
Abstract
Intercellular communication sets the pace for transformed cells to survive and to thrive. Extracellular vesicles (EVs), such as exosomes, microvesicles and large oncosomes, are involved in this process shuttling reciprocal signals and other molecules between transformed and stromal cells, including fibroblasts, endothelial and immune cells. As a result, these cells are adapted or recruited to a constantly evolving cancer microenvironment. Moreover, EVs take part in the response to anticancer therapeutics not least by promoting drug resistance throughout the targeted tumor. Finally, circulating EVs can also transport important molecules to remote destinations in order to prime metastatic niches in an otherwise healthy tissue. Although the understanding of EV biology remains a major challenge in the field, their characteristics create new opportunities for advances in cancer diagnostics and therapeutics.
Similar content being viewed by others
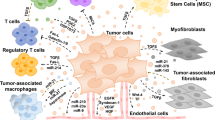
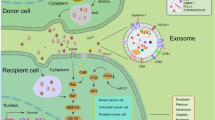

EVs at the interface of stromal communication
Instigated by malignant cells the surrounding stroma undergoes a shake-up in its organization that supports cancerous growth. Crucial parts of this self-organization process include induction of metabolic changes, modifications of cell identities, initiation of neo-vascularization and reprogramming of inert immune cells. In order to achieve these defining properties of the tumor microenvironment, cancer and non-cancer cells continuously exchange information brought together through cell–cell traversing gap junctions, tunneling nanotubes and the secretion of effector molecules. One way to guarantee coordinated release of multiple ‘game’-changing molecules relies on their packaging into membrane-enclosed vesicles widely known as extracellular vesicles (EVs). ‘EVs’ is a general term coined to denominate vesicle carriers that in fact hugely differ in their subcellular origin (Figure 1). They contain cargo such as lipids, proteins, various RNAs and DNA fragments and metabolic products. EVs may shuttle these molecules between neighboring cells or via systemic transport to distant anatomic sites where they may induce signaling pathways or directly alter the phenotype of specified recipient cells.

EV biogenesis. EVs can form from the endomembrane system or through budding/blebbing from the plasma membrane. The best-described pathway for the production of exosomes starts at the plasma membrane through endocytosis at cholesterol-enriched lipid raft domains. The subsequently generated early endosomes (EE) fuse in a number of fusion events and concomitantly mature to late endosomes (LE) that can then form intraluminal vesicles (ILVs) by invaginations and pinching of the limiting membrane. The product is referred to as a multi-vesicular body (MVB). MVBs are then either destined for the fusion with the lysosomal compartment leading to cargo degradation or tagged for fusion with the plasma membrane, thereby releasing ILVs, thereafter called exosomes. The orchestrated redistribution of membrane lipids, sphingosine metabolites84, 85 and/or the endosomal sorting complex required for transport machinery86 have been reported to have crucial functions in exosomes and microvesicle biogenesis. Large oncosomes derived by membrane blebbing can be artificially induced through knockdown of the cytoskeletal protein DIAPH3.26 CCV, clathrin coated vesicles; PM, plasma membrane.
One kind of EVs finds its origin in secretory multi-vesicular bodies that fuse with the plasma membrane-releasing intraluminal vesicles, thereafter called exosomes (50–150nm in diameter). Another kind of EVs derive from vesicle budding at the plasma membrane. These are commonly called microvesicles and are more heterogeneous in size (>100nm–1μm in diameter). Finally, large oncosomes (>1μm) have been described that differ in their buoyant density from the aforementioned vesicle types, which are produced by plasma membrane blebbing (reviewed in Abels and Breakefield1 and Colombo et al.2). All of those nanovesicles can be found in and isolated from conditioned tissue culture medium of cancer and stromal cells but also from diverse body fluids such as cerebrospinal liquid, breast milk, urine or blood plasma.
Owing to their cargo specificity and their easy sourcing, circulating EVs are being evaluated for the early diagnosis of various cancers. Indeed, EV cargo such as survivin may serve as a marker for the early diagnosis of prostate cancer,3 caveolin-1 for melanoma,3 Glypican-1 for early pancreatic cancers,4 and various miRNA profiles in colorectal5 and lung cancer.6
EVs have recently also been implicated as direct mediators of the response of solid tumors to cytotoxic chemotherapy, and as putative ‘real-time’ biomarkers to assess individual drug responses. The evidence demonstrating modulation of drug sensitivity has centered on the EV-mediated transfer of proteins, mRNAs and miRNAs with the capacity to influence key antiapoptotic or proliferative pathways between tumor cells or from the endothelium to tumor cells (see below).
Navigating across these different aspects, this review will focus on the latest functional insights that EVs bear in intercellular communication during cancer progression.
Modulation of EV composition
Both exogenous as well as endogenous factors can modulate type, content and the number of released EVs. As discussed in more detail further below, hypoxia appears to be a strong driving force in the enhancement of EV shedding, resulting in pro-angiogenic effects. Furthermore, intratumoral hypoxic conditions augment microvesicle release leading to increased risks of metastasis and mortality in patients with advanced breast cancer.7 PH changes in the tumor microenvironment can also contribute to changes of the lipid composition of EVs.8 In addition, the cellular stress-regulated protein TSAP6 that is under the control of the p53 tumor suppressor was shown to enhance exosome production with possible effects on adjacent cells and the immune system.9 Although our understanding of changes observed in EV composition under different physiological conditions is still minimal, they nevertheless may pave the way to novel, exciting avenues in diagnosis and treatment of cancers.
In breast cancer for instance, the overexpression of oncogenes such as ERBB2/HER2 in the mammary luminal epithelial cell line (HB4a) can shift the bias of EV content towards a malignant phenotype, as defined by the detection of oncodriver signaling components, including HER2, cell adhesion and cytoskeleton-remodeling components and sphingosine-1-phosphate.10 Similarly, oncogenic Ras-transformed NIH3T3 cells showed an increase of over 34 proteins in EVs, including milk fat globule EGF factor 8 (lactadherin), collagen alpha-1 (VI), 14-3-3 isoforms, guanine nucleotide-binding proteins (G proteins), the eukaryotic translation initiation factors elF-3gamma and elF-5A accumulated (>2-fold).11 Mutated KRAS in colon cancer cells has also been reported to effect EV cargo composition towards tumor-promoting factors including mutated KRAS itself as well as EGFR, SRC family kinases and integrins, when compared to its isogenically matched wild-type KRAS cells.12 Importantly, mutant cell-line-derived EVs positively enhanced cell growth of wild-type cells.12 Another oncogene, the melanoma cells-secreted Wnt5A, was also reported to induce the release of EVs.13 Finally, tumorigenic viruses such as EBV can manipulate the secretion of EV-bound cellular components, namely integrins, actin, IFN and nuclear factor κB that subsequently activate cellular signaling in the surrounding stroma.14
Although we now have evidence that oncogenes can directly modify cargo load, the knowledge of its consequences is still stuck in its infancy.
EVs reprogram cancer cell metabolism
The development of cancers as a multi-stage process is often ignored in in vitro studies. As a result, we obtain a picture of cancer signaling and oncodriver activity that is blind to the spatiotemporal context of our observations, leaving us with the egg-chicken problem. EV composition presumably reflects the cellular physiology of their parent cells and can transport ‘seeding’ information to recipient cells. This implies that EVs carry the capacity to reprogram the cellular metabolism and re-wire cellular interactions (Figure 2). Therefore, EVs provide the rare opportunity to analyze the direct and causal effect that fractionated information has on oncogenic transformation.

Schematic representation of the flow of information regulated by EVs and large oncosomes (LOs) during metabolic reprogramming. EVs from glycolytic cancer cells can contain information that is fed to malignant or non-transformed cells (of cancer or stromal origin) and cause metabolic changes. For instance, significant alterations can be induced in CAFs that in turn respond by the release of EVs containing sufficient material to sustain the cancer cell metabolism. This intercellular reprogramming evidences the dependency between the tumor and its adapted microenvironment, whereby EVs can be seen as outsourced ‘investments’ undertaken to deliver metabolites and other material that promote tumor growth.
In this context, it is useful to understand that during the lifetime of a solid tumor its cells are subjected to enormous microenvironmental shifts, some of which are large enough to induce permanent transformations, may these be post-transcriptional and/or epigenetic or indeed metabolic, such as the Warburg effect. Additionally, during cancer development cell populations become increasingly heterogeneous. The extent to which an initial population is clonally diverse is still under debate; however, a hostile environment prompts malignant cells to adapt, primarily, by changes to their metabolic profiles, thus reprogramming the energetics of biosynthesis. For instance, the effect of hypoxia on hypoxia-inducible factor 1α, carbonic anhydrases (such as CAIX), the sodium/proton exchanger NHE1 and the glucose transporter Glut1 have been portrayed exquisitely in most solid tumors and paved the way for the discovery of metabolite import/export pumps demonstrating cancer cell plasticity by recycling their ‘waste material’. The best understood of such systems is provided by the proton-lactate symporters belonging to the family of monocarboxylate transporters15, 16 and their co-chaperone, the glycoprotein CD147.17 Under regimes of high glycolytic flux, lactic acid is initially exported in response to intracellular pH regulators. These alter cellular acid export providing the cell with an alkaline pH that in turn favors glycolysis and the import of glucose. However, the acidic burden resulting from glycolysis can eventually result in toxicity prompting the emergence of invasive cells.18 Lactate can then be re-imported through the monocarboxylate transporters, a process known as lactate shuttling, and used as a source of energy in OXPHOS active cells via the lactate dehydrogenases (LDHA/LDHB) that convert lactate to pyruvate.19 Therefore, it is of great interest that exosomes have been shown to contain high levels of Glut1, MCT4 and CD147 as well as reduced phosphoglycerate kinase levels20 because this finding appears consistent with the key elements characterizing the ‘reverse Warburg effect’ shown to occur in stromal cells. In this scenario, metabolic EV content could ‘highjack’ the existing cellular program and re-wire it, presumably mimicking the cell of origin. The uptake and release of EVs is considered an energetically unfavorable event; cancer cells notoriously show reduced or lack of OXPHOS-derived ATP, elicit increased reliance on glycolysis, the pentose phosphate pathway and alternative energy sources such as lactate and acetate. However, recent evidence has shown that EVs originating from prostate cancer cells can actually produce ATP from glycolysis and show reduced ATPase activity, when compared with EV populations released by normal prostate tissue (or prostasomes),20 making their reception, rather than their release, the energetically favorable event. In many ways, EV formation by cancer cells appears more similar to an energetic investment made towards future re-homing by outsourcing their energy requirements. It would be of significant interest, and presumably possible, to re-engineer this machinery in the opposite direction and deliver tumor suppressor information from the microenvironment (such as fibroblasts, T-lymphocytes or neutrophils) to the cancer cells. Instead, cancer-associated fibroblasts (CAFs)-derived EVs shuttle a range of metabolites to prostate and pancreatic cells, including lactate, glutamine, lipids, tricarboxylic acid cycle intermediates, resulting in reduced OXPHOS and increased reliance on glutamine and glycolysis.21 This is at odds with the current understanding of metabolic reprogramming being an autonomous event occurring in cancer cells in response to nutrient deprivation. In this light, it appears that metabolic re-wiring is enhanced and could even be initiated by the tumor microenvironment, questioning much of the theoretical framework elaborated to explain malignant transformation and progression.
KRAS activating mutations have been associated with oncodriver activity along the MAPK signaling pathway and have recently been shown to drive a glycolytic switch in NSCLC cells.22 During PanIN de-differentiation, KRAS mutations in acinar cells have been shown to drive PKD1-dependent mitochondrial ROS increase and that this event is the leading factor responsible for EGFR-mediated ADAM17 shedding.23 Similarly, in KRAS mutant colorectal cancer, inhibition of the PI3K/mTOR pathway sensitizes cells to EGFR inhibitors.24 Indeed, it has been reported that some EV populations form through diacylglycerol-controlled fission and the secretion of which is dependent on the combined action of DGKα, which releases phosphatidic acid from diacylglycerol, and PKD1/2.25 Furthermore, pancreatic cancer patient-derived EVs contain oncogenic KRAS and subsequent analysis showed that the KRAS mutation status of EVs matched the primary tumor.4 It is reasonable to hypothesize that EVs shuffle a diverse pool of signaling elements belonging to the KRAS pathway, as well as metabolites such as diacylglycerol, lactate and glutamine satisfying sufficient requirements to drive malignant transformation in healthy recipient cells. Proteomic profiling of EVs using stable isotope labeled amino acids in cell culture (SILAC) has further shown that exosomal cargo content is dependent on vesicle size.26 Large oncosomes preferentially contain protein cargo targeted to mitochondrial metabolic processes including VDAC1/2, the solute carriers SLC25A6 and SLC25A5 that are mitochondrial ADP/ATP translocators as well as the ATP synthase subunit ATP5B. Nano-sized EV cargo on the other hand contained higher amounts of proteins clustered towards glucose and glutamine metabolism and gluconeogenesis.26 Because EV content seems size-dependent, it is plausible that release and uptake of small EVs are coordinated separately from large oncosomes. Cholesterol flux and functional lipid rafts affect the uptake of EVs in A375 melanoma cells.27 We speculate that these and other mechanisms may in part help explain why certain cargo is tailored in an organotropic manner, thus favoring a tissue-specific metastatic phenotype. Metabolic reprogramming under stress appears to be one of the primary functions of EVs and hypoxia-inducible factor 1α has been detected in nasopharyngeal carcinoma exosomes where LMP1-induced transmission of transcriptionally active hypoxia-inducible factor 1α drives oncogenic processes.28
Our current understanding of metabolic reprogramming events during cancer development is still widely elusive; in particular, the spatiotemporal order with which cells undergo metabolic reprogramming has not been fully evaluated. Further characterization of the feedback loops initiated by EVs on tumor cells and the stromal environment might provide critical missing pieces in this picture.
Stromal effects of EVs
EVs mediate fibroblasts and cancer cell changes
Fibroblasts make up the bulk of stromal cells. Although hugely variable, even within the same kind of tumor, fibroblasts are in most cases the main contributor to the stroma. For instance, in invasive ductal carcinoma the average number of fibroblasts/myofibroblasts may reach up to 50–70% of the total stroma cell number. Transforming growth factor-β (TGFβ), platelet derived growth factor and fibroblast growth factor 2 signaling ligands in conjunction with other molecules including miRNAs can induce a cancer-activated or associated fibroblasts (CAFs)/myofibroblast phenotype characterized by increased proliferation rate, migratory properties and heightened deposition of extracellular matrix. CAFs originate from resident fibroblasts, through induction of epithelial-to-mesenchymal transition or via recruited and reprogrammed mesenchymal stem cells and produce several growth factors such as hepatocyte growth factor, vascular endothelial growth factor and TGF.29 Breast cancer cells-derived TGFβ-EVs show the ability to differentiate adipose tissue-derived mesenchymal stem cells into α-smooth muscle actin positive CAFs utilizing the TGFβ/Smad pathway.30 Furthermore, prostate cancer-derived EVs may induce CAFs from bone-marrow mesenchymal stem cells with pro-angiogenic and invasive functions.31 This could be in part explained by the abundance of miR-1227 in large oncosomes from the prostate cancer cell line RWPE-2 that enhances CAF migration properties.32 EVs appear to induce CAFs, as recently substantiated by the findings that bladder cancer-derived EVs induce epithelial-to-mesenchymal transition in urothelial cells.33 However, EVs from non-solid cancer chronic lymphocytic leukemia can also turn stromal endothelial cells and mesenchymal stem cells into CAFs.34 On the other hand, stromal cells themselves are known to secrete EVs. In a human/mouse tissue culture system, Wnt11-EVs activated the Wnt-planar cell-polarity signaling pathway at the leading edge of breast cancer cells eliciting cell migration. In that case, cancer cells and fibroblasts work together to assemble fibroblast EVs that are internalized by breast cancer cells, loaded with Wnt11 protein and then re-released for paracrine signaling.35
In a different context, CAF EVs with increased levels of miRNA-21 profoundly impact ovarian cancer growth by suppressing apoptosis through binding to its novel target, APAF1.36 Finally, as discussed above CAF-derived EVs directly participate in metabolic reprogramming. In aggregate, these few examples add to an increasing number of described EV functions in bidirectional cell interactions between fibroblasts and cancer cells.
EVs set the place and time for neo-angiogenesis
Neo-angiogenesis allows tumors to get their own constant vascular supply of nutrients and oxygen, enabling them to grow above 2 mm3 and become much more aggressive. One of the most recent advances in this field is the involvement of EVs in tumor-associated neo-angiogenesis.37, 38 Indeed, several groups reported the pro-angiogenic effect of tumor cell-derived EVs on endothelial cells in different types of cancer such as glioblastoma,39 leukemia,40 melanoma41 and ovarian cancer.42 Since EVs can be taken up by endocytic-like processes, they may evade the ligand-receptor system on the cell surface influencing intracellular signaling and protein expression in endothelial cells.43 As mentioned above, EVs can exert functions over short and long distances. In this way, pro-angiogenic EVs influence the neo-angiogenic program in the proximal tumor microenvironment but can also prime metastatic niches for angiogenetic events.41, 44
Pro-angiogenic factors such as VEGF, fibroblast growth factor 2, platelet derived growth factor, interleukins, matrix metalloproteinase (MMPs), EGFR or signaling proteins including Rac1 and Cdc42/Pak2 can be found among other proteins in tumor cell-derived EVs.39, 43, 45, 46 The presence of these proteins in EVs brought novel aspects of tumor-associated neo-angiogenesis into the limelight. For instance, Al Nedawi et al.47 reported that upon uptake of tumor cell-derived EVs that contained oncogenic EGFR, endothelial cells establish a VEGF-dependent autocrine loop, a main mechanism in tumor neo-angiogenesis. Such a process re-programs endothelial cells and consequently strongly enhances neo-angiogenesis. More recently, Gopal et al.43 showed that tumor cell-derived EVs are able to deliver signaling factors, such as Rac1 or Pak2, or receptor proteins such as neuropilin 1, a co-receptor for VEGF, directly to endothelial cells promoting neo-angiogenesis. In comparison with the classical ‘ligand/receptor’ process, the authors called this phenomenon a ‘more direct avenue to induce angiogenesis’ and suggest that it could be involved in metastatic spread43 (Figure 3).

EV-mediated transfer versus the secretion of soluble molecules bound for ligand/receptor interactions. Local diffusion of proteins such as cytokines, chemokines or growth factors (exemplified for tumor to endothelial cells delivery) allows the engagement with their respective receptors on proximal located cells. In contrast, tumor cell-derived EVs allow the transfer of diffusible factors but also that of receptors, intracellular signaling mediators and RNAs all protected from degrading enzymes in the microenvironment allowing systemic transport via bodily fluids such as blood or the lymph for their distribution. Thus, EVs can transfer their content not only to neighboring stromal cells but also to potentially remote locations of future metastatic sites. The delivery of EV cargo to target cells may circumvent the necessity of specific ligand/receptor interactions.
Some mRNAs and miRNAs found in EVs are thought to be specifically involved in neo-angiogenesis.46 For example in colorectal cancer, tumor-derived EVs can promote proliferation of endothelial cells and enhance their cell-cycle activities through M-phase-related mRNAs, such as those coding for the centromere protein E (CENPE), PDZ binding kinase (PBK) or cyclin-dependent kinase 8 (CDK8).48 Additionally, the involvement of vesicular miRNAs in neo-angiogenesis has been studied such as miRNA-210 that exhibited strong pro-angiogenic activity.49, 50, 51 Furthermore, miRNA-210 has been observed to suppress the expression of specific genes such as EFNA3 (coding for Ephrin-A3) in endothelial cells, resulting in enhanced neo-angiogenesis.52, 53, 54 Colorectal carcinoma cell-derived vesicular miRNA-9 shows pro-angiogenic effects through inhibiting the expression of suppressor of cytokine signaling 5 (SOCS 5), promoting the activation of the janus kinase/signal transducers and activators of transcription signaling, a driver of endothelial cell migration.55 Leukemia cell-derived exosomal miRNA-92a has also been shown to stimulate tumor-associated neo-angiogenesis, through the inhibition of integrin α5 expression.56
Despite the direct pro-angiogenic effect of cancer cell-derived EVs on endothelial cells, such vesicles also promote neo-angiogenesis through indirect effects on other stromal resident cells. For example, leukemia-derived EVs can induce a CAF phenotype in stromal cells in the surrounding microenvironment, hence leading to increased expression of pro-angiogenic factors in such cells.34, 50 Finally, EV-mediated crosstalk occurs also between endothelial cells themselves.57
On the other hand, EVs may act on tumor cells during neo-angiogenic processes since endothelial cells themselves have been shown to release EVs that can target tumor cells. Indeed, endothelial human umbilical vein endothelial cells were shown to secrete EVs containing miRNA such as miRNA-503 that were taken up by co-cultured tumor cells in vitro. MiRNA-503 was subsequently linked to response to neo-adjuvant chemotherapy in breast cancer.58
Several reports suggested that the increased number of tumor cells-derived EVs during neo-angiogenesis could be a reaction to a hypoxic condition, a key event in promoting neo-angiogenesis.8, 52, 59 In addition, recent data showed that the composition of EVs may also depend on the hypoxic status of glioma cells.45 Using glioma cell lines and patient-derived cells EV signature composition was positively correlated to hypoxia. This led to the observation that hypoxic tumor cell-derived EVs are more potent neo-angiogenesis inducers than EVs derived from normoxic populations. Interestingly, hypoxic tumor cell-derived EVs execute this function by PI3K/Akt signaling modulation.45 Furthermore, vesicular miRNA-135b from hypoxic multiple myeloma cells can directly contribute to enhanced neo-angiogenesis under chronic hypoxia through the inhibition of the factor inhibiting hypoxia-inducible factor 1 expression, promoting the activity of hypoxia-inducible factor 1.56 Other groups also reported on special selection processes for proteins and RNA content of tumor cell-derived EVs in response to hypoxia, providing them with specific pro-angiogenic functions.54, 60, 61, 62 Finally, WNT5A signaling protein induces mechanisms that lead to the release of EVs from tumor cells containing pro-angiogenic factors such as VEGF.13
These data also suggest that different tumor types can release different EVs with variable outcome for neo-angiogenesis. For instance, tumor cells undergoing complete epithelial–mesenchymal transition release EVs that are more effective at enhancing neo-angiogenesis than those undergoing intermediate epithelial-to-mesenchymal transition.43 Similarly, for renal cancer, EVs with the most powerful pro-angiogenic activity were those derived from cancer stem cells and contained different angiogenic factors, compared with non-cancer stem cells.42
EVs tune the immune response
EVs, as mediators of intercellular communication, can modulate the activity and therefore the nature and vigor of diverse cellular immune response systems. Early data demonstrated the ability of dendritic cell-derived EVs to stimulate an antitumor immune response as well as documented the presence of key MHC1 and MHCII proteins in EVs.63 More rigid functional evidence of intercellular shuttling of miRNAs with the ability to epigenetically affect target genes in a variety of dendritic cells was first obtained from EVs from different dendritic cell populations that showed varying miRNA signatures depending on their maturation state;64 miRNA transfer has been demonstrated in both in vitro and in vivo settings and can effect a range of diverse processes. Transmission occurs sometimes in a unidirectional fashion for instance at the immune synapse from T-cell to antigen-presenting cell, in an antigen-driven fashion.65 T-cell-derived exosomes containing specific miRNA signatures have been recently shown to suppress T-H1-mediated immune responses in systemic diseases.66 There is now a growing body of evidence that suggests that cancer cells use EV transmitted nucleic acids and proteins as a way of enacting an immune escape.
Colorectal cancer cell-derived microvesicle content such as TNF-related apoptosis-inducing ligand and fas cell surface death receptor ligand has been demonstrated to induce T-cell death through the activation of the FAS ligand.48 This has also been demonstrated for other tumor types.67 In the context of hepatocellular carcinoma the release of heat-shock protein chaperones from EVs was shown to act as a decoy enabling an NK cell response to be directed away from tumor cells. In contrast, in resistant cell lines these HSP-bearing EVs were upregulated.68 Circulating EVs in breast cancer similarly enable tumor growth by downregulating NK cell activity.69 Tumor-derived EVs in nasopharyngeal cancer were found to induce T-reg activity and inhibit T-cell proliferation in vitro.
While the above examples demonstrate that tumor-derived EVs can downregulate the immune response, it appears that EVs from activated immune cells can also influence the tumor phenotype. For example, EVs from activated CD8+ T-cells can increase tumor immunogenicity by activating ERK and nuclear factor κB signaling through TNF-related signaling, leading ultimately to the upregulation of matrix metalloproteinase-9. This chain of events increases the metastatic potential in melanoma and lung cancer.70 In another chain of events, pancreatic ductal adenocarcinomas cell-derived EVs can lead to pre-metastatic niche formation in sequential steps of induction of TGFβ signaling in Kupffer cells leading to extracellular matrix modification and subsequently an influx of bone-marrow-derived macrophages to the liver, providing a favorable niche for liver metastasis.71
EVs as ‘real-time’ biomarkers during cancer therapies
Some of the most promising studies involving EV cargo modulation during drug treatment have been performed in glioblastoma multiforme. Levels of the DNA repair enzymes APNG and MGMT are inversely correlated to response to the gold standard chemotherapeutic temozolomide.72 EVs containing MGMT mRNA have been demonstrated to accurately reflect the levels of these enzymes in parental cells and in patients throughout treatment and therefore could serve as a potential ‘real-time’ biomarker of chemotherapy response during drug treatment.73 Similarly, circulating EVs containing the EGFRvIII splice variant that is thought to be predictive of response to EGFR inhibition were detectable in the serum of glioblastoma multiforme patients but not in the 30 matched controls.39
In the context of the neo-adjuvant treatment of breast carcinoma, elevated levels of the EV-bound MDR-glycoprotein BCRP were detected in non-responders compared with responders or treatment naïve patients.74 In addition, the receptor channel protein TRCP5, a known regulator of multidrug resistance glycoprotein-P, was required for EV formation in anthracycline-resistant breast carcinoma cell lines. Moreover, EVs containing TRCP5 protein from the same chemoresistant cells can enter chemosensitive cells and transmit resistance to cytotoxic chemotherapy. The same group also demonstrated elevated levels of TRCP5 mRNA in circulating EVs from patients who did not respond to chemotherapy.75
Horizontal transfer of nucleic acids has been postulated as one mechanism that can alter apoptotic and proliferative cell responses during cancer treatment. Indeed, EVs from triple negative breast cancer cells in vitro can evoke proliferative and angiogenic properties in recipient cells that are similar to those seen in the parental cell line.76 A recent study elaborating on this work additionally demonstrated transfer of miRNAs including mir-100, miR-222, miR -17 and miR-30a through exosomes in breast cancer cell lines with the effect of modulating target genes that can be critical to drug response. For instance, the transfer of miR-222 specifically caused PTEN mRNA downregulation in recipient cells. The subsequent apoptotic response to doxorubicin was also reduced.77 In addition to miRNAs, proteins transported by EVs have also been shown to modify the apoptotic response. The key negative regulator of AKT/PI3 kinase signaling PTEN for instance has been identified as EV cargo eliciting active phosphatase function in the recipient cell.78
Only a few studies have been published on the role of EVs in modulating a response to more specific targeted treatments. One such study explored the role of EV transfer between cetuximab resistant and sensitive colorectal cancer cell lines in vitro. Although an effect on cell viability was observed, this effect turned out to be rather modest.79 Recently, IncARSR (Inc RNA Activated in renal cell carcinoma with Sunitinib Resistance) has been shown to promote sunitinib resistance via its EV-bound transfer to sensitive renal cell carcinoma cells where it competitively binds miR-34/miR-449. Decreasing the levels of those miRNAs facilitates AXL and c-MET expression in renal cell carcinoma cells, rendering IncARSR as a hopeful predictor for sunitinib resistance. Although these few examples seem quite promising it remains widely unexplored and elusive whether EVs are indeed significant contributors to either intrinsic or acquired resistance to the plethora of Food and Drug Administration-approved small-molecule inhibitors currently in clinical use.
For antiangiogenic therapies more data are available overall concluding on positive effects of EVs in modulating drug response. Raimondo et al.61 analyzed the occurring changes in EV composition and evaluated their effects on drug treatment responses. Interestingly, angiogenic factors present in EVs correlated with patients who were likely to benefit from a particular antiangiogenic therapy. In addition, EV-dependent mechanisms could be implicated in the refractoriness of some tumor cells to current antiangiogenic therapies, as observed for glioblastomas in response to bevacizumab. Finally, antiangiogenic therapies could alter the pro-angiogenic properties of EVs, suggesting this as a new strategy to decrease tumor-associated vasculature and tumor resistance.80 Taken together the interference with EV communication could potentially have a strong antiangiogenic effect.47, 50
Studying EV-based therapies, some groups have explored the utilization of EVs as therapeutic delivery systems. Taking advantage of EVs in delivering specific RNAs designed to alter the phenotype of malignant cells could prove an attractive prospect. Such a prospect was successfully executed by engineering let-7a miRNA containing EVs to modify EGFR expression in breast cancer cell lines leading to dramatic effects on tumor growth.81 Similarly, delivery of extrinsically administered siRNA using exosomes in a murine setting has been demonstrated recently82 to be effective in knocking down a central nervous system specific protein. These promising sets of data suggest that this technology is now emerging allowing targeted use of extrinsically generated EVs in order to counteract tumors.
Conclusion and outlook
Cumulatively, the studies briefly described make a resounding case for the involvement of EVs in all stages during cancer development. However, most of the aforementioned results are gathered from tissue culture experiments generating non-physiological vesicle concentration levels. Therefore, it would be vital to substantiate these findings in more rigorous in vivo settings. These undertakings are currently hampered by considerable gaps in our knowledge of EV biogenesis and a lack of available in vivo tools.83 It is interesting to note that, although EV formation occurs in all cells, most of our knowledge about their function stems from cells that have adapted to malignant transformation, while our knowledge about their roles in healthy tissue homeostasis lags behind. We have discussed the release and reception of cargo containing signaling molecules, as well as metabolic and growth regulators, shuttled between tumor cells and their surrounding microenvironment. In this regard, it is the abundance or rather the delicate mixture of these molecules that charge EVs with cell transforming ‘superpowers’. Like Trojan horses they may cross the cell barrier and reprogram cellular functions in favor of the malignant cells. However, these properties also make them formidable candidates for cancer diagnostics as well as for novel therapeutic approaches. Firstly, their composition may hold important clues about the type and stage of various types of cancers and also reveal possible new targets. Secondly, they could potentially be designed for the purpose of targeted intervention, including the stimulation of local autoimmune responses or for the ‘trapping’ of disseminating cancer cells. Thirdly, during cancer treatment, EVs may switch their composition and may therefore exhibit traits for ‘real-time’ monitoring of therapeutic efficiency. However, while we make incremental progress in exploring all those possibilities many questions remain still unresolved. In particular, those concerning their biogenesis, cargo selection and loading, as well as the mechanisms involved in their uptake, cargo liberation and incorporation into the context of the recipient cells. The incentives to investigate the functional connotations of EVs promise to change our understanding of cancer biology and potentially of how to tackle this complex set of diseases.
References
Abels ER, Breakefield XO . Introduction to extracellular vesicles: biogenesis, RNA cargo selection, content, release, and uptake. Cell Mol Neurobiol 2016; 36: 301–312.
Colombo M, Raposo G, Thery C . Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 2014; 30: 255–289.
Khan S, Jutzy JM, Valenzuela MM, Turay D, Aspe JR, Ashok A et al. Plasma-derived exosomal survivin, a plausible biomarker for early detection of prostate cancer. PLoS One 2012; 7: e46737.
Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST, Kaye J et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 2015; 523: 177–182.
Ogata-Kawata H, Izumiya M, Kurioka D, Honma Y, Yamada Y, Furuta K et al. Circulating exosomal microRNAs as biomarkers of colon cancer. PLoS One 2014; 9: e92921.
Cazzoli R, Buttitta F, Di Nicola M, Malatesta S, Marchetti A, Rom WN et al. microRNAs derived from circulating exosomes as noninvasive biomarkers for screening and diagnosing lung cancer. J Thorac Oncol 2013; 8: 1156–1162.
Wang T, Gilkes DM, Takano N, Xiang L, Luo W, Bishop CJ et al. Hypoxia-inducible factors and RAB22A mediate formation of microvesicles that stimulate breast cancer invasion and metastasis. Proc Natl Acad Sci USA 2014; 111: E3234–E3242.
Parolini I, Federici C, Raggi C, Lugini L, Palleschi S, De Milito A et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J Biol Chem 2009; 284: 34211–34222.
Yu X, Harris SL, Levine AJ . The regulation of exosome secretion: a novel function of the p53 protein. Cancer Res 2006; 66: 4795–4801.
Amorim M, Fernandes G, Oliveira P, Martins-de-Souza D, Dias-Neto E, Nunes D . The overexpression of a single oncogene (ERBB2/HER2) alters the proteomic landscape of extracellular vesicles. Proteomics 2014; 14: 1472–1479.
Ji H, Erfani N, Tauro BJ, Kapp EA, Zhu HJ, Moritz RL et al. Difference gel electrophoresis analysis of Ras-transformed fibroblast cell-derived exosomes. Electrophoresis 2008; 29: 2660–2671.
Clark DJ, Fondrie WE, Yang A, Mao L . Triple SILAC quantitative proteomic analysis reveals differential abundance of cell signaling proteins between normal and lung cancer-derived exosomes. J Proteomics 2016; 133: 161–169.
Ekstrom EJ, Bergenfelz C, von Bulow V, Serifler F, Carlemalm E, Jonsson G et al. WNT5A induces release of exosomes containing pro-angiogenic and immunosuppressive factors from malignant melanoma cells. Mol Cancer 2014; 13: 88.
Meckes Jr DG, Gunawardena HP, Dekroon RM, Heaton PR, Edwards RH, Ozgur S et al. Modulation of B-cell exosome proteins by gamma herpesvirus infection. Proc Natl Acad Sci USA 2013; 110: E2925–E2933.
Halestrap AP . The monocarboxylate transporter family—structure and functional characterization. IUBMB Life 2012; 64: 1–9.
Halestrap AP, Wilson MC . The monocarboxylate transporter family—role and regulation. IUBMB Life 2012; 64: 109–119.
Le Floch R, Chiche J, Marchiq I, Naiken T, Ilc K, Murray CM et al. CD147 subunit of lactate/H+ symporters MCT1 and hypoxia-inducible MCT4 is critical for energetics and growth of glycolytic tumors. Proc Natl Acad Sci USA 2011; 108: 16663–16668.
Webb BA, Chimenti M, Jacobson MP, Barber DL . Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer 2011; 11: 671–677.
Porporato PE, Payen VL, Baselet B, Sonveaux P . Metabolic changes associated with tumor metastasis, part 2: mitochondria, lipid and amino acid metabolism. Cell Mol Life Sci 2016; 73: 1349–1363.
Ronquist KG, Sanchez C, Dubois L, Chioureas D, Fonseca P, Larsson A et al. Energy-requiring uptake of prostasomes and PC3 cell-derived exosomes into non-malignant and malignant cells. J Extracell Vesicles 2016; 5: 29877.
Zhao H, Yang L, Baddour J, Achreja A, Bernard V, Moss T et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016; 5.
Kerr EM, Gaude E, Turrell FK, Frezza C, Martins CP . Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature 2016; 531: 110–113.
Liou GY, Doppler H, DelGiorno KE, Zhang L, Leitges M, Crawford HC et al. Mutant KRas-induced mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Rep 2016; 14: 2325–2336.
Belmont PJ, Jiang P, McKee TD, Xie T, Isaacson J, Baryla NE et al. Resistance to dual blockade of the kinases PI3K and mTOR in KRAS-mutant colorectal cancer models results in combined sensitivity to inhibition of the receptor tyrosine kinase EGFR. Sci Signal 2014. 7ra107.
Mazzeo C, Calvo V, Alonso R, Merida I, Izquierdo M . Protein kinase D1/2 is involved in the maturation of multivesicular bodies and secretion of exosomes in T and B lymphocytes. Cell Death Differ 2016; 23: 99–109.
Minciacchi VR, You S, Spinelli C, Morley S, Zandian M, Aspuria PJ et al. Large oncosomes contain distinct protein cargo and represent a separate functional class of tumor-derived extracellular vesicles. Oncotarget 2015; 6: 11327–11341.
Plebanek MP, Mutharasan RK, Volpert O, Matov A, Gatlin JC, Thaxton CS . Nanoparticle targeting and cholesterol flux through scavenger receptor type B-1 inhibits cellular exosome uptake. Sci Rep 2015; 5: 15724.
Aga M, Bentz GL, Raffa S, Torrisi MR, Kondo S, Wakisaka N et al. Exosomal HIF1alpha supports invasive potential of nasopharyngeal carcinoma-associated LMP1-positive exosomes. Oncogene 2014; 33: 4613–4622.
Kalluri R, Zeisberg M . Fibroblasts in cancer. Nat Rev Cancer 2006; 6: 392–401.
Cho JA, Park H, Lim EH, Lee KW . Exosomes from breast cancer cells can convert adipose tissue-derived mesenchymal stem cells into myofibroblast-like cells. Int J Oncol 2012; 40: 130–138.
Chowdhury R, Webber JP, Gurney M, Mason MD, Tabi Z, Clayton A . Cancer exosomes trigger mesenchymal stem cell differentiation into pro-angiogenic and pro-invasive myofibroblasts. Oncotarget 2015; 6: 715–731.
Morello M, Minciacchi VR, de Candia P, Yang J, Posadas E, Kim H et al. Large oncosomes mediate intercellular transfer of functional microRNA. Cell Cycle 2013; 12: 3526–3536.
Franzen CA, Blackwell RH, Todorovic V, Greco KA, Foreman KE, Flanigan RC et al. Urothelial cells undergo epithelial-to-mesenchymal transition after exposure to muscle invasive bladder cancer exosomes. Oncogenesis 2015; 4: e163.
Paggetti J, Haderk F, Seiffert M, Janji B, Distler U, Ammerlaan W et al. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood 2015; 126: 1106–1117.
Luga V, Zhang L, Viloria-Petit AM, Ogunjimi AA, Inanlou MR, Chiu E et al. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell 2012; 151: 1542–1556.
Au Yeung CL, Co NN, Tsuruga T, Yeung TL, Kwan SY, Leung CS et al. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat Commun 2016; 7: 11150.
Frydrychowicz M, Kolecka-Bednarczyk A, Madejczyk M, Yasar S, Dworacki G . Exosomes—structure, biogenesis and biological role in non-small-cell lung cancer. Scand J Immunol 2015; 81: 2–10.
Park JE, Tan HS, Datta A, Lai RC, Zhang H, Meng W et al. Hypoxic tumor cell modulates its microenvironment to enhance angiogenic and metastatic potential by secretion of proteins and exosomes. Mol Cell Proteomics 2010; 9: 1085–1099.
Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol 2008; 10: 1470–1476.
Taverna S, Flugy A, Saieva L, Kohn EC, Santoro A, Meraviglia S et al. Role of exosomes released by chronic myelogenous leukemia cells in angiogenesis. Int J Cancer 2012; 130: 2033–2043.
Hood JL, Pan H, Lanza GM, Wickline SA, Consortium for Translational Research in Advanced Imaging and Nanomedicine (C-TRAIN).. Paracrine induction of endothelium by tumor exosomes. Lab Invest 2009; 89: 1317–1328.
Millimaggi D, Mari M, D'Ascenzo S, Carosa E, Jannini EA, Zucker S et al. Tumor vesicle-associated CD147 modulates the angiogenic capability of endothelial cells. Neoplasia 2007; 9: 349–357.
Gopal SK, Greening DW, Hanssen EG, Zhu HJ, Simpson RJ, Mathias RA . Oncogenic epithelial cell-derived exosomes containing Rac1 and PAK2 induce angiogenesis in recipient endothelial cells. Oncotarget 2016, e-pub ahead of print 22 February 2016 doi:10.18632/oncotarget.7573.
Grange C, Tapparo M, Collino F, Vitillo L, Damasco C, Deregibus MC et al. Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res 2011; 71: 5346–5356.
Kucharzewska P, Christianson HC, Welch JE, Svensson KJ, Fredlund E, Ringner M et al. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc Natl Acad Sci USA 2013; 110: 7312–7317.
Vader P, Breakefield XO, Wood MJ . Extracellular vesicles: emerging targets for cancer therapy. Trends Mol Med 2014; 20: 385–393.
Al-Nedawi K, Meehan B, Kerbel RS, Allison AC, Rak J . Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc Natl Acad Sci USA 2009; 106: 3794–3799.
Huber V, Fais S, Iero M, Lugini L, Canese P, Squarcina P et al. Human colorectal cancer cells induce T-cell death through release of proapoptotic microvesicles: role in immune escape. Gastroenterology 2005; 128: 1796–1804.
Fasanaro P, D'Alessandra Y, Di Stefano V, Melchionna R, Romani S, Pompilio G et al. MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand Ephrin-A3. J Biol Chem 2008; 283: 15878–15883.
Kosaka N . Decoding the secret of cancer by means of extracellular vesicles. J Clin Med 2016; 5.
Zhang Y, Liu D, Chen X, Li J, Li L, Bian Z et al. Secreted monocytic miR-150 enhances targeted endothelial cell migration. Mol Cell 2010; 39: 133–144.
Fan GC . Hypoxic exosomes promote angiogenesis. Blood 2014; 124: 3669–3670.
Kosaka N, Iguchi H, Hagiwara K, Yoshioka Y, Takeshita F, Ochiya T . Neutral sphingomyelinase 2 (nSMase2)-dependent exosomal transfer of angiogenic microRNAs regulate cancer cell metastasis. J Biol Chem 2013; 288: 10849–10859.
Tadokoro H, Umezu T, Ohyashiki K, Hirano T, Ohyashiki JH . Exosomes derived from hypoxic leukemia cells enhance tube formation in endothelial cells. J Biol Chem 2013; 288: 34343–34351.
Zhuang G, Wu X, Jiang Z, Kasman I, Yao J, Guan Y et al. Tumour-secreted miR-9 promotes endothelial cell migration and angiogenesis by activating the JAK-STAT pathway. EMBO J 2012; 31: 3513–3523.
Umezu T, Tadokoro H, Azuma K, Yoshizawa S, Ohyashiki K, Ohyashiki JH . Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood 2014; 124: 3748–3757.
van Balkom BW, de Jong OG, Smits M, Brummelman J, den Ouden K, de Bree PM et al. Endothelial cells require miR-214 to secrete exosomes that suppress senescence and induce angiogenesis in human and mouse endothelial cells. Blood 2013; 121: 3997–4006; S3991-3915.
Bovy N, Blomme B, Freres P, Dederen S, Nivelles O, Lion M et al. Endothelial exosomes contribute to the antitumor response during breast cancer neoadjuvant chemotherapy via microRNA transfer. Oncotarget 2015; 6: 10253–10266.
Sun L, Wang HX, Zhu XJ, Wu PH, Chen WQ, Zou P et al. Serum deprivation elevates the levels of microvesicles with different size distributions and selectively enriched proteins in human myeloma cells in vitro. Acta Pharmacol Sin 2014; 35: 381–393.
King HW, Michael MZ, Gleadle JM . Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer 2012; 12: 421.
Raimondo S, Corrado C, Raimondi L, De Leo G, Alessandro R . Role of extracellular vesicles in hematological malignancies. Biomed Res Int 2015; 2015: 821613.
Salomon C, Ryan J, Sobrevia L, Kobayashi M, Ashman K, Mitchell M et al. Exosomal signaling during hypoxia mediates microvascular endothelial cell migration and vasculogenesis. PLoS One 2013; 8: e68451.
Zitvogel L, Regnault A, Lozier A, Wolfers J, Flament C, Tenza D et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med 1998; 4: 594–600.
Montecalvo A, Larregina AT, Shufesky WJ, Stolz DB, Sullivan ML, Karlsson JM et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood 2012; 119: 756–766.
Mittelbrunn M, Gutierrez-Vazquez C, Villarroya-Beltri C, Gonzalez S, Sanchez-Cabo F, Gonzalez MA et al. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat Commun 2011; 2: 282.
Okoye IS, Coomes SM, Pelly VS, Czieso S, Papayannopoulos V, Tolmachova T et al. MicroRNA-containing T-regulatory-cell-derived exosomes suppress pathogenic T helper 1 cells. Immunity 2014; 41: 89–103.
Andreola G, Rivoltini L, Castelli C, Huber V, Perego P, Deho P et al. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J Exp Med 2002; 195: 1303–1316.
Lv LH, Wan YL, Lin Y, Zhang W, Yang M, Li GL et al. Anticancer drugs cause release of exosomes with heat shock proteins from human hepatocellular carcinoma cells that elicit effective natural killer cell antitumor responses in vitro. J Biol Chem 2012; 287: 15874–15885.
Liu C, Yu S, Zinn K, Wang J, Zhang L, Jia Y et al. Murine mammary carcinoma exosomes promote tumor growth by suppression of NK cell function. J Immunol 2006; 176: 1375–1385.
Cai Z, Yang F, Yu L, Yu Z, Jiang L, Wang Q et al. Activated T cell exosomes promote tumor invasion via Fas signaling pathway. J Immunol 2012; 188: 5954–5961.
Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol 2015; 17: 816–826.
Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005; 352: 997–1003.
Shao H, Chung J, Lee K, Balaj L, Min C, Carter BS et al. Chip-based analysis of exosomal mRNA mediating drug resistance in glioblastoma. Nat Commun 2015; 6: 6999.
Chen Y, Wang L, Zhu Y, Chen Z, Qi X, Jin L et al. Breast cancer resistance protein (BCRP)-containing circulating microvesicles contribute to chemoresistance in breast cancer. Oncol Lett 2015; 10: 3742–3748.
Ma X, Chen Z, Hua D, He D, Wang L, Zhang P et al. Essential role for TrpC5-containing extracellular vesicles in breast cancer with chemotherapeutic resistance. Proc Natl Acad Sci USA 2014; 111: 6389–6394.
O'Brien K, Rani S, Corcoran C, Wallace R, Hughes L, Friel AM et al. Exosomes from triple-negative breast cancer cells can transfer phenotypic traits representing their cells of origin to secondary cells. Eur J Cancer 2013; 49: 1845–1859.
Chen WX, Liu XM, Lv MM, Chen L, Zhao JH, Zhong SL et al. Exosomes from drug-resistant breast cancer cells transmit chemoresistance by a horizontal transfer of microRNAs. PLoS One 2014; 9: e95240.
Putz U, Howitt J, Doan A, Goh CP, Low LH, Silke J et al. The tumor suppressor PTEN is exported in exosomes and has phosphatase activity in recipient cells. Sci Signal 2012; 5: ra70.
Ragusa M, Statello L, Maugeri M, Barbagallo C, Passanisi R, Alhamdani MS et al. Highly skewed distribution of miRNAs and proteins between colorectal cancer cells and their exosomes following Cetuximab treatment: biomolecular, genetic and translational implications. Oncoscience 2014; 1: 132–157.
Hannafon BN, Carpenter KJ, Berry WL, Janknecht R, Dooley WC, Ding WQ . Exosome-mediated microRNA signaling from breast cancer cells is altered by the anti-angiogenesis agent docosahexaenoic acid (DHA). Mol Cancer 2015; 14: 133.
Ohno S, Takanashi M, Sudo K, Ueda S, Ishikawa A, Matsuyama N et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther 2013; 21: 185–191.
Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ . Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol 2011; 29: 341–345.
Tkach M, Thery C . Communication by extracellular vesicles: where we are and where we need to go. Cell 2016; 164: 1226–1232.
Budnik V, Ruiz-Canada C, Wendler F . Extracellular vesicles round off communication in the nervous system. Nat Rev Neurosci 2016; 17: 160–172.
Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F et al. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008; 319: 1244–1247.
Hurley JH . ESCRTs are everywhere. EMBO J 2015; 34: 2398–2407.
Acknowledgements
This work was supported by Action Against Cancer and NIHR grant #NIHR-RP-011-053.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Wendler, F., Favicchio, R., Simon, T. et al. Extracellular vesicles swarm the cancer microenvironment: from tumor–stroma communication to drug intervention. Oncogene 36, 877–884 (2017). https://doi.org/10.1038/onc.2016.253
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2016.253
- Springer Nature Limited
This article is cited by
-
Radiogenomic analysis of cellular tumor-stroma heterogeneity as a prognostic predictor in breast cancer
Journal of Translational Medicine (2023)
-
Extracellular vesicles remodel tumor environment for cancer immunotherapy
Molecular Cancer (2023)
-
The potential applications of microparticles in the diagnosis, treatment, and prognosis of lung cancer
Journal of Translational Medicine (2022)
-
The critical role of STAT3 in biogenesis of tumor-derived exosomes with potency of inducing cancer cachexia in vitro and in vivo
Oncogene (2022)
-
Tumor-produced and aging-associated oncometabolite methylmalonic acid promotes cancer-associated fibroblast activation to drive metastatic progression
Nature Communications (2022)