Kumaki, J. & Yashima, E. Two-dimensional hierarchical self-assembly of one-handed helical polymers on graphite. Angew. Chem. Int. Ed. 45, 1245–1248 (2006).
Sakurai, S. -i., Okoshi, K., Kumaki, J. & Yashima, E. Two-dimensional surface chirality control by solvent-induced helicity inversion of a helical polyacetylene on graphite. J. Am. Chem. Soc. 128, 5650–5651 (2006).
Sakurai, S. -i., Ohsawa, S., Nagai, K., Okoshi, K., Kumaki, J. & Yashima, E. Two-dimensional helix-bundle formation of a dynamic helical poly(phenylacetylene) with achiral pendant groups on graphite. Angew. Chem. Int. Ed. 46, 7605–7608 (2007).
Kajitani, T., Okoshi, K., Sakurai, S.-i., Kumaki, J. & Yashima, E. Helix-Sense controlled polymerization of a single phenyl isocyanide enantiomer leading to diastereomeric helical polyisocyanides with opposite helix-sense and cholesteric liquid crystals with opposite twist-sense. J. Am. Chem. Soc. 128, 708–709 (2006).
Onouchi, H., Okoshi, K., Kajitani, T., Sakurai, S. -i., Nagai, K., Kumaki, J., Onitsuka, K. & Yashima, E. Two- and three-dimensional smectic ordering of single-handed helical polymers. J. Am. Chem. Soc. 130, 229–236 (2008).
Maeda, T., Furusho, Y., Sakurai, S. -i., Kumaki, J., Okoshi, K. & Yashima, E. Double-stranded helical polymers consisting of complementary homopolymers. J. Am. Chem. Soc. 130, 7938–7945 (2008).
Ohsawa, S., Sakurai, S. -i., Nagai, K., Banno, M., Maeda, K., Kumaki, J. & Yashima, E. Hierarchical amplification of macromolecular helicity of dynamic helical poly(phenylacetylene)s composed of chiral and achiral phenylacetylenes in dilute solution, liquid crystal, and two-dimensional crystal. J. Am. Chem. Soc. 133, 108–114 (2011).
Mullin, N. & Hobbs, J. K. Direct imaging of polyethylene films at single-chain resolution with torsional tapping atomic force microscopy. Phys. Rev. Lett. 107, 197801-1–197801-5 (2011).
Kim, J. H., Jang, J. & Zin, W. C. Thickness dependence of the melting temperature of thin polymer films. Macromol. Rapid. Commun. 22, 386–389 (2001).
Wang, Y., Rafailovich, M., Sokolov, J., Gersappe, D., Araki, T., Zou, Y., Kilcoyne, A. D. L., Ade, H., Marom, G. & Lustiger, A. Substrate effect on the melting temperature of thin polyethylene films. Phys. Rev. Lett. 96, 028303 (2006).
Muthukumar, M. Molecular modelling of nucleation in polymers. Philos. Trans. R. Soc. Lond. Ser. A 361, 539–556 (2003).
Spěváček, J. & Schneider, B. Aggregation of stereoregular poly(methyl methacrylates). Adv. Colloid Interface Sci. 27, 81–150 (1987).
Schomaker, E. & Challa, G. Complexation of stereoregular poly(methyl methacrylates). 14. The basic structure of the stereocomplex of isotactic and syndiotactic poly(methyl methacrylate). Macromolecules 22, 3337–3341 (1989).
Brinkhuis, R. H. G. & Shouten, A. J. Thin-film behavior of poly(methyl methacrylates). 4. Stereocomplexation of isotactic and syndiotactic poly(methyl methacrylate) at the air-water interface. Macromolecules 25, 2725–2731 (1992).
Kusuyama, H., Miyamoto, N., Chatani, Y. & Tadokoro, H. Crystalline complexes of syndiotactic poly(methyl methacrylate) with some organic solvents. Polymer 24, 119–122 (1983).
Christofferson, A. J., Yiapanis, G., Ren, J. M., Qiao, G. G., Satoh, K., Kamigaito, M. & Yarovsky, I. Molecular mapping of poly(methyl methacrylate) super-helix stereocomplexes. Chem. Sci. 6, 1370–1378 (2015).
Hatada, K., Kitayama, T., Ute, K & Nishiura, T. Uniform polymer in synthetic polymer chemistry. J. Polym. Sci. Part A 42, 416–431 (2004).
Krämer, R., Lehn, J. -M. & Marquis-Rigault, A. Self-recognition in helicate self-assembly: spontaneous formation of helical metal complexes from mixtures of ligands and metal ions. Proc. Natl Acad. Sci. USA 90, 5394–5398 (1993).
Caulder, D. L. & Raymond, K. N. Superamolecular self-recognition and self-assembly in gallium(III) catecholamide triple helices. Angew. Chem. Int. Ed. 36, 1440–1442 (1997).
Wittung, P., Nielsen, P. E., Buchardt, O., Egholm, M. & Nordén, B. DNA-like double helix formed by peptide nucleic acid. Nature 368, 561–563 (1994).
de Gennes, P.-G. Scaling Concepts In Polymer Physics, (Cornell University Press, London, UK, 1979).
Aoki, H., Morita, S., Sekine, R. & Ito, S. Conformation of single poly(methyl methacrylate) chains in an ultra-thin film studied by scanning near-field optical microscopy. Polym J. 40, 274–280 (2008).
Mayer, H., Kreer, T., Aichele, A., Cavallo, A., Johner, A., Baschnagel, J. & Wittmer, J. P. Perimeter length and form factor in two-dimensional polymer melts. Phys. Rev. E 79, 050802-1–050802-4 (2009).
Yethiraj, A. Computer simulation study of two-dimensional polymer solution. Macromolecules 36, 5854–5862 (2003).
Wang, X. & Folts, V. J. Chain conformation in two-dimensional dense state. J. Chem. Phys. 121, 8158–8162 (2004).
Kumaki, J. & Hashimoto, T. Two-dimensional microphase separation of a block copolymer in a Langmuir-Blodgett film. J. Am. Chem. Soc. 120, 423–424 (1998).
Gallyamov, M. O., Tartsch, B., Potemkin, I. I., Börner, H. G., Matyjaszewski, K., Khokhlov, A. R. & Möller, M. Individual bottle brush molecules in dense 2D layers restoring high degree of extension after collapse-decollapse cycle: directly measured scaling exponent. Eur. Phys. J. E 29, 73–85 (2009).
Sasaki, Y., Aiba, N., Hashimoto, H. & Kumaki, J. Reversible hierarchical phase separation of a poly(methyl methacrylate) and poly(n-nonyl acrylate) blend in a Langmuir monolayer. Macromolecules 43, 9077–9086 (2010).
Morita, S., Giessible, F. J. & Wiesendanger, R. (eds). Noncontact Atomic Force Microscopy, Vol. 2 (Springer, Berlin, Germany, 2009).
Acknowledgements
We thank our co-workers for their excellent contributions reported in this review. In particular, Emeritus Professors Naoya Ogata and Takeji Hashimoto and Professor Eiji Yashima are highly appreciated. Financial support from the Japan Science and Technology Agency and MEXT/JSPS KAKENHI Grand Numbers 20106009, 21350059, 23655208, 24655091, 24350113, 25107706 and 26620092 is greatly appreciated.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Polymer Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Kumaki, J. Observation of polymer chain structures in two-dimensional films by atomic force microscopy. Polym J 48, 3–14 (2016). https://doi.org/10.1038/pj.2015.67
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pj.2015.67
- Springer Nature Limited
This article is cited by
-
Screw sense excess and reversals of helical polymers in solution
Nature Communications (2023)
-
Chain movements of a molecularly flat PMMA substrate surface prepared by thermal imprinting with mica and isolated PMMA chains deposited on the PMMA substrate observed by AFM around the bulk Tg
Polymer Journal (2022)
-
Extended-chain crystallization and stereocomplex formation of polylactides in a Langmuir monolayer
Polymer Journal (2020)
-
Ultra-high resolution imaging of thin films and single strands of polythiophene using atomic force microscopy
Nature Communications (2019)
-
Metastable interface formation in isotactic poly(methyl methacrylate)/alumina nanoparticle mixtures
Polymer Journal (2018)