
Abstract
Arginine, one among the 20 most common natural amino acids, has a pivotal role in cellular physiology as it is being involved in numerous cellular metabolic and signaling pathways. Dependence on arginine is diverse for both tumor and normal cells. Because of decreased expression of argininosuccinate synthetase and/or ornithine transcarbamoylase, several types of tumor are auxotrophic for arginine. Deprivation of arginine exploits a significant vulnerability of these tumor cells and leads to their rapid demise. Hence, enzyme-mediated arginine depletion is a potential strategy for the selective destruction of tumor cells. Arginase, arginine deiminase and arginine decarboxylase are potential enzymes that may be used for arginine deprivation therapy. These arginine catabolizing enzymes not only reduce tumor growth but also make them susceptible to concomitantly administered anti-cancer therapeutics. Most of these enzymes are currently under clinical investigations and if successful will potentially be advanced as anti-cancer modalities.
Similar content being viewed by others


Introduction
Amino acids play a major role in regulating important cellular events in both normal and malignant cells. Besides their role in the synthesis of hormones and peptides, amino acids also function as cell signaling molecules, playing a modulatory role in gene expression.1 Amino acids regulate RNA synthesis by diverse mechanisms ranging from regulating transcription factors assembly,2 to total mRNA turnover.3, 4 Amino acids are major determinants of a normal cellular physiology, therefore potential signaling pathways such as amino acid response (AAR) pathway sense their altered metabolism (Figure 1). Hence, amino acid levels in the body are critical for important cellular functions.5, 6, 7, 8, 9

AAR pathway. Restriction of essential amino acids activates the general control nondepressible protein 2 (GCN2) kinase by increasing uncharged tRNA pool.196 Activated GCN2 kinase phosphorylates the translation initiation factor eIF2α. Phosphorylated eIF2α binds more tightly to eIF2β, inhibiting the exchange of GDP for GTP. Inhibition of GDP exchange for GTP further inhibits the binding of eIF2 complex to methionine aminoacyl tRNA, leading to inhibition of translational initiation.197 Recently, SLC38A9 has been identified as an upstream positive regulator of the mTOR pathway. Amino acids activate the RAG GTPases, which then recruit mTOR to the lysosomal surface. Rheb also localizes to lysosomal membrane. mTOR activation occurs only when both RAG GTPases and Rheb are active. Upon amino acid deprivation, tuberous sclerosis complex translocates to lysosomal surface and promotes GTP hydrolysis by Rheb and thereby inhibiting mTOR complex.164
There is a significant difference between the metabolism of normal and malignant cells.10 For instance, bio-energetic requirements for homeostasis in normal cells are fulfilled by catabolic metabolism. On the other hand, the majority of the tumor cells alter their metabolic program ('metabolic remodeling') and consume additional nutrients in order to maintain a balance between elevated macromolecular biosynthesis11 and adequate levels of ATP for survival.12, 13 However, the endogenous supply of nutrients becomes inadequate during intense growth. Thus tumor cells depend on exogenous nutrients in their microenvironment to fulfill the elevated energy requirements, that is, they become auxotrophic for nutrient and energy sources.14, 15, 16 Deprivation of amino acids results in growth inhibition or death of tumor cells by the modulation of various signaling cascades.6, 7, 8, 9, 17, 18
Exogenously incorporated enzymes that deprive amino acids could be a novel strategy for the treatment of auxotrophic tumors. The first Food and Drug Administration approved heterologous enzyme for the treatment of cancer was Escherichia coli l-asparaginase.19 l-asparaginase exploits the differences on their dependence of normal and leukemic cells toward l-asparagine.20 l-asparaginase has been proven to be a promising agent for the treatment of l-asparagine auxotrophic T-cell acute lymphoblastic lymphoma (T-ALL). Use of l-asparaginase in T-ALL opened up new windows of ‘amino acid-depriving therapy’. Currently, there is a resurgence of interest in enzyme-mediated amino acid deprivation as a new therapeutic approach for cancer treatment.6, 7, 21, 22 For example, arginine depletion can inhibit tumor cell proliferation and induce cell death pathways. Here we endeavor to provide a basic understanding of the roles of arginine in normal and tumor cell with emphasis on current knowledge and developments in the application of enzyme-mediated arginine-depriving therapy as a potential anti-cancer approach.
Enzyme-mediated arginine deprivation: a potential anti-cancer approach
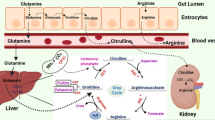
Arginine is involved in the regulation of various molecular pathways and thus the availability of arginine can modulate key metabolic, immunological, neurological and signaling pathways of the cells (Figures 2 and 3).23, 24 Auxotrophy toward arginine by certain tumor cells (particularly that of hepatocellular carcinoma and melanoma) has been well characterized.25, 26 Normal cells, when deprived of arginine, undergo cell cycle arrest at Go/G1 phase and become quiescent. If reinstated with arginine, the majority of the normal cells recover to their normal proliferation status. However, arginine deprivation in tumor cells does not arrest cell cycle at G1 phase and continue to be in a cell cycle, leading tumor cells to undergo unbalanced growth and eventually lead to the activation of apoptotic pathways.27, 28

Involvement of arginine in human physiology. Arginine is a dibasic, cationic amino acid and is considered as ‘conditionally essential’ amino acid. Arginine plays a crucial role in innate and adaptive immunity. For example, increased role of arginine in myeloid-derived suppressor cells results in the impairment of T-cell proliferation and function.190 Arginine has been identified as the sole physiological precursor for NO, a key performer in many cellular regulatory functions. Arginine also is a precursor of two important amino acids, proline and glutamate.198 One of the most important roles of arginine is its implication in the synthesis of polyamines through the diversion from NO synthesis pathway. Polyamines are known to promote tumor growth, invasion and metastasis.199 Arginine also has a vital role in the synthesis of nucleotides, creatine, agmatine and hormones such as insulin and prolactin.200

Arginine synthesis and homeostasis pathways. Arginine is synthesized as an intermediate in the urea cycle. Arginine homeostasis is mainly achieved by catabolism. In neonates, the gene expression of arginine anabolic enzymes such as 1-pyrroline-5-carboxylase, ASS and ASL is low. Thus, arginine is considered as an essential amino acid in neonates. After birth, the expression of ASS and ASL increases and expression of arginase is found undetectable at this stage.201 Arginine can be degraded by arginase, ADC, ADI and NOSs (please note that ADI is not a mammalian enzyme). The products of arginine catabolism have important roles in tumor cell biology. For example, ornithine, the product of arginase, is diverted to polyamine synthesis via ornithine decarboxylase. NOSs degrade arginine into citrulline and NO. Citrulline is recycled to urea cycle, while NO is as a modulator of important metabolic and signaling cascades. Agmatine is synthesized by decarboxylation of arginine via ADC and has an important role in neurotransmission.
Owing to the involvement of arginine in a plethora of cellular pathways, arginine dependence of tumor cells has rapidly emerged as a potential target for cancer.29 However, dietary restriction results in the reduction of only 30% of plasma arginine.30 Thus, arginine degrading enzyme-mediated arginine deprivation has been proposed as a potential anti-cancer therapy by various research groups.27, 28, 29, 30, 31, 32, 33, 34, 35 Enzymes that can be used for arginine deprivation therapy (ADT) include arginine deiminase (ADI), arginase and arginine decarboxylase (ADC) as discussed below (Figure 3).
Arginine deiminase
ADI (E.C.3.5.3.6) is a prokaryotic enzyme originally isolated from Mycoplasma, which catalyzes an irreversible deimination of the guanidine group of l-arginine to citrulline and ammonium ion.36 Normal cells are able to convert citrulline into arginine through argininosuccinate synthetase (ASS) and ASL, expression of which are tightly regulated. However, the expression of ASS/ASL is downregulated in certain tumor cells by unknown mechanisms and these cells are unable to convert citrulline to arginine.30, 31, 32, 33, 37 This makes the tumor cells auxotrophic for arginine for their growth and cellular functioning. ADI-mediated arginine deprivation leads to apoptotic cell death, selectively of arginine auxotrophic ASS (−) tumor cells sparing the ASS (+) ADI resistant normal cells38 (Table 1). Incidence of ASS deficiency varies depending on the tumor type and expression level of ASS has been proposed as a biomarker for identification of ADI sensitive tumors.24, 25, 39, 40, 41, 42
In 1990, Miyazaki et al.43 were the first to report the growth inhibition of Mycoplasma infected human tumor cells. The cause of growth inhibition of human tumor cell lines was identified as a ADI produced by Mycoplasma. In vitro growth-inhibitory dose of Mycoplasmal ADI appeared to be 1000 times lower than that of bovine liver arginase. Subsequently in 1992, growth-inhibitory activity of ADI was demonstrated in ASS-downregulated human melanoma cells.44 These pioneering studies established ADI as a potential anti-cancer enzyme (Figure 4).

Timeline of important advancement in arginine deprivation therapy of cancer.
PEGylated ADI
Being microbial in origin, ADI has serious disadvantages of eliciting strong antigenicity and rapid plasma clearance (half-life of 4 h). To circumvent these limitations, several studies have aimed to extend the plasma half-life of ADI and to minimize its antigenicity. In 1993, Takaku et al.45 addressed these problems for the first time by polyethylene glycol (PEG) modification. Remarkably, PEGylation of Mycoplasma arginini ADI enhanced its cytotoxic potential in vivo and once a week intravenous injection of PEG-ADI at a dose of 5 U per mouse (10 mg protein per kg) depleted plasma arginine to an undetectable level at least for a week, whereas native enzyme required 10 daily injections to achieve similar effects. Nevertheless, PEGylation of Mycoplasma hominis ADI also resulted in significant enhancement of arginine lowering potential of native M. hominis ADI.46, 47 Recently, PEGylation and pharmacological properties of an engineered ADI originated from Pseudomonas plecoglossicida have been studied. PEGylated P. plecoglossicida ADI remarkably improves the stystemic half-life (by 11-folds) and found to exhibit superior efficacy than native ADI in depleting plasma arginine.48
PEG-ADI has also shown promising outcomes for the treatment of human malignancies. In March 1999, ADI-PEG20, PEGylated recombinant Mycoplasmal ADI was approved as an orphan drug by the US Food and Drug Administration for the treatment of hepatocellular carcinoma (HCC) and malignant melanomas. Subsequently in July 2005, European Agency for the Evaluation of Medicinal Products granted orphan drug status to ADI-PEG20 for the treatment of HCCs.49
ADI-PEG20 is currently undergoing clinical investigation as a randomized double-blind phase III trial in patients with advanced HCC (NCT 01287585), phase II studies in patients with ASS-negative metastatic melanoma (NCT 01279967) and phase II studies in patients with relapsed small-cell lung cancer (NCT 01266018)50 (Table 2). Outcomes of the previous clinical studies were also encouraging, achieving response rates of 25 and 47% in melanoma and HCC, respectively (Table 2). Moreover, grades III and IV toxicities have not been observed in clinical investigations involving ADI-PEG20 in metastatic melanoma and HCC patients.51, 52 Therefore, clinicians are looking forward to the establishment of ADI-PEG20 as a potent anti-cancer modality.
Tumor sensitivity toward ADI
The auxotrophicity of tumors toward arginine and their sensitivity toward it can be attributed to the lack or reduced expression of ASS in tumors.25, 37, 38, 39, 53 Notably, numerous tumor cells that are deficient in ASS expression, are sensitive toward ADI treatment (Table 1). Transfection of an expression plasmid containing human ASS cDNA in HCC and melanoma cells confers severe resistance to ADI treatment compared with ASS-negative cells.47 Till date, most promising targets for ASS expression-dependent ADT identified are human melanoma and HCCs. Other promising targets include malignant pleural mesothelioma, renal cell carcinoma, prostate cancer, T-ALL and osteosarcoma.50 However, molecular mechanisms underlying tumor sensitivity toward ADI treatment, by downregulation of ASS expression in tumor cells, are still elusive. Promoter hypermethylation-dependent silencing of ASS gene is an endorsed mechanism of ASS gene repression.37, 54, 55, 56 Methylation frequency of the ASS promoter upto 50–80% level at the CpG loci is documented across a broad range of lymphomas. In contrast, normal lymphoid samples were found unmethylated.26 Treatment of ADI-PEG20 to ASS-methylated lymphoma cell lines revealed dramatic decrease in the proliferation rate and viability count, by inducing caspase-dependent apoptosis, without affecting normal lymphoblastoid cell lines. Demethylation-induced resistance to ADI-PEG20 treatment has also been confirmed in cutaneous T-cell lymphoma cell lines, as their incubation with 5-Aza-dC (demethylating agent) for 8 days which resulted in partial demethylation, followed by transcriptional activation and synthesis of ASS protein.26
Recently Rabinovich et al.57 have confirmed that proliferation of the osteosarcoma cells is supported by downregulation of ASS, by facilitating pyrimidine synthesis via activation of CAD (carbamoyl-phosphate synthase 2, aspartate transcarbamylase and dihydroorotase) complex. As cytosolic aspartate serves as a substrate for both ASS and for CAD complex, ASS downregulation can enhance aspartate availability for CAD for the synthesis of pyrimidine nucleotides to promote proliferation. Thus, aspartate transport can be exploited as an additional therapeutic target in tumors with ASS downregulation, especially in those ones which develop resistance to arginine-depriving enzymes.
Tumor resistance toward ADI
ASS-deficient tumors are sensitive to ADI treatment; however, arginine deprivation eventually upregulates ASS expression in tumor cells and thereby confers resistance toward ADI.25, 58 Transcriptional induction of ASS expression and increase in ASS mRNA level is reported in human embryonic kidney cells and melanoma cells during arginine starvation.59, 60 Transcription factors such as c-Myc and HIF-1α are involved in the upregulation of ASS expression under arginine-depleted conditions.60 E-box and GC-box are the important sequences located between −85 and −35 nucleotides in the ASS promoter region that modulate ASS expression through their interactions with c-Myc and HIF-1α. Under the normal concentrations of arginine, HIF-1α (but not c-Myc) binds to E-box and thus acts as a negative regulator of ASS expression. Under the conditions of arginine depletion, HIF-1α is degraded and replaced by up-regulated c-Myc, which directly binds to E-box; thus, c-Myc acts as a positive regulator of ASS expression (Ref. 60; Figure 6). Recently reported in melanoma cells, inhibition of ubiquitin-mediated protein degradation is a molecular mechanism responsible for the stabilization and accumulation of c-Myc.61 Furthermore, various cellular pathways, such as Ras and its downstream ERK/PI3K/AKT kinase cascade are associated with the post-translational modifications of c-Myc, leading to its phosphorylation and stabilization during ADI-PEG20-mediated arginine deprivation conditions. Involvement of Ras/PI3K/ERK signaling pathway in the development of resistance toward ADI treatment suggests that combination of ADI with Ras/ERK, PI3K/AKT inhibitors is a potential therapeutic strategy to improve the anti-cancer response.62, 63
Development of anti-drug neutralizing antibodies is another possible mechanism of resistance toward ADI-PEG20 treatment.64 Arginine concentrations were recovered upto pre-treatment levels in a patient with malignant pleural mesothelioma and in Asian patients with advanced hepatocellular carcinoma following the ADI-PEG20 treatment. This recovery in arginine concentration was found concomitant with an increase in anti–ADI-PEG20 antibody titer.65 These studies suggest the involvement of drug-associated resistance i.e. anti-drug neutralizing antibodies, rather than tumor-related factors as another possible mechanism of resistance of some tumor cell types toward ADI-PEG20 treatment.62, 63
Anti-tumor mechanisms of ADI treatment
Role of autophagy and apoptosis in ADI-mediated arginine deprivation therapy
Due to the involvement of arginine in numerous cellular pathways (Figure 2), the exact anti-proliferative mechanisms of ADI treatment, besides that of arginine depletion, are still elusive. One of the potential pathways involved in the cytostatic and cytotoxic potential of ADI is TRAIL (tumor necrosis factor-related apoptosis-inducing ligand).66, 67, 68 TRAIL has an important role in the cleavage of Beclin-1 (Atg6) and Atg5 in arginine-deprived melanoma cells.69 Beclin-1 and Atg5 are essential for the formation of autophagosomes and thus crucial for autophagy. Since autophagy serves as a mean to evade apoptosis in arginine-depleted cells, TRAIL induced cleavage of Beclin-1 and Atg5 leads to decreased autophagy, thereby increasing apoptosis.69 In addition, these two drugs (ADI and TRAIL) complement each other by activating the intrinsic apoptosis pathways. ADI-PEG20 increases cell surface receptors DR4/5 for TRAIL thereby binding TRAIL to these death receptors. As a result, caspase-8 or 10 are activated.66 ADI-PEG20 treatment also modulates different autophagic pathways involved in the cell survival. Adenosine 5′-monophosphate-activated protein kinase and ERK pathways are activated in ADI-treated prostate cancer cells; while AKT, mTOR and S6K pathways are attenuated. ADI-PEG20 treatment to CWR22Rv1 prostate cancer cells induced autophagy, as revealed by the appearance of LC-II only after 30 min exposure continues its persistence after 24 h following ADI-PEG20 treatment.70, 71 Additionally, inhibition of autophagy by chloroquine, a clinically approved anti-malarial agent which inactivates lysosomal functions, accelerates the ADI-induced apoptotic cell death of prostate cancer70, 71 and small-cell lung cancers.39 Thus autophagy has been proposed as a pro-survival mechanism of tumor cells during arginine deprivation.71
ADI-mediated arginine deprivation is also known to induce caspase-dependent apoptotic pathways in many of the tumor cells types. ADI-PEG20 treatment activates caspase-3 in ASS-methylated malignant lymphoma cells, whereas ASS-positive normal lymphoblastoid cells are resistant to it.26 Similarly, cell death has been attributed to caspases activation in glioblastoma,54 melanoma,38, 72 leukemia73 and pancreatic cancer cells.74 Moreover, all these studies indicate that inhibition of autophagy leads to further advancement in the ADI-PEG20-mediated demise of tumor cells, suggesting the induction of autophagy as a mechanism of tumor resistance to ADI-PEG20 treatment.
Cumulative pieces of evidence suggest that the activation of caspases is not a sole decisive phenomenon in programmed cell death pathways. Caspase-dependent apoptosis is a major mode of cell death, but in its absence or failure, there are other pathways which can also execute cell death.75, 76, 77 ADI-PEG20 treatment to small-cell lung cancer, leukemia, retinoblastoma and prostate cancer cells induces apoptotic cell death pathways; however, without activation of caspases, suggesting the role of caspase-independent apoptosis as a cell death pathway.33, 39, 69, 70, 78 The inter-membrane space of mitochondrion contains proteins such as apoptosis-inducing factor (AIF) and endonuclease G (EndoG), which can induce apoptotic cell death in a caspase-independent fashion.79 EndoG is one of the predominant endonucleases that are involved in the regulation of cellular functions such as mitochondrial biogenesis, DNA synthesis and repair. AIF is an FAD-containing flavoprotein which plays an important role in the stability of an electron transport chain.80 Nutrient deficiency-mediated stress signals induce mitochondrial outer membrane permeabilization, which consequently releases inter-membrane space proteins such as AIF, EndoG and cytochrome c. AIF has a role of central mediator in caspase-independent cell death pathway.81 AIF, once released into the cytosol, interacts with EndoG and cyclophilin A before its translocation into the nucleus.82 Subsequently after translocation into the nucleus, it triggers cell death either directly, through interaction with DNA, or indirectly, through the production of reactive oxygen species.73, 74, 79, 80 Mitochondrial outer membrane permeabilization promotes both, caspase-dependent and caspase-independent apoptotic pathways, but with different kinetics.83 Although, the upstream signaling stimulus for both, a caspase-dependent and caspase-independent pathway is the same, that is, via induction of mitochondrial outer membrane permeabilization, their downstream pathways are different. Moreover, nuclear alterations and the changes occurring in mitochondrial trans-membrane potential during caspase-independent pathways are different than those observed in a caspase-dependent apoptotic pathway.84
To summarize, growing evidence suggests that autophagy is a prevailing cell survival mechanism in tumor cells undergoing ADI-mediated arginine deprivation. The overall cellular response to ADI-mediated arginine deprivation in different tumor cells operates through a complex cascade, initiating with induction of autophagy and followed by the activation of either caspase-dependent or caspase-independent cell death pathways. It is worth emphasizing that the discrepancy of cellular responses of tumor cells to ADI-mediated arginine depletion in activation of either caspases-dependent or caspases-independent cell death pathways can vary depending on tumor cell type.38, 39, 70, 71, 74 As a result, the precise mechanisms of tumor cell death—consequential of cellular response to ADI-mediated arginine depletion—appear to be complex and variable, and need to be further elucidated.
Inhibition of de novo protein synthesis by ADI-mediated arginine deprivation
Inhibition of de novo protein synthesis is another mechanism, which can be attributed to the anti-tumor potential of ADI. As extracellular arginine pool is responsible for 40% of de novo protein synthesis, ADI treatment to human lung carcinoma cells results in an anti-proliferative effect, mediated by inhibition of protein synthesis.85 Arginine is present in various compartments such as extracellular, intracellular and citrulline-arginine regeneration, that is, cytosolic compartment and it is known to regulate various cellular pathways differently. Protein synthesis mainly utilizes arginine either from the intracellular pool or the citrulline-arginine regeneration mechanism, while polyamines synthesis largely utilizes arginine pool from the intracellular origin.86, 87 Polyamines are synthesized through the methionine salvage pathway via decarboxylation of S-adenosylmethionine. S-adenosylmethionine is a donor metabolite necessary for the transfer of methyl group to DNA and proteins. Human colon cancer (HCT116) cells treated with short hairpin CD44 RNA interference showed a decrease in the total amount of methionine-pool metabolites including polyamines, suggesting the role of polyamines in cancer proliferation.88
ADI treatment toward human mammary adenocarcinoma and lung carcinoma cells differently modulates polyamine synthesis and the global protein synthesis. Interestingly, inhibition of protein synthesis has been correlated with the ASS-mediated regeneration of arginine. Cells expressing low levels of ASS (A549) result in decreased protein synthesis (without affecting polyamine synthesis) and those expressing higher ASS levels (MCF-7) are resistant to ADI treatment, as the decreased arginine levels can be replaced by citrulline-arginine regeneration pathway.85
Anti-angiogenic effects of ADI-mediated arginine deprivation
As a tumor grows beyond a certain size (2 mm in diameter for most solid tumors), available vasculature within the tumor becomes inadequate to supply sufficient quantities of essential nutrients for their growth.89 This results in the generation of hypoxic tumor microenvironment and leads to the development of new blood vessels (angiogenesis) as a colossal requisite of the developing tumors.90 Accordingly, neovascularization can be stated as one of the decisive phenomena during tumor growth and metastasis.91 Emerging studies now indicate that not only molecular signals but also metabolic mechanisms regulate angiogenesis.92 Under stress conditions such as hypoxia, tumor cells secrete angiogenic factors such as vascular endothelial growth factor (VEGF).93 Increased levels of VEGF activate VEGF receptor 2 (VEGFR2) signaling in the quiescent endothelial cells which in turn initiate angiogenesis.94, 95, 96 Endothelial cells produce 85% of their total amount of ATP via glycolysis. Addiction of endothelial cells on anaerobic rather than aerobic pathway enables them for the formation of vascular sprouts in hypoxic areas.97, 98 Metabolism of tumor endothelial cells resembles that of highly activated endothelial cells because of the tumor induced switch from quiescence to proliferation due to metabolically regulated migration during sprouting.99, 100
Besides ADI’s role in modulation of apoptotic pathways, it has an anti-angiogenic activity that contributes to its anti-tumor potential. The growth, migration and differentiation of human umbilical vein endothelial cells are strongly impaired in a medium containing recombinant ADI.101 As a consequence; it results in decreased tube formation with intermittent and incomplete microvascular network. Similarly, Park et al.102 found that E. coli ADI inhibits angiogenesis by inhibiting tube formation of endothelial cells and neovascularization in Chick Chorioallantoic membrane and Matrigel plug assay.
Suppression of nitric oxide (NO) generation is also another possible mechanism for anti-angiogenic activity of ADI. Since l-arginine is required for nitric oxide synthases (NOSs) to generate NO, the depletion of arginine by ADI suppresses NO synthesis.102 Potential role of ADI-mediated arginine depletion in inhibition of NO synthesis has been reported.103, 104 We and others have previously reported that NO promotes tumor growth through the stimulation of angiogenesis105, 106, 107 and regulates cellular interaction by controlling adhesion molecule expression and ultimately cell adhesion.108, 109 NO directly, or indirectly through NO-mediated reactive nitrogen species, induces the activation of certain angiogenic signaling pathways in the endothelial cells.110 NO acts as an autocrine mediator in endothelial cell functioning and as a final modulator in VEGF-stimulated angiogenesis.109, 111 NO not only mediates angiogenesis but also subsequent vessel maturation112, 113 Moreover, NO is known to inhibit angiostatin and thrombospondin-1, two main inhibitors of angiogenesis.114 Owing to the important role of NO in angiogenesis, ADI inhibits tumor growth not only by draining the supply of arginine but also by its anti-angiogenic activity via suppression of NO generation.
To summarize, certain tumor cell types such as, HCCs and metastatic melanomas are invariably deficient in ASS expression and can be specifically targeted by ADI-mediated ADT. It is worth noting that more than one pathway may be attributed to the cytotoxic potential of ADI-mediated ADT (Figure 5). The anti-tumor potential of ADI may not only be simply accredited to its action as arginine degrading enzyme but also to several other mechanisms important in the cellular functioning of tumor cells. Induction of apoptotic pathways, inhibition of angiogenesis and inhibition of de novo protein synthesis are the important mechanisms attributed to the cytotoxic potential of ADI. Moreover, studies have revealed the ADI-mediated modulations in tumor cell cycle. The fundamental difference of cell cycle modulations in normal and malignant cells should be exploitable as a means of selective demise of tumor cells and ADI, in combination with other anti-cancer chemotherapeutic agents, which can be a potential strategy to improve chemo-sensitization against tumor cells.115, 116, 117, 118

Schematic representation of cytostatic and cytotoxic pathways involved in arginine deprivation therapy. ADT can potentially modulate numerous cellular and signaling pathways rendering their cytotoxic and cytostatic pathways. Induction of apoptotic pathways, inhibition of angiogenesis and inhibition of de novo protein synthesis are the important mechanisms attributed to the cytotoxic potential of ADT. Moreover, ADT-mediated modulations in tumor cell cycle can be exploited as a means of tumor growth arrest.
Arginase
Arginase (E.C.3.5.3.1) is a mammalian enzyme which catalyzes the conversion of arginine to ornithine and urea. Arginase is considered as an enzyme responsible for the cyclic nature of urea cycle, since only the organisms containing arginase are able to carry out the complete urea cycle.119 Two distinct isoforms of mammalian arginase have been identified that are encoded by two separate genes.120 Type I arginase (arginase I) is located in the cytosol and is mainly expressed in liver. Type II arginase is located in the mitochondrial matrix and is expressed in extra-hepatic tissues.121, 122 Intracellular regulation of arginase expression is of immense importance as it has crucial implications for the synthesis of essential cellular metabolites,123 For example, cytosolic co-localization of arginase I with ornithine decarboxylase (ODC) preferentially utilizes ornithine for the biosynthesis of polyamine. On the other hand, due to its co-localization with ornithine aminotransferase in the mitochondria, arginase II directs ornithine for the production of proline and glutamine.124, 125
PEGylated recombinant human arginase I
Elevated requirements of arginine by tumor cells were first identified in 1947 and preferential utilization of arginine by tumor bearing animals was revealed in 1953.126, 127 The use of bovine and murine arginase in ADT was prevailing until the advent of recombinant DNA technology,128, 129, 130 followed by the pervasive use of recombinant human arginase in subsequent decades.131, 132 Arginase from bovine and murine sources has been extensively used for the ADT in vitro. However, limited success was achieved in vivo because of its alkaline optimum pH and very low affinity for the substrate. Human arginase I also has a serious limitation of very short circulatory half-life (~30 min).
To extend plasma half-life of arginase, PEGylation has been applied successfully. PEGylated recombinant human arginase I (rhArg-Peg5000mw) had efficient catalytic activity at physiological pH with improved in vivo half-life of 3 days. Furthermore, rhArg-Peg5000mw was found to have significant tumor inhibitory activity in BALB/c nude mice bearing HCC xenografts.131 Notably, these results were consistent with those demonstrated by Tsui et al.133 Recently, a bioengineered form of human arginase I was developed by the co-factor replacement, the replacement of two Mn2+ ions by Co2+ ions. The modified Co2+-arginase I resulted in 10-fold increase in the catalytic activity and five-fold greater stability at the physiological pH. Nevertheless, IC50 values for killing human HCC and melanoma cell lines were lowered by 12-15 folds.134 More recently, modifications in bioengineered Co2+-arginase I were performed by conjugating 5-kDa PEG to enhance plasma half-life. This modified version of bioengineered arginase I (Co-hArgI–PEG) was proven to be cytotoxic by significantly increasing the expression of caspases-3 in HCC and pancreatic carcinoma tumor xenografts.135 Lately, the cytotoxic potential of Co-hArgI–PEG was identified in acute myeloid leukemia and glioblastoma cells. Acute myeloid leukemia cell lines were found sensitive toward Co-hArgI–PEG-mediated arginine deprivation with very low (58-722 PM) IC50 values, suggesting a very high potential of Co-hArgI–PEG-mediated arginine depletion in acute myeloid leukemia cells.136 Moreover, Co-hArgI–PEG-mediated arginine deprivation has been demonstrated to induce caspase-independent, non-apoptotic cell death in human glioblastoma cells.137 Alternative method to extend the plasma half-life of recombinant human arginase also has been established. Plasma half-life of a fusion protein form of a recombinant human arginase (rhArg-Fc, constructed by linking rhArg to the Fc region of human immunoglobulin IgG1), was evidenced to significantly extend upto ~4 days.138 In addition, rhArg-Fc was confirmed to conspicuously inhibit the cell growth of human HCC cells in vitro and in vivo.138
Past decade has evidenced a prevalent use of recombinant human arginase-mediated ADT in numerous cancer cell types, mainly metastatic HCC and melanomas.131, 139, 140 Currently, PEGylated derivative of recombinant human arginase I is undergoing clinical trials for the treatment of human HCC.141, 142 Moreover, initiatives are now being taken to overcome the possible problem of accumulation of PEGylated products in the liver by impending approaches such as fusion proteins.138
Anti-tumor mechanisms of arginase-mediated arginine deprivation
Selective starvation of l-arginine in tumor cells, which are auxotrophic for l-arginine, is one of the most important anti-tumor mechanisms of ADT. Arginase can render its cytostatic effect as a result of modulations in the cell cycle proteins, whereas, cytotoxic effects rendered by arginase I-mediated arginine deprivation have been proposed as a result of induction of potential cell death pathways namely apoptosis and probably by ‘autophagic cell death’. Summarized below are the current understandings of the molecular mechanisms of cytostatic and cytotoxic effects rendered by arginase-mediated ADT.
Role of autophagy in arginase-mediated arginine deprivation
Autophagy is a key sensing and regulatory mechanism of cells in nutrient deprived conditions. Under stress conditions, autophagy functions as a bio-energy management system by recycling cell organelles and damaged and/or long-lived proteins.143 Although autophagy seems to be a survival mechanism of the cells, there is a growing evidence of accumulation of autophagosomes and other autophagic markers in dying cells unable to process apoptosis, raising the term ‘autophagic cell death’.144, 145, 146, 147 However, the term ‘autophagic cell death’ is based on morphological features rather than the causative role of autophagy in cell death. New definition of ‘autophagic cell death’ has been proposed, implying that cell death must occur without the involvement of apoptotic machinery, (caspase activation) but with an increase in autophagic flux.148, 149
Mammalian target of rapamycin (mTOR) is a key regulator of coupling cell growth and nutritional status of the cell.150, 151 Autophagy is induced by the inhibition of mTOR-signaling pathway.152 During nutrient affluent conditions, mTOR is involved in the negative regulation of Atg1 (autophagy-related gene 1) which inhibits autophagy.153, 154 Arginase-mediated arginine deprivation leads to decreased levels of ATP, which in turn activates the adenosine 5′-monophosphate-activated protein kinase. Activated adenosine 5′-monophosphate-activated protein kinase eventually inhibits the mTOR-signaling pathway, manifested by the reduced phosphorylation of key downstream molecules, such as 4E-BP1 (eukaryotic translation initiation factor 4E-binding protein-1). Dephosphorylation of 4E-BP1 is observed in Chinese hamster ovary (CHO), human melanoma cells and human prostate cancer cells following their exposure to recombinant human arginase I.65, 155, 156 Phagosome/lysosome activity is also significantly increased following an incubation of human tumor cells in l-arginine-deficient medium.157 Additionally, studies carried out by Hsueh et al.156 evidenced no significant induction of apoptotic mechanisms in prostate cells after their exposure to rhArgI, suggesting the role of autophagic cell death, rather than apoptosis, as an alternative cell death mechanism. In addition, autophagy has often accompanied damaged mitochondria and higher levels of reactive oxygen species.158, 159 Acute generation of reactive oxygen species has been attributed to causing severe damages to the cellular macromolecules, which in consequence, leads to necrosis of the tumor cells.160, 161 Overall, arginase leads to deprivation of arginine, in consequence, it inhibits mTOR pathway during the deprivation and thus forcing tumor cells to undergo ‘autophagic cell death’ pathway.162
SLC38A9, a member 9 of the solute carrier family 38, has been recently identified as an integral component of the lysosomal machinery that controls amino acid-induced mTOR activation.163, 164 Amino acid starvation in human embryonic kidney (HEK293T) cells with stable expression of SLC38A9 has been shown to activate mTOR in a sustained manner. Moreover, shRNA-mediated silencing of SLC38A9 results in a reduction of arginine-induced mTOR activation. Also, depletion of SLC38A9 impaired mTOR activation induced by cycloheximide (a protein synthesis inhibitor which induces accumulation of intracellular amino acids), further suggests the role of SLC38A9 in mTOR activation at the lysosomal rather than at the plasma membrane. These studies have demonstrated that SLC38A9 acts as an upstream positive regulator tor mTOR functioning and thereby modulating autophagy in arginine-deprived tumor cells.
Although some studies have advocated autophagy as a cell death mechanism of arginase-mediated ADT,156, 157 many groups have explained it as a pro-survival mechanism; mainly by postponing the activation of apoptosis.38, 161 Thus, understanding the exact role of autophagy in arginase-mediated cell death pathways is a complicated episode.162, 165 Therefore, much need to be elucidated about these new findings related to ‘autophagic cell death’ and caution must be taken to assign autophagy as a cell death pathway in arginase-mediated ADT.
Role of apoptosis in arginase-mediated arginine deprivation
The role of autophagy, either in cell survival or in cell death, depends on many factors such as cell type, nature and severity of the stimuli and so on.166 If the attempt of the cells to survive through autophagy fails, apoptotic pathways take over and ultimately cause cell death.143 Inhibition of autophagy in amino acid deficient conditions induces tumor cell death, mainly because of further exacerbation of energy dearth.167, 168 Also, longer persistence of autophagy is proposed to eventually lead the activation of caspase-dependent cell death pathways, as autophagy and apoptotic cell death pathways are interconnected and also share some common pathways through the induction of the membrane permeability transitions.169, 170, 171 Induction of apoptotic pathways is another consequence of arginine depletion and anti-tumor mechanism of arginase I-mediated arginine deprivation.
Involvement of apoptosis as a cell death mechanism in arginase-mediated ADT has been illustrated in various literature reports. Annexin V is known to selectively stain the cells, which are destined for apoptosis or in the process of apoptosis. 33% of human melanoma cell population was destined for apoptotic cell death following rhArg treatment.139 Arginase I-mediated arginine deprivation led to the transcriptional upregulation of caspase 3, the intrinsic mitochondrial pathway of apoptosis, which is marked by the change in mitochondrial membrane potential.172 Recently, an anti-leukemic potential of PEGylated-arginase has been attributed to kinases general control nonderepressible 2 (GCN2)-mediated induction of apoptosis in T-ALL cells.173
Cell cycle arrest by arginase-mediated arginine deprivation and combination approaches
rhArg-Peg5000mw-mediated arginine deprivation in various HCC cells results in their cell cycle arrest at G2/M phase, by decreased expression levels of cyclin B1 and cdc2, or in S phase, by a transcriptional upregulation of cyclin A1.140 rhArg-Peg5000mw-mediated arginine depletion was witnessed to impair the expression of cyclin D3 in T-ALL cells, which was followed by an arrest of the cells in the G0-G1 phase of the cell cycle and induction of apoptosis.172 Recent investigations of rhArg-Fc-mediated arginine deprivation in human HCC cells exhibited cell cycle arrest at S phase.138 The exact mechanisms of these findings are still elusive, but the possible reasons seem to be the increased expression of cyclin A and declined transcription levels of p27 and p21 (the key cyclin kinase inhibitors).
Owing to the evidence of cell cycle arrest, a combination of arginase and other cell-cycle specific anti-cancer chemotherapeutics as potential anti-tumor approaches have been established. Synergistic effects of rhArg-Peg5000mw with 5-fluorouracil (5-FU, uracil analog which interferes with RNA and DNA synthesis) and cytarabine (Ara-C, anti-metabolic chemotherapeutic agent) have been investigated on the inhibition of proliferation of HCC and T-ALL cells, respectively.131, 172 Treatment of either rhArg-Peg5000mw or Ara-C alone induces a heterogeneous anti-tumor effect in vivo, whereas, combined treatment of rhArg-Peg5000mw and Ara-C induces a homogenous prevention of spleen growth, leading to the prolonged survival in all of the T-ALL bearing mice.172 Moreover, combined treatment of PEGylated recombinant human arginase I and oxaliplatin has been demonstrated to synergize the inhibiting effect on tumor growth and enhanced overall survival probability as compared with PEGylated recombinant human arginase I or oxaliplatin treatment alone.174
Altogether, arginase has an advantage over ADI that it is efficacious in both ASS-negative and ornithine transcarbamoylase (OTC) negative tumors,59 whereas ADI is efficacious only in ASS-negative tumors. The tumor cell types expressing ASS are resistant to arginine deprivation treatment by ADI.25, 26, 54, 61, 131 Even though arginase has been considered as a potential drug candidate over a period of six decades, low substrate specificity (high km of 2–4 mm), short plasma life and optimum alkaline pH (pH 9.3) limit in vivo applications of arginase.131, 140 In addition, robust homeostatic mechanisms in the body allow faster restoration of plasma-free arginine, making in vivo arginine deprivation by arginase more difficult. Most of the scientific efforts nowadays pay attention to these limiting characteristics of arginase.134, 175, 176
Arginine decarboxylase
ADC (E.C. 4.1.1.19) metabolizes arginine to agmatine, one of the minor metabolic products of arginine. ADC is mainly found in plants, bacteria and mammalian liver and brain membranes.177, 178 The mammalian ADC is different from other sources and distinct but related to ODC.179 Although, arginine decarboxylation by ADC is a minor metabolic route, its product i.e. agmatine has a significant role in numerous cellular pathways.180 Agmatine modulates the polyamine metabolism through its negative interaction with ODC.181 Agmatine also confers an inhibitory effect on intracellular polyamine content by inhibiting polyamine uptake182 and probably by increased polyamine catabolism.183 Mayeur et al.,184 has reported the effect of agmatine accumulation on polyamine metabolism, cell proliferation and cell cycle distribution in human colon adenocarcinoma epithelial cell lines. Because of the agmatine-mediated reduction in polyamine synthetic capacity of the cells, agmatine markedly inhibits the cell proliferation of HT-29 and Caco-2 cells in a dose dependent manner, without affecting cell membrane integrity. Moreover, agmatine modulates the cell cycle progression by decreasing ODC activity and expression.181, 185 As ODC plays an important role in the G1/S progression of the cells, agmatine-mediated modulations in ODC expression lead to modifications in the cell cycle progression.186 Additionally, agmatine also has been shown to delay the expression of cyclins in tumor cells, leading to the modifications in the cell cycle progression.184
ADC has been investigated for the enzymatic degradation of arginine in normal and malignant cell cultures.187 Arginine deprivation in human diploid fibroblasts (normal cells), achieved using human recombinant ADC, resulted in the cell cycle arrest at G1/G0. While treatment of 0.1 unit ml−1 ADC to HeLa (Human cervical cancer) cells resulted in cell cycle arrest with an initiation of cell death after 2 days.187 Similar results were evidenced in the studies by Wheatley et al.,188 where 5 units per ml ADC was found as effective as arginase in the inhibition of HeLa cells and cell cycle arrest at G1 (quiescence) in fibroblasts.
Although some research groups have exhibited ADC as a potential anti-tumor enzyme, only a few reports are available to support this fact (Table 1).187, 188 Even though ADC possesses low Km and can degrade arginine very rapidly, the serious problem is related to its product, that is, agmatine. Agmatine is toxic to normal cells when its concentration reaches to millimolar level, particularly when free arginine levels are low. Additionally, agmatine is not converted back to arginine under normal physiological conditions, which may lead to its accumulation and toxicity to normal cells.189 Though recombinant human ADC expressed in E. coli has been evidenced more active than Sigma enzymes prepared from other sources, its PEGylation has been shown to result in the loss of its entire activity.187, 189 To consider the further rational use of this prospective enzyme as potential anti-cancer modality, it clearly warrants further evaluation (Table 3).
Concluding remarks
Sufficient evidence has been accumulated indicating that arginine catabolic enzymes-based approaches may be an effective way to target malignant cells. These enzymes control tumor cell proliferation as well as make them highly vulnerable to cell-cycle-specific chemotherapeutic agents. This combinatorial approach is one of the potential strategies to maximize the efficacy to obliterate the tumor cells. Extensive research of the arginine metabolic pathways led to the establishment of arginine-depriving enzymes as a potential anti-cancer strategy against arginine auxotrophic tumors. However, many of these enzymes can be co-expressed in the cells, which results in complex interactions. For example, arginine is a common substrate for arginase as well as NOS. The specific role of NO, either in inhibition or induction of cell proliferation is dependent on numerous factors like its interaction with other free radicals, cellular makeup, tumor milieu, proteins present the cellular microenvironment and also upon the chemical and biological heterogeneity of NO. NO has been known to demonstrate bipolar cellular effects and often termed as ‘double-edged sword’. Although, NOS remains a viable candidate for cancer treatment, the precise role of NO in the tumor microenvironment is extremely complex and conflicting. Also, the preferential utilization of arginine by arginase and/or NOS pathway is not fully understood. Thus, many of these pathways warrant further research to understand the arginine metabolism at cellular and molecular levels involving upstream and downstream pathways of the enzymes involved.
It should be noted that modulation of the immunological responses is one of the major roles of arginine availability. Arginine metabolism in myeloid-derived suppressor cells via arginase and/or NOS markedly impairs the T-cell responses that would eradicate and remove tumor cells.190 Many excellent articles are available which focus on the role of arginine in immunological aspects of the tumors.191, 192, 193, 194 It would suffice to say here that the ADT may have further anti-tumor effect through restoration of anti-tumor immunity.
Arginine dependence of the tumor cells has been considered as the ‘Achilles heel’ of tumor cells.195 Inability of tumor cells to proliferate in the absence of arginine can be targeted for their selective destruction by arginine-depriving enzymes. Large numbers of enzyme-based anti-cancer therapies are currently undergoing clinical evaluation. It is encouraging that arginase and ADI already have achieved considerable success, without causing detrimental side effects and with high tolerability.51, 63, 141 The knowledge acquired about the PEGylation has helped in the generation of adducts of potential value, overcoming the serious limitations of the anti-cancer enzymes of the non-human origin. The approach of enzyme-mediated ADT is highly challenging, however rewarding upon success because of the provision of overturning the cancer dogma.
References
Wu G . Functional amino acids in nutrition and health. Amino Acids 2013; 45: 407–411.
Loreni F, Mancino M, Biffo S . Translation factors and ribosomal proteins control tumor onset and progression: how? Oncogene 2014; 33: 2145–2156.
Proud CG . Control of the translational machinery by amino acids. Am J Clin Nutr 2014; 99: 231s–236s.
Luo J-Q, Chen D-W, Yu B . Upregulation of amino acid transporter expression induced by l-leucine availability in L6 myotubes is associated with ATF4 signaling through mTORC1-dependent mechanism. Nutrition 2013; 29: 284–290.
Palii SS, Kays CE, Deval C, Bruhat A, Fafournoux P, Kilberg MS . Specificity of amino acid regulated gene expression: analysis of genes subjected to either complete or single amino acid deprivation. Amino Acids 2009; 37: 79–88.
Qie S, Liang D, Yin C, Gu W, Meng M, Wang C et al. Glutamine depletion and glucose depletion trigger growth inhibition via distinctive gene expression reprogramming. Cell Cycle 2012; 11: 3679–3690.
Agrawal V, Alpini SEJ, Stone EM, Frenkel EP, Frankel AE . Targeting methionine auxotrophy in cancer: discovery and exploration. Expert Opin Biol Ther 2012; 12: 53–61.
Wu G, Wu Z, Dai Z, Yang Y, Wang W, Liu C et al. Dietary requirements of ‘‘nutritionally non-essential amino acids’’ by animals and humans. Amino Acids 2013; 44: 1107–1113.
Fu YM, Yu Z-X, Li Y-Q, Ge X, Sanchez PJ, Fu X et al. Specific amino acid dependency regulates invasiveness and viability of androgen-independent prostate cancer cells. Nutr Cancer 2003; 45: 60–73.
Icard P, Lincet H . A global view of the biochemical pathways involved in the regulation of the metabolism of cancer cells. Biochim Biophys Acta Rev Cancer 2012; 1826: 423–433.
Cantor JR, Sabatini DM . Cancer cell metabolism: one hallmark, many faces. Cancer Disc 2012; 2: 881–898.
Locasale JW, Cantley LC . Metabolic flux and the regulation of mammalian cell growth. Cell Metab 2011; 14: 443–451.
Ferreira LMR, Hebrant A, Dumont JE . Metabolic reprogramming of the tumor. Oncogene 2012; 31: 3999–4011.
Yamamoto T, Takano N, Ishiwata K, Ohmura M, Nagahata Y, Matsuura T et al. Reduced methylation of PFKFB3 in cancer cells shunts glucose towards the pentose phosphate pathway. Nat Commun 2014; 5: 3480.
Matthew G, Heiden V . Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Disc 2011; 10: 671–684.
Ward PS, Thompson CB . Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell 2012; 21: 297–308.
Gelb T, Pshenichkin S, Rodriguez OC, Hathaway HA, Grajkowska E, DiRaddo JO et al. Metabotropic glutamate receptor 1 acts as a dependence receptor creating a requirement for glutamate to sustain the viability and growth of human melanomas. Oncogene 2015; 34: 2711–2720.
Cetinbas N, Daugaard M, Mullen AR, Hajee S, Rotblat B, Lopez A et al. Loss of the tumor suppressor Hace1 leads to ROS-dependent glutamine addiction. Oncogene 2015; 34: 4005–4010.
Graham ML . Pegaspargase:a review of clinical studies. Adv Drug Deliv Rev 2003; 55: 1293–1302.
Durden DL, Distasio JA . Characterization of the effects of asparaginase from Escherichia coli and a glutaminase-free asparaginase from Vibrio succinogenes on specific cell-mediated cytotoxicity. Int J cancer 1981; 27: 59–65.
Dodd KM, Tee AR . Leucine and mTORC1: a complex relationship. Am J Physiol Endocrinol Metab 2012; 302: E1329–E1342.
Takano N, Sarfraz Y, Gilkes DM, Chaturvedi P, Xiang L, Suematsu M et al. Mol Cancer Res 2014; 12: 1398–1406.
Wu G, Bazer FW, Davis TA, Kim SW, Li P, Rhoads JM et al. Arginine metabolism and nutrition in growth, health and disease. Amino Acids 2009; 37: 153–168.
Appleton J . Arginine: clinical potential of a semi-essential amino acid. Altern Med Rev 2002; 7: 512–522.
Feun LG, Marini A, Walker G, Elgart G, Moffat F, Rodgers SE et al. Negative argininosuccinate synthetase expression in melanoma tumors may predict clinical benefit from arginine-depleting therapy with pegylated arginine deiminase. Br J Cancer 2012; 106: 1481–1485.
Delage B, Luong P, Maharaj L, O’Riain C, Syed N, Crook T et al. Promoter methylation of argininosuccinate synthetase-1 sensitises lymphomas to arginine deiminase treatment, autophagy and caspase-dependent apoptosis. Cell Death Dis 2012; 3: e342.
Wheatley DN . Controlling cancer by restricting arginine availability-arginine catabolizing enzymes as anticancer agents. Anti-Cancer Drugs 2004; 15: 825–833.
García-Navas R, Munder M, Mollinedo F . Depletion of L-arginine induces autophagy as a cytoprotective response to endoplasmic reticulum stress in human T lymphocytes. Autophagy 2012; 8: 1557–1576.
Synakiewicz A, Stachowicz-Stencel T, Adamkiewicz-Drozynska E . The role of arginine and the modified arginine deiminase enzyme ADI-PEG 20 in cancer therapy with special emphasis on phase I/II clinical trials. Expert Opin Inv Drug 2014; 23: 1517–1529.
Dillon BJ, Holtsberg FW, Ensor CM, Bomalaski JS, Clark MA . Biochemical characterization of the arginine degrading enzymes arginase and arginine deiminase and their effect on nitric oxide production. Med Sci Monit 2002; 8: BR248–BR253.
Qui F, Huang J, Sui M . Targeting arginine metabolism pathway to treat arginine-dependent cancers. Cancer Lett 2015; 364: 1–7.
Phillips MM, Sheaff MT, Szlosarek PW . Targeting arginine-dependent cancers with arginine-degrading enzymes: Opportunities and challenges. Cancer Res Treat 2013; 45: 251–262.
Noh EJ, Kang SW, Shin YJ, Choi SH, Kim CG, Park IS et al. Arginine deiminase enhances dexamethasone-induced cytotoxicity in human T-lymphoblastic leukemia CCRF-CEM cells. Int J Cancer 2004; 112: 502–508.
Stasyk OV, Boretsky YR, Gonchar MV, Sibirny AA . Recombinant arginine-degrading enzymes in metabolic anticancer therapy and bioanalytics. Cell Biol Int 2015; 39: 246–252.
Feun LG, Kuo MT, Savaraj N . Arginine deprivation in cancer therapy. Curr Opin Clin Nutr 2015; 18: 78–82.
Shirai H, Blundell TL, Mizuguchi K . A novel superfamily of enzymes that catalyse the modification of guanidine groups. Trends Biochem Sci 2001; 26: 465–468.
Huang H-Y, Wu H-Y, Wang Y-H, Wang J-W, Fang F-M, Tsai J-W et al. ASS1 as a novel tumor suppressor gene in myxofibrosarcomas: aberrant loss via epigenetic DNA methylation confers aggressive phenotypes, negative prognostic impact, and therapeutic relevance. Clin Cancer Res 2013; 19: 2861–2872.
Savaraj N, You M, Wu C, Wangpaichitr M, Kuo MT, Feun LG . Arginine deprivation, autophagy, apoptosis (AAA) for the treatment of melanoma. Curr Mol Med 2010; 10: 405–412.
Kelly MP, Jungbluth AA, Wu B-W, Bomalaski J, Old LJ, Ritter G . Arginine deiminase PEG20 inhibits growth of small cell lung cancers lacking expression of argininosuccinate synthetase. Br J Cancer 2012; 106: 324–332.
Manca A, Sini MC, Izzo F, Ascierto P, Tatangelo F, Botti G et al. Induction of arginosuccinate synthetase (ASS) expression affects the antiproliferative activity of arginine deiminase (ADI) in melanoma cells. Oncol Rep 2011; 25: 1495–1502.
Jungbluth AA, Tassello J, Frosina D, Hanson N, Ritter G, Wu B-W et al. Expression pattern of argininosuccinate-synthetase (ASS) in normal and tumor tissue as a marker for susceptibility to arginine-deiminase (ADI) therapy. Mod Pathol 2010; 23: 387 A.
Savaraj N, Wu C, Li YY, Wangpaichitr M, You M, Bomalaski J . Targeting argininosuccinate synthetase negative melanomas using combination of arginine degrading enzyme and cisplatin. Oncotarget 2015; 6: 6295–6309.
Miyazaki K, Takaku H, Umeda M, Fujita T, Huang W, Kimura T et al. Potent growth inhibition of human tumor cells in culture by arginine deiminase purified from a culture medium of a Mycoplasma-infected cell line. Cancer Res 1990; 50: 4522–4527.
Sugimura K, Ohno T, Kusuyama T, Azuma I . High sensitivity of human melanoma cell lines to the growth inhibitory activity of mycoplasmal arginine deiminase in vitro. Melanoma Res 1992; 2: 191–196.
Takaku H, Misawa S, Hayashi H, Miyazaki K . Chemical modification by polyethylene glycol of the anti-tumor enzyme arginine deiminase from Mycoplasma arginini. Jpn J Cancer Res 1993; 84: 1195–1200.
Holtsberg FW, Ensor CM, Steiner MR, Bomalaski JS, Clark MA . Poly (ethylene glycol) (PEG) conjugated arginine deiminase: effects of PEG formulations on its pharmacological properties. J Control Release 2002; 80: 259–271.
Ensor CM, Holtsberg FW, Bomalaski JS, Clark MA . Pegylated arginine deiminase (ADI-SS PEG20,000mw) inhibits human melanomas and hepatocellular carcinomas in vitro and i n vivo. Cancer Res 2002; 62: 5443–5450.
Zhang L, Liu M, Jamil S, Han R, Xu G, Ni Y . PEGylation and pharmacological characterization of a potential anti-tumor drug, an engineered arginine deiminase originated from Pseudomonas plecoglossicida. Cancer Lett 2015; 357: 346–354.
Ni Y, Schwaneberg U, Sun ZH . Arginine deiminase, a potential anti-tumor drug. Cancer Lett 2008; 261: 1–11.
Yoon J, Frankel AE, Feun LG, Ekmekcioglu S, Kim KB . Arginine deprivation therapy for malignant melanoma. Clin Pharmacol 2013; 5: 11–19.
Glazer ES, Piccirillo M, Albino V, Di Giacomo R, Palaia R, Mastro AA et al. Phase II study of pegylated arginine deiminase for nonresectable and metastatic hepatocellular carcinoma. J Clin Oncol 2010; 28: 2220–2226.
Ascierto PA, Scala S, Castello G, Daponte A, Simeone E, Ottaiano A et al. Pegylated arginine deiminase treatment of patients with metastatic melanoma: results from phase I and II studies. J Clin Oncol 2005; 23: 7660–7668.
Kobayashi E, Masuda M, Nakayama R, Ichikawa H, Satow R, Shitashige M et al. Reduced argininosuccinate synthetase is a predictive biomarker for the development of pulmonary metastasis in patients with osteosarcoma. Mol Cancer Ther 2010; 9: 535–544.
Syed N, Langer J, Janczar K, Singh P, Lo Nigro C, Lattanzio L et al. Epigenetic status of argininosuccinate synthetase and argininosuccinate lyase modulates autophagy and cell death in glioblastoma. Cell Death Dis 2013; 4: e458.
Nicholson LJ, Smith PR, Hiller L, Szlosarek PW, Kimberley C, Sehouli J et al. Epigenetic silencing of argininosuccinate synthetase confers resistance to platinum-induced cell death but collateral sensitivity to arginine auxotrophy in ovarian cancer. Int J Cancer 2009; 125: 1454–1463.
Szlosarek PW, Klabatsa A, Pallaska A, Sheaff M, Smith P, Crook T et al. In vivo loss of expression of argininosuccinate synthetase in malignant pleural mesothelioma is a biomarker for susceptibility to arginine depletion. Clin Cancer Res 2006; 12: 7126–7131.
Rabinovich S, Adler L, Yizhak K, Sarver A, Silberman A, Agron S et al. Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature 2015; 527: 379–383.
Feun L, Savaraj N . Pegylated arginine deiminase: a novel anticancer enzyme agent. Expert Opin Investig Drugs 2006; 15: 815–822.
Bobak YP, Vynnytska BO, Kurlishchuk YV, Sibirny AA, Stasyk OV . Cancer cell sensitivity to arginine deprivation in vitro is not determined by endogenous levels of arginine metabolic enzymes. Cell Biol Int 2010; 34: 1085–1089.
Tsai WB, Aiba I, Lee SY, Feun L, Savaraj N, Kuo MT . Resistance to arginine deiminase treatment in melanoma cells is associated with induced argininosuccinate synthetase expression involving c-Myc/HIF-1α/Sp4. Mol Cancer Ther 2009; 8: 3223–3233.
Tsai WB, Aiba I, Long Y, Lin HK, Feun L, Savaraj N et al. Activation of Ras/PI3K/ERK pathway induces c-Myc stabilization to upregulate argininosuccinate synthetase, leading to arginine deiminase resistance in melanoma cells. Cancer Res 2012; 72: 2622–2633.
Long Y, Tsai W-B, Wangpaichitr M, Tsukamoto T, Savaraj N, Feun LG et al. Arginine deiminase resistance in melanoma cells is associated with metabolic reprogramming, glucose dependence and glutamine addiction. Mol Cancer Ther 2013; 12: 2581–2590.
Feun L, You M, Wu CJ, Kuo MT, Wangpaichitr M, Spector S et al. Arginine deprivation as a targeted therapy for cancer. Curr Pharm Des 2008; 14: 1049–1057.
Szlosarek PW, Luong P, Phillips MM, Baccarini M, Ellis S, Szyszko T et al. Metabolic response to pegylated arginine deiminase in mesothelioma with promoter methylation of argininosuccinate synthetase. J Clin Oncol 2013; 31: e111–e113.
Yang TS, Lu SN, Chao Y, Sheen IS, Lin CC, Wang TE et al. A randomised phase II study of pegylated arginine deiminase (ADI-PEG 20) in Asian advanced hepatocellular carcinoma patients. Br J Cancer 2010; 103: 954–960.
You M, Savaraj N, Wangpaichitr M, Wu C, Kuo TM, Varona-Santos J et al. The combination of ADI-PEG20 and TRAIL effectively increases cell death in melanoma cell lines. Biochem Biophys Res Commun 2010; 394: 760–766.
Feun LG, Wu G, Clark M, Bombalaski J, Holtsberg F, Wangpaijit M et al. Mechanism of anti-tumor effect of arginine deiminase-polyethylene (ADI-PEG20) and the possible mechanism of resistance in melanoma. Proc Am Assoc Cancer Res 2004; 45: 4565.
Wangpaichitr M, Wu C, Bigford G, Theodoropoulos G, You M, Li YY et al. Combination of arginine deprivation with TRAIL treatment as a targeted-therapy for mesothelioma. Anticancer Res 2014; 34: 6991–7000.
You M, Savaraj N, Kuo MT, Wangpaichitr M, Varona-Santos J, Wu C et al. TRAIL induces autophagic protein cleavage through caspase activation in melanoma cell lines under arginine deprivation. Mol Cell Biochem 2013; 374: 181–190.
Kim RH, Coates JM, Bowles TL, McNerney GP, Sutcliffe J, Jung JU et al. Arginine deiminase as a novel therapy for prostate cancer induces autophagy and caspase-independent apoptosis. Cancer Res 2009; 69: 700–708.
Kim RH, Bold RJ, Kung HJ . ADI, autophagy and apoptosis: metabolic stress as a therapeutic option for prostate cancer. Autophagy 2009; 5: 567–568.
Savaraj N, Wu C, Kuo MT, You M, Wangpaichitr M, Robles C et al. The relationship of arginine deprivation, argininosuccinate synthetase and cell death in melanoma. Drug Target Insights 2007; 2: 119–128.
Gong H, Zölzer F, Recklinghausen G, Havers W, Schweigerer L . Arginine deiminase inhibits proliferation of human leukemia cells more potently than asparaginase by inducing cell cycle arrest and apoptosis. Leukemia 2000; 14: 826–829.
Bowles TL, Kim R, Galante J, Parsons CM, Virudachalam S, Kung HJ et al. Pancreatic cancer cell lines deficient in argininosuccinate synthetase are sensitive to arginine deprivation by arginine deiminase. Int J Cancer 2008; 123: 1950–1955.
Surova O, Zhivotovsky B . Various modes of cell death induced by DNA damage. Oncogene 2013; 32: 3789–3797.
Changou CA, Chen Y-R, Xing L, Yen Y, Chuang FYS, Cheng RH et al. Arginine starvation-associated atypical cellular death involves mitochondrial dysfunction, nuclear DNA leakage, and chromatin autophagy. Proc Natl Acad Sci USA 2014; 111: 14147–14152.
Kung H-J, Changou CA, Li C-F, Ann DK . Chromatophagy: autophagy goes nuclear and captures broken chromatin during arginine-starvation. Autophagy 2015; 11: 419–421.
Gong H, Zölzer F, Recklinghausen GV, Rössler J, Breit S, Havers W et al. Arginine deiminase inhibits cell proliferation by arresting cell cycle and inducing apoptosis. Biochem Biophys Res Commun 1999; 261: 10–14.
Lorenzo HK, Susin SA . Mitochondrial effectors in caspase-independent cell death. FEBS Lett 2004; 557: 14–20.
Polster BM . AIF, reactive oxygen species, and neurodegeneration: a ‘‘complex’’ problem. Neurochem Int 2013; 62: 695–702.
Norberg E, Orrenius S, Zhivotovsky B . Mitochondrial regulation of cell death: processing of apoptosis-inducing factor (AIF). Biochem Biophys Res Commun 2010; 396: 95–100.
Zhu C, Wang X, Deinum J, Huang Z, Gao J, Modjtahedi N et al. Cyclophilin A participates in the nuclear translocation of apoptosis-inducing factor in neurons after cerebral hypoxia-ischemia. J Exp Med 2007; 204: 1741–1748.
Pradelli LA, Bénéteau M, Ricci J-E . Mitochondrial control of caspase-dependent and -independent cell death. Cell Mol Life Sci 2010; 67: 1589–1597.
Ulukaya E, Acilan C, Yilmaz Y . Apoptosis: why and how does it occur in biology? Cell Biochem Funct 2011; 29: 468–480.
Shen LJ, Beloussow K, Shen WC . Modulation of arginine metabolic pathways as the potential anti-tumor mechanism of recombinant arginine deiminase. Cancer Lett 2006; 231: 30–35.
Mandal S, Mandal A, Johansson HE, Orjalo AV, Park MH . Depletion of cellular polyamines, spermidine and spermine, causes a total arrest in translation and growth in mammalian cells. Proc Natl Acad Sci USA 2013; 110: 2169–2174.
Gerner EW, Meyskens FL Jr . Polyamines and cancer: old molecules, new understanding. Nat Rev Cancer 2004; 4: 781–792.
Ohmura M, Hishiki T, Yamamoto T, Nakanishi T, Kubo A, Tsuchihashi K et al. Impacts of CD44 knockdown in cancer cells on tumor and host metabolic systems revealed by quantitative imaging mass spectrometry. Nitric Oxide 2015; 46: 102–113.
Folkman J . What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst 1990; 82: 4–6.
Jain RK . Tumor angiogenesis and accessibility: role of vascular endothelial growth factor. Semin Oncol 2002; 29: 3–9.
Cao Z, Shang B, Zhang G, Miele L, Sarkar FH, Wang F et al. Tumor cell-mediated neovascularisation and lymphangiogenesis contrive tumor progression and cancer metastasis. Biochim Biophys Acta Rev Cancer 2013; 1836: 273–286.
Stapor P, Wang X, Goveia J, Moens S, Carmeliet P . Angiogenesis revisited – role and therapeutic potential of targeting endothelial metabolism. J Cell Sci 2014; 127: 4331–4341.
Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev 2011; 91: 1071–1121.
Cantelmo AR, Brajic A, Carmeliet P . Endothelial metabolism driving angiogenesis: emerging concepts and principles. Cancer J 2015; 21: 244–249.
Chamorro-Jorganes A, Lee MY, Araldi E, Landskroner-Eiger S, Fernández-Fuertes M, Sahraei M et al. VEGF-induced expression of miR-17~92 cluster in endothelial cells is mediated by ERK/ELK1 activation and regulates angiogenesis. Circ Res 2015; 118: 38–47.
Zhao D, Pan C, Sun J, Gilbert C, Drews-Elger K, Azzam DJ et al. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene 2015; 34: 3107–3119.
Kalucka J, Missiaen R, Georgiadou M, Schoors S, Lange C, De Bock K . Metabolic control of the cell cycle. Cell cycle 2015; 14: 3379–3388.
Moens S, Goveia J, Stapor PC, Cantelmo AR, Carmeliet P . The multifaceted activity of VEGF in angiogenesis-Implications for therapy responses. Cytokine Growth Factor Rev 2014; 25: 473–482.
Eelen G, Zeeuw P, Simons M, Carmeliet P . Endothelial cell metabolism in normal and diseased vasculature. Circ Res 2015; 116: 1231–1244.
Zecchin A, Stapor PC, Goveia J, Carmeliet P . Metabolic pathway compartmentalization: an underappreciated opportunity? Curr Opin Biotech 2015; 34: 73–81.
Beloussow K, Wang L, Wu J, Ann D, Shen W-C . Recombinant arginine deiminase as a potential anti-angiogenic agent. Cancer Lett 2002; 183: 155–162.
Park I-S, Kang S-W, Shin Y-J, Chae K-Y, Park M-O, Kim M-Y et al. Arginine deiminase: a potential inhibitor of angiogenesis and tumor growth. Br J Cancer 2003; 89: 907–914.
Thomas JB, Holtsberg FW, Ensor CM, Bomalaski JS, Clark MA . Enzymic degradation of plasma arginine using arginine deiminase inhibits nitric oxide production and protects mice from the lethal effects of tumor necrosis factor α and endotoxin. Biochem J 2002; 363: 581–587.
Noh EJ, Kang SW, Shin YJ, Kim DC, Park I-S, Kim MY et al. Characterization of Mycoplasma arginini deiminase expressed in E. coli and its inhibitory regulation of nitric oxide synthesis. Mol Cells 2002; 13: 137–143.
Fraisl P . Crosstalk between oxygen- and nitric oxide-dependent signaling pathways in angiogenesis. Exp Cell Res 2013; 319: 1331–1339.
Morbidelli L, Donnini S, Ziche M . Role of nitric oxide in tumor angiogenesis. Cancer Treat Res 2004; 117: 155–167.
Jurasz P, Sawicki G, Duszyk M, Sawicka J, Miranda C, Mayers I et al. Matrix metalloproteinase 2 in tumor cell-induced platelet aggregation: regulation by nitric oxide. Cancer Res. 2001; 61: 376–382.
Carreau A, Kieda C, Grillon C . Nitric oxide modulates the expression of endothelial cell adhesion molecules involved in angiogenesis and leukocyte recruitment. Exp Cell Res 2011; 317: 29–41.
Fukumura D, Kashiwagi S, Jain RK . The role of nitric oxide in tumour progression. Nat Rev Cancer 2006; 6: 521–534.
Lee MY, Luciano AK, Ackah E, Rodriguez-Vita J, Bancroft TA, Eichmann A et al. Endothelial Akt1 mediates angiogenesis by phosphorylating multiple angiogenic substrates. Proc Natl Acad Sci USA 2014; 111: 12865–12870.
Fukumura D, Gohongi T, Kadambi A, Izumi Y, Ang J, Yun CO et al. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc Natl Acad Sci USA 2001; 98: 2604–2609.
Kashiwagi S, Izumi Y, Gohongi T, Demou ZN, Xu L, Huang PL et al. NO mediates mural cell recruitment and vessel morphogenesis in murine melanomas and tissue-engineered blood vessels. J Clin Invest 2005; 115: 1816–1827.
Kashiwagi S, Tsukada K, Xu L, Miyazaki J, Kozin SV, Tyrrell JA et al. Perivascular nitric oxide gradients normalize tumor vasculature. Nat Med 2008; 14: 255–257.
Roberts DD, Isenberg JS, Ridnour LA, Wink DA . Nitric oxide and its gatekeeper thrombospondin-1 in tumor angiogenesis. Clin Cancer Res 2007; 13: 795–798.
McAlpine JA, Lu H-T, Wu KC, Knowles SK, Thomson JA . Down-regulation of argininosuccinate synthetase is associated with cisplatin resistance in hepatocellular carcinoma cell lines: implications for PEGylated arginine deiminase combination therapy. BMC Cancer 2014; 14: 621.
Liu J, Ma J, Wu Z, Li W, Zhang D, Han L et al. Arginine deiminase augments the chemosensitivity of argininosuccinate synthetase-deficient pancreatic cancer cells to gemcitabine via inhibition of NF-κB signaling. BMC Cancer 2014; 14: 686.
Allen MD, Luong P, Hudson C, Leyton J, Delage B, Ghazaly E et al. Prognostic and therapeutic impact of argininosuccinate synthetase 1 control in bladder cancer as monitored longitudinally by PET imaging. Cancer Res 2013; 74: 896–907.
Daylami R, Muilenburg DJ, Virudachalam S, Bold RJ . Pegylated arginine deiminase synergistically increases the cytotoxicity of gemcitabine in human pancreatic cancer. J Exp Clin Cancer Res 2014; 33: 102.
Jenkinson CP, Grody WW, Cederbaum SD . Comparative properties of arginases. Comp Biochem Physiol 1996; 114B: 107–132.
Wu G, Morris SM Jr . Arginine metabolism: nitric oxide and beyond. Biochem J 1998; 336: 1–17.
Morris SM Jr . Regulation of enzymes of urea and arginine synthesis. Annu Rev Nutr 1992; 12: 81–101.
Gotoh T, Sonoki T, Nagasaki A, Tereda K, Takiguchi M, Mori M . Molecular cloning of cDNA for nonhepatic mitochondrial arginase (arginase II) and comparison of its induction with nitric oxide synthase in a murine macrophage-like cell line. FEBS Lett 1996; 395: 119–122.
Elms S, Chen F, Wang Y, Qian J, Askari B, Pandey D et al. Insights into the arginine paradox: evidence against the importance of subcellular location of arginase and eNOS. Am J Physiol Heart Circ Physiol 2013; 305: H651–H666.
Morris SM Jr . Arginine metabolism: boundaries of our knowledge. J Nutr 2007; 137: 1602S–1609S.
Li H, Meininger CJ, Hawker JR Jr, Haynes TE, Kepka-Lenhart D, Mistry SK et al. Regulatory role of arginase I and II in nitric oxide, polyamine, and proline syntheses in endothelial cells. Am J Physiol Endocrinol Metab 2001; 280: E75–E82.
Bach SJ, Lasnitzki I . Some aspects of the role of arginine and arginase in mouse carcinoma. Enzymologia 1947; 12: 198–205.
Bach SJ, Maw GA . Creatine synthesis by tumor-bearing rats. Biochim Biophys Acta 1953; 11: 69–78.
Koji T, Terayama H . Arginase as one of the inhibitory principles in the density-dependent as well as plasma membrane-mediated inhibition of liver cell growth in vitro. Exp Cell Res 1984; 155: 359–370.
Terayama H, Koji T, Kontani M, Okumoto T . Arginase is an inhibitory principle in liver growth of various mammalian cells in vitro Plasma membranes arresting the growth of various mammalian cells in vitro. Biochim Biophys Acta 1982; 720: 188–192.
Huang M-H, Yang C-C, Wang C-C . Inhibition of lymphocyte proliferation by liver arginase. Life Sci 1992; 51: 1725–1730.
Cheng PNM, Lam TL, Lam WM, Tsui SM, Cheng AWM, Lo WH et al. Pegylated recombinant human arginase (rharg-peg 5,000mw) inhibits the in vitro and in vivo proliferation of human hepatocellular carcinoma through arginine depletion. Cancer Res 2007; 67: 309–317.
Wheatley DN, Campbell E . Arginine deprivation, growth inhibition and tumor cell death: 3. Deficient utilisation of citrulline by malignant cells. Br J Cancer 2003; 89: 573–576.
Tsui SM, Lam WM, Lam TL, Chong HC, So PK, Kwok SY et al. Pegylated derivatives of recombinant human arginase (rhArg1) for sustained in vivo activity in cancer therapy: preparation, characterization and analysis of their pharmacodynamics in vivo and in vitro and action upon hepatocellular carcinoma cell (HCC). Cancer Cell Int 2009; 9: 9.
Stone EM, Glazer ES, Chantranupong L, Cherukuri P, Breece RM, Tierney DL et al. Replacing Mn2+ with Co2+ in human arginase I enhances cytotoxicity toward L-arginine auxotrophic cancer cell lines. ACS Chem Biol 2010; 5: 333–342.
Glazer ES, Stone EM, Zhu C, Massey KL, Hamir AN, Curley SA . Bioengineered human arginase I with enhanced activity and stability controls hepatocellular and pancreatic carcinoma xenografts. Transl Oncol 2011; 4: 138–146.
Tanios R, Bekdash A, Kassab E, Stone E, Georgiou G, Frankel AE et al. Human recombinant arginase I(Co)-PEG5000 [HuArgI(Co)-PEG5000]-induced arginine depletion is selectively cytotoxic to human acute myeloid leukemia cells. Leukemia Res 2013; 37: 1565–1571.
Khoury O, Ghazale N, Stone E, El-Sibai M, Frankel AE, Abi-Habib RJ . Human recombinant arginase I (Co)-PEG5000 [HuArgI(Co)-PEG5000]-induced arginine depletion is selectively cytotoxic to human glioblastoma cells. J Neurooncol 2015; 122: 75–85.
Li L, Wang Y, Chen J, Cheng B, Hu J, Zhou Y et al. An engineered arginase FC protein inhibits tumor growth in vitro and in vivo. Evid Based Complement Alternat Med 2013; 2013: 423129.
Lam TL, Wong GKY, Chow HY, Chong HC, Chow TL, Knok SY et al. Recombinant human arginase inhibits the in vitro and in vivo proliferation of human melanoma by inducing cell cycle arrest and apoptosis. Pigment Cell Melanoma Res 2010; 24: 366–376.
Lam TL, Wong GKY, Chong HC, Cheng PNM, Choi SC, Chow TL et al. Recombinant human arginase inhibits proliferation of human hepatocellular carcinoma by inducing cell cycle arrest. Cancer Lett 2009; 277: 91–100.
Yau T, Cheng PNM, Chan P, Chan W, Chen L, Yuen J et al. A phase 1 dose-escalating study of pegylated recombinant human arginase 1 (Peg-rhArg1) in patients with advanced hepatocellular carcinoma. Invest New Drugs 2013; 31: 99–107.
Yau CC, Chan P, Pang R, Chan W, Cheng PNM, Poon R . A phase I study of recombinant human arginase I (rhArgI) for patients with advanced hepatocellular carcinoma. J Clin Oncol ASCO Annual Meeting Abstracts 2010; 28: e13503.
Ferraro E, Cecconi F . Autophagic and apoptotic response to stress signals in mammalian cells. Arch Biochem Biophys 2007; 462: 210–219.
Jiang P, Mizushima N . Autophagy and human diseases. Cell Res 2014; 24: 69–79.
Gozuacik D, Kimchi A . Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004; 23: 2891–2906.
Macintosh RL, Ryan KM . Autophagy in tumour cell death. Semin Cancer Biol 2013; 23: 344–351.
Fulda S, Köge D . Cell death by autophagy: emerging molecular mechanisms and implications for cancer therapy. Oncogene 2015; 34: 5105–5113.
Denton D, Nicolson S, Kumar S . Cell death by autophagy: facts and apparent artefacts. Cell Death Differ 2012; 19: 87–95.
Shen H-M, Codongo P . Autophagic cell death: Loch Ness monster or endangered species? Autophagy 2011; 7: 457–465.
Efeyan A, Zoncu R, Sabatini DM . Amino acids and mTORC1: from lysosomes to disease. Trends Mol Med 2012; 18: 524–533.
Beauchamp EM, Platanias LC . The evolution of the TOR pathway and its role in cancer. Oncogene 2013; 32: 3923–3932.
Yan L, Lamb RF . Amino acid sensing and regulation of mTORC1. Semin Cell Dev Biol 2012; 23: 621–625.
Ryter SW, Cloonan SM, Choi AMK . Autophagy: a critical regulator of cellular metabolism and homeostasis. Mol Cells 2013; 36: 7–16.
Cui J, Gong Z, Shen H-M . The role of autophagy in liver cancer: molecular mechanisms and potential therapeutic targets. Biochim Biophys Acta Rev Cancer 2013; 1836: 15–26.
Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, Avruch J . Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J Biol Chem 1998; 273: 14484–14494.
Hsueh EC, Knebel SM, Lo W-H, Leung Y-C, Cheng PNM, Hsueh CT . Deprivation of arginine by recombinant human arginase in prostate cancer cells. J Hematol Oncol 2012; 5: 17–22.
Scott L, Lamb J, Smith S, Wheatley DN . Single amino acid (arginine) deprivation: rapid and selective death of cultured transformed and malignant cells. Br J Cancer 2000; 83: 800–810.
Wang Y, Nartiss Y, Steipe B, McQuibban GA, Kim PK . ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy 2012; 8: 1462–1476.
Li Z-Y, Yang Y, Ming M, Liu B, Mitochondrial ROS . generation for regulation of autophagic pathways in cancer. Biochem Biophys Res Commun 2011; 414: 5–8.
Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB . Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ 2008; 15: 171–182.
Wang Z, ShiX LiY, Zeng X, Fan J, Sun Y et al. Involvement of autophagy in recombinant human arginase-induced cell apoptosis and growth inhibition of malignant melanoma cells. Appl Microbiol Biotechnol 2014; 98: 2485–2494.
Wu WKK, Coffelt SB, Cho CH, Wang XJ, Lee CW, Chan FKL et al. The autophagic paradox in cancer therapy. Oncogene 2012; 31: 939–953.
Rebsamen M, Pochini L, Stasyk T, de Arau'jo MEG, Galluccio M, Kandasamy RK et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 2015; 519: 477–481.
Wang S, Tsun Z-Y, Wolfson RL, Shen K, Wyant GA, Plovanich ME et al. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 2015; 347: 188–194.
Eng CH, Abraham RT . The autophagy conundrum in cancer: influence of tumorigenic metabolic reprogramming. Oncogene 2011; 30: 4687–4696.
Pérez E, Das G, Bergmann A, Baehrecke EH . Autophagy regulates tissue overgrowth in a context-dependent manner. Oncogene 2015; 34: 3369–3376.
Tsujimoto Y, Shimizu S . Another way to die: autophagic programmed cell death. Cell Death Differ 2005; 12: 1528–1534.
Wang Z, Shi X, Li Y, Fan J, Zeng X, Xian Z et al. Blocking autophagy enhanced cytotoxicity induced by recombinant human arginase in triple-negative breast cancer cells. Cell Death Dis 2014; 5: e1563.
Booth LA, Tavallai S, Hamed HA, Cruickshanks N, Dent P . The role of cell signalling in the crosstalk between autophagy and apoptosis. Cellular Signal 2014; 26: 549–555.
Fimia GM, Corazzari M, Antonioli M, Piacentini M . Ambra1 at the crossroad between autophagy and cell death. Oncogene 2013; 32: 3311–3318.
Djavaheri-Mergny M, Maiuri MC, Kroemer G . Cross talk between apoptosis and autophagy by caspase-mediated cleavage of Beclin 1. Oncogene 2010; 29: 1717–1719.
Hernandez CP, Morrow K, Lopez-Barcons LA, Zabaleta J, Sierra R, Velasco C et al. Pegylated arginase I: a potential therapeutic approach in T-ALL. Blood 2010; 115: 5214–5221.
Morrow K, Hernandez CP, Raber P, Del Valle L, Wilk AM, Majumdar S et al. Anti-leukemic mechanisms of pegylated arginase I in acute lymphoblastic T-cell leukemia. Leukemia 2013; 27: 569–577.
Chow AK, Ng L, Sing LiH, Cheng CW, Lam CS, Yau TC, Cheng PN et al. Anti-tumor efficacy of a recombinant human arginase in human hepatocellular carcinoma. Curr Cancer Drug Targets 2012; 12: 1233–1243.
Marino T, Russo N, Toscano M . What occurs by replacing Mn2+ with Co2+ in human arginase I: first- principles computational analysis. Inorg Chem 2013; 52: 655–659.
Glazer ES, Kaluarachchi WD, Massey KL, Zhu C, Curley SA . Bioengineered arginase I increases caspase-3 expression of hepatocellular and pancreatic carcinoma cells despite induction of argininosuccinate synthetase-1. Surgery 2010; 148: 310–318.
Regunathan S, Reis DJ . Characterization of arginine decarboxylase in rat brain and liver: distinction from ornithine decarboxylase. J Neurochem 2000; 74: 2201–2208.
Li G, Regunathan S, Barrow CJ, Eshraghi J, Cooper R, Reis DJ . Agmatine: an endogenous clonidine-displacing substance in the brain. Science 1994; 263: 966–969.
Zhu MY, Iyo A, Piletz JE, Regunathan S . Expression of human arginine decarboxylase, the biosynthetic enzyme for agmatine. Biochim Biophys Acta 2004; 1670: 156–164.
Molderings GJ, Haenisch B . Agmatine (decarboxylated l-arginine): physiological role and therapeutic potential. Pharmacol Ther 2012; 133: 351–365.
Dudkowska M, Lai J, Gardini G, Stachurska A, Grzelakowska-Sztabert B, Colombatto S et al. Agmatine modulates the in vivo biosynthesis and interconversion of polyamines and cell proliferation. Biochim Biophys Acta 2003; 1619: 159–166.
Satriano J, Matsufuji S, Murakami Y, Lortie MJ, Schwartz D, Kelly CJ et al. Agmatine suppresses proliferation by frameshift induction of antizyme and attenuation of cellular polyamine levels. J Biol Chem 1998; 273: 15313–15316.
Choi YS, Cho YD . Effects of agmatine on polyamine metabolism and the growth of prostate tumor cells. J Biochem Mol Biol 1999; 32: 173–180.
Mayeur C, Veuillet G, Michaud M, Raul F, Blottière H, Blachier F . Effects of agmatine accumulation in human colon carcinoma cells on polyamine metabolism, DNA synthesis and the cell cycle. Biochim Biophys Acta 2005; 1745: 111–123.
Moinard C, Cynober L, Bandt JPD . Polyamines: metabolism and implications in human diseases. Clin Nutr 2005; 24: 184–197.
Satriano J . Arginine pathways and the inflammatory response: interregulation of nitric oxide and polyamines. Amino Acids 2004; 26: 321–329.
Philip R, Campbell E, Wheatley DN . Arginine deprivation, growth inhibition and tumor cell death: 2. Enzymatic degradation of arginine in normal and malignant cell cultures. Br J Cancer 2003; 88: 613–623.
Wheatley DN, Scott L, Lamb J, Smith S . Single amino acid (arginine) restriction: growth and death of cultured HeLa and human diploid fibroblasts. Cell Physiol Biochem 2000; 10: 37–55.
Wheatley DN, Campbell E . Arginine catabolism, liver extracts and cancer. Pathol Oncol Res 2002; 8: 18–25.
Bronte V, Zanovello P . Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol 2005; 5: 641–654.
Sikalidis AK . Amino acids and immune response: a role for cysteine, glutamine, phenylalanine, tryptophan and arginine in T-cell function and cancer? Pathol Oncol Res 2015; 21: 9–17.
Raber P, Ochoa AC, Rodriguez PC . Metabolism of L-arginine by myeloid-derived suppressor cells in cancer: Mechanisms of T-cell suppression and therapeutic perspectives. Immunol Invest 2012; 41: 614–634.
Peranzoni E, Marigo I, Dolcetti L, Ugel S, Sonda N, Taschin E et al. Role of arginine metabolism in immunity and immunopathology. Immunobiol 2008; 212: 795–812.
Popovic PJ, Zeh HJ III, Ochoa JB . Arginine and immunity. J Nutr 2007; 137: 1681s–1686s.
Wheatley DN, Campbell E, Lai PBS, Cheng PNM . A rational approach to the systemic treatment of cancer involving medium-term depletion of arginine. Gene Ther Mol Biol 2005; 9: 33–40.
Kilberg MS, Balasubramanian M, Fu L, Shan J . The transcription factor network associated with the amino acid response in mammalian cells. Adv Nutr 2012; 3: 295–306.
Jackson RJ, Hellen CU, Pestova TV . The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 2010; 11: 113–127.
Zang H, Forman HJ . Glutathione synthesis and its role in redox signaling. Semin Cell Dev Biol 2012; 23: 722–728.
Weiger TM, Hermann A . Cell proliferation, potassium channels, polyamines and their interactions: a mini review. Amino Acids 2014; 46: 681–688.
Lind D . Arginine and cancer. J Nutr 2004; 134: 2837s–2841s.
Kohler ES, Sankaranarayanan S, Van Ginneken CJ, Van Dijk P, Vermeulen JLM, Ruijter JM et al. The human neonatal small intestine has the potential for arginine synthesis; developmental changes in the expression of arginine-synthesizing and catabolizing enzymes. BMC Dev Biol 2008; 8: 107.
Kim JE, Kim SY, Lee KW, Lee HJ . Arginine deiminase originating from Lactobacillus lactis ssp. lactis American Type Culture Collection (ATCC) 7962 induces G1phase cellcycle arrest and apoptosis in SNU1 stomach adenocarcinoma cells. Br J Nutr 2003; 102: 1469–1476.
Gill P, Pan J . Inhibition of cell division in L5178Y cells by arginine-degrading mycoplasmas: the role of arginine deiminase. Can J Microbiol 1970; 16: 415–419.
Kim JH, Kim JH, Yu YS, Kim DH, Min BH, Kim KW . Anti-tumor activity of arginine deiminase via arginine deprivation in retinoblastoma. Oncol Rep 2007; 18: 1373–1377.
Huang CC, Tsai ST, Kuo CC, Chang JS, Jin YT, Chang JY et al. Arginine deprivation as a new treatment strategy for head and neck cancer. Oral Oncol 2012; 48: 1227–1235.
Tan B, Yin Y, Kong X, Li P, Li X, Gao H et al. L-Arginine stimulates proliferation and prevents endotoxin-induced death of intestinal cells. Amino Acids 2010; 38: 1227–1235.
Wheatley DN, Philip R, Campbell E . Arginine deprivation and tumor cell death: arginase and its inhibition. Mol Cell Biochem 2003; 244: 177–185.
Izzo F, Marra P, Beneduce G, Castello G, Vallone P, De Rosa V et al. Pegylated arginine deiminase treatment of patients with unresectable hepatocellular carcinoma: results from phase I/II studies. J Clin Oncol 2004; 22: 1815–1822.
Feun LG, You M, Wu C, Wangpaichitr M, Kuo MT, Marini A et al. Final results of phase II trial of pegylated arginine deiminase (ADI-PEG20) in metastatic melanoma (MM) [abstract]. J Clin Oncol 2010; 28: 8528.
Ott PA, Carvajal RD, Pandit-Taskar N, Jungbluth AA, Hoffman EW, Wu BW et al. Phase I/II study of pegylated arginine deiminase (ADI-PEG 20) in patients with advanced melanoma. Invest New Drugs 2013; 31: 425–434.
Szlosarek PW, Steele J, Sheaff M, Szyszko T, Ellis S, Nolan L A randomised phase II trial of pegylated arginine deiminase in patients with malignant pleural mesothelioma. 15th World Conference on Lung Cancer, Sydney, Australia, 2013, Abstr no. MO09.02.
Tomlinson BK, Bomalaski JS, Diaz M, Akande T, Mahaffey N, Li T et al. Phase I trial of ADI-peg 20 plus docetaxel (DOC) in patients (pts) with advanced solid tumors. J Clin Oncol (ASCO Meeting Abstracts) 2013; 31: 2569.
Tomlinson BK, Thomson JA, Bomalaski JS, Diaz M, Akande T, Mahaffey N et al. Phase I trial of arginine deprivation therapy with ADI-PEG 20 plus Docetaxel in patients with advanced malignant solid tumors. Clin Cancer Res 2015; 21: 2480–2486.
Bach SJ, Swaine D . The effect of arginase on the retardation of tumor growth. Br J Cancer 1965; 19: 379–386.
Currie GA . Activated macrophages kill tumor cells by releasing arginase. Nature 1978; 273: 758–759.
Acknowledgements
MP gratefully acknowledges Department of Biotechnology (DBT), New Delhi, India for the award of Senior Research Fellowship. We are thankful to Professor Rakesh K Jain, Department of Radiation Oncology, Massachusetts General Hospital, Harvard Medical School, USA; Dr Utpal Mohan, Department of Biotechnology, NIPER, Guwahati, India; and Dr Umesh Patil, School of Chemical Sciences, North Maharashtra University, Maharashtra, India for the assistance provided during preparation of the manuscript. We apologize to those authors whose work could not be cited owing to space limitations. Funding: This work was supported in part by National Institutes of Health (CA080124, CA096915, CA126642, CA197743).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Patil, M., Bhaumik, J., Babykutty, S. et al. Arginine dependence of tumor cells: targeting a chink in cancer’s armor. Oncogene 35, 4957–4972 (2016). https://doi.org/10.1038/onc.2016.37
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2016.37
- Springer Nature Limited
This article is cited by
-
FOXO3a-regulated arginine metabolic plasticity adaptively promotes esophageal cancer proliferation and metastasis
Oncogene (2024)
-
Effects of Exercise Training and L-Arginine Loaded Chitosan Nanoparticles on Hippocampus Histopathology, β-Secretase Enzyme Function, APP, Tau, Iba1and APOE-4 mRNA in Aging Rats
Neurotoxicity Research (2024)
-
Arginase-1 inhibition reduces migration ability and metastatic colonization of colon cancer cells
Cancer & Metabolism (2023)
-
Integrative analysis of transcriptomic and metabolomic profiles reveals enhanced arginine metabolism in androgen-independent prostate cancer cells
BMC Cancer (2023)
-
Impact of context-dependent autophagy states on tumor progression
Nature Cancer (2023)