
Abstract
Insulin-like growth factor binding protein 2 (IGFBP2) is a pleiotropic oncogenic protein that has both extracellular and intracellular functions. Despite a clear causal role in cancer development, the tumor-promoting mechanisms of IGFBP2 are poorly understood. The contributions of intracellular IGFBP2 to tumor development and progression are also unclear. Here we present evidence that both exogenous IGFBP2 treatment and cellular IGFBP2 overexpression lead to aberrant activation of epidermal growth factor receptor (EGFR), which subsequently activates signal transducer and activator of transcription factor 3 (STAT3) signaling. Furthermore, we demonstrate that IGFBP2 augments the nuclear accumulation of EGFR to potentiate STAT3 transactivation activities, via activation of the nuclear EGFR signaling pathway. Nuclear IGFBP2 directly influences the invasive and migratory capacities of human glioblastoma cells, providing a direct link between intracellular (and particularly nuclear) IGFBP2 and cancer hallmarks. These activities are also consistent with the strong association between IGFBP2 and STAT3-activated genes derived from The Cancer Genome Atlas database for human glioma. A high level of all three proteins (IGFBP2, EGFR and STAT3) was strongly correlated with poorer survival in an independent patient data set. These results identify a novel tumor-promoting function for IGFBP2 of activating EGFR/STAT3 signaling and facilitating EGFR accumulation in the nucleus, thereby deregulating EGFR signaling by two distinct mechanisms. As targeting EGFR in glioma has been relatively unsuccessful, this study suggests that IGFBP2 may be a novel therapeutic target.
Similar content being viewed by others


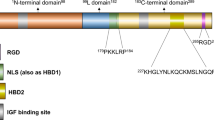
Introduction
Secreted proteins such as growth factors and hormones exert their function by binding to the extracellular domain of membrane receptors. Secreted factors can also enter the cell through receptor-mediated endocytosis.1, 2, 3, 4 Once internalized, these proteins can regulate intracellular cytoplasmic signal transduction and transcriptional activity in the nucleus.5, 6, 7, 8, 9, 10, 11 Insulin-like growth factor (IGF) binding protein 2 (IGFBP2) is a secreted protein that was initially characterized as binding and modulating the activity of IGF-I and -II.12, 13, 14 IGFBP2 can also function independently of IGF binding, and its versatility as a secreted or cytoplasmic signaling effector has been widely characterized. IGFBP2 can bind integrins15, 16, 17 and activate phosphatidylinositol 3′-kinase/AKT,18 nuclear factor-κB15 and extracellular signal–regulated kinase.19 Recently, a classic nuclear localization signal sequence that is responsible for nuclear entry has been identified in IGFBP2.20 However, the functional and clinical significance of nuclear IGFBP2 has not been clearly elucidated.21, 22, 23, 24
In mammals, IGFBP2 is expressed at high levels in embryonic tissues, but the expression is drastically decreased after birth. However, IGFBP2 expression has been observed postnatally in hematopoietic stem cells and in the liver and spleen progenitor cell populations.25, 26, 27, 28, 29, 30 IGFBP2 is reactivated during progression of a wide spectrum of cancer types, including glioma and prostate, breast and lung cancers.18, 30, 31, 32 IGFBP2 has an oncogenic role in tumor initiation and progression to high-grade glioma33 and is reported as one of the nine genes in a signature associated with poor clinical outcome in high-grade glioma.34 IGFBP2 mediates cell expansion and survival of glioma stem cells.35, 36 Despite the clear role for IGFBP2 in tumorigenesis, the mechanisms underlying nuclear IGFBP2’s contribution to the tumorigenic program remain unknown.
EGFR/IGFBP2 and EGFR/STAT337, 38 are concurrently co-expressed in glioma. EGFR, a cell surface tyrosine kinase receptor, is activated in 30–50% of high-grade gliomas through amplification, overexpression or mutation.39, 40, 41 EGFR signal transduction can be mediated by STAT3. STAT3 interacts with EGFR at two autophosphorylation sites in the cytoplasmic domain, tyrosine 1068 or tyrosine 108642 and is activated by phosphorylation at tyrosine 705 (Y705).43 In addition to this cytoplasmic interaction, EGFR and STAT3, after translocation into the nucleus, can form a complex to activate transcription of genes such as cyclooxygenase 2 (COX2),44 iNOS45 and c-MYC.46 Nuclear EGFR expression in glioma and other cancers, such as breast carcinoma,47 esophageal squamous cell carcinoma 48 and ovarian cancer,49 is associated with poor survival and linked to an aggressive tumor phenotype.50 Furthermore, IGFBP2 regulates the expression of VEGF, MMP2, TIMP1, TWIST, BCL2 and HIF1A genes,20, 51 which are known transcriptional targets of STAT3. Recent research implicated nuclear IGFPB2 in angiogenesis through activation of VEGF, a STAT3 target gene.20 These observations suggest that there is a functional connection between IGFBP2, EGFR and STAT3 in glioma. Here we tested this hypothesis and provide evidence that IGFBP2 mediates the tumorigenic program through a tightly linked IGFBP2–EGFR–STAT3 regulatory signaling network.
Results
IGFBP2 activates the STAT3 signaling pathway via an EGFR-dependent mechanism
To explore the functional interaction between IGFBP2, EGFR and STAT3, we stimulated SNB19 parental (SNB19.par) glioma cells, which had been serum-starved overnight, with increasing amounts of exogenous IGFBP2 protein. Immunoblotting analysis demonstrated increased expression of both total EGFR and EGFR activated via phosphorylation at tyrosine Y1068, or pEGFR(Y1068), in parallel with IGFBP2 uptake into the cell (Figure 1a). STAT3 activation via phosphorylation at tyrosine 705, designated pSTAT3(Y705), and the expression of STAT3 transcriptional targets Bcl-xL, cyclin D1 and c-MYC also increased in response to IGFBP2 stimulation.

IGFBP2 activates STAT3 through EGFR. (a) Immunoblot analysis of SNB19 cells starved of serum overnight and then stimulated with exogenous IGFBP2 protein at the indicated dosages (0, 50, 100, 250 ng/ml) for 60 min. Densitometric analysis shown below the immunoblot indicates fold-change relative to unstimulated control cells (normalized to beta-actin loading control or total protein for phosphorylated proteins). (b) Immunoblot analysis of U87 cells starved of serum overnight and then stimulated with exogenous IGFBP2 (100 ng/ml) for the indicated time points (0, 5, 10, 15, 30, 60 min). Densitometric analysis shown below the immunoblot indicates fold-change relative to unstimulated control cells (normalized to loading control or total protein for phosphorylated proteins). (c) Immunoblot analysis comparing stable SNB19 empty vector cells (SNB19.EV) to SNB19 cells stably overexpressing IGFBP2 (SNB19.BP2). Densitometric analysis shown below the immunoblot indicates fold-change relative to SNB19.EV after normalization to beta-tubulin loading control (or total protein for phosphorylated proteins). (d) Immunoblot analysis comparing SNB19.EV and SNB19.BP2 cells depleted of EGFR via two independent pools of EGFR siRNA (EGFR sir#1, EGFR sir#2) to cells transfected with scrambled negative control siRNA (ctrl siR). The intensity of pSTAT3(Y705), quantified by densitometry, is shown below the immunoblot as fold-change relative to control siRNA, normalized to total STAT3.
Next, we performed a time-course experiment in which U87 glioma cells, which lack endogenous IGFBP2 expression, were stimulated with exogenous IGFBP2 after overnight serum starvation. Immunoblotting analysis revealed induction of EGFR, STAT3 and Bcl-xL expression as early as 5 min following addition of exogenous IGFBP2 (Figure 1b). In comparison with EGF (EGFR ligand) stimulation, IGFBP2 stimulation of EGFR activation was substantially less than that produced by EGF (data not shown). Furthermore, immunoblotting analysis of SNB19 cells stably overexpressing IGFBP2 (SNB19.BP2) demonstrated that, compared with SNB19 cells stably transfected with empty vector (SNB19.EV), IGFBP2 overexpression resulted in the increased expression of EGFR and phosphorylated STAT3, along with Bcl-xL, cyclin D1 and c-MYC (Figure 1c).
To examine the involvement of EGFR in IGFBP2-mediated STAT3 activation, we depleted EGFR by using two different pools of small interfering RNA (siRNA) in SNB19.BP2 cells and observed decreases in STAT3 activation (Figure 1d), supporting the hypothesis that IGFBP2 mediates STAT3 activation through EGFR. To rule out the possibility of off-target effects of EGFR siRNA-mediated knockdown, we knocked down EGFR in SNB19.BP2 cells and stimulated the cells with recombinant interleukin-6. We observed STAT3 phosphorylation in these cells, confirming that EGFR knockdown impairs STAT3 activation by IGFBP2 without compromising alternate STAT3 activation pathways (Supplementary Figure S1).
EGFR can be indirectly activated through transactivation, which involves a disintegrin and metalloproteinases (ADAMs).52 To determine whether ADAMs are involved in IGFBP2-mediated EGFR activation, we inhibited ADAMs by treatment with two different ADAM inhibitors, TAPI2 and marimastat.53, 54 U87 cells serum-starved overnight were pretreated with 20 μM TAPI-2 or marimastat and then stimulated with exogenous IGFBP2 for 5 min (Supplementary Figure S2A). Immunoblotting analysis demonstrated that exogenous IGFBP2 stimulated EGFR and STAT3 activation despite ADAM inhibition. Furthermore, because ADAM17 is essential to regulation of EGFR transactivation,52 we knocked down ADAM17 using two different pools of siRNA to evaluate whether IGFBP2-mediated EGFR activation involves ADAM17 (Supplementary Figure S2B). Immunoblotting analysis showed that ADAM17 knockdown did not affect EGFR and STAT3 activation in SNB19.BP2 cells. These data demonstrate that ADAMs are not involved in IGFBP2-mediated EGFR/STAT3 activation.
IGFBP2 is significantly correlated with STAT3 pathway activation in glioma
Previous studies showed that IGFBP2 regulates the expression of many STAT3 target genes,20, 51 and our results demonstrate that IGFBP2 can stimulate STAT3 activation through EGFR. To gain a comprehensive view of the relationship between IGFBP2 and STAT3 signaling, we analyzed the whole-genome gene expression profiling data from The Cancer Genome Atlas (TCGA) low-grade glioma (LGG) database. Gene set enrichment analysis (GSEA) revealed that STAT3-activated genes were significantly and positively correlated with IGFBP2 (Figure 2a), suggesting that IGFBP2 expression is associated with STAT3 activation. To further substantiate the IGFBP2–STAT3 link, we performed hierarchical clustering on the 157 experimentally validated STAT3 target genes across all samples in the REpository for Molecular BRAin Neoplasia DaTa (REMBRANDT) data set. Two distinct clusters were formed, associated with tumor grade and IGFBP2 and STAT3 expression but not with other transcription factors, such as beta-catenin (CTNNB1) or Forkhead box protein M1 (FOXM1) (Supplementary Figure S3). This finding further validates that the expression of IGFBP2 and STAT3 are tightly linked.

IGFBP2 is strongly and significantly correlated with STAT3 pathway genes. (a) GSEA demonstrated enrichment for STAT3 target genes based on correlation with IGFBP2 expression in the TCGA low-grade glioma database. The top of the panel shows the enrichment score (ES) for genes associated with STAT3 signaling pathway targets. The blue lines indicate where the STAT3 target genes appear in the ranked gene list, and the black lines represent the top 45 highly correlated targets. The bottom of the panel shows the ranking scores (correlation of all genes associated with the STAT3 signaling pathway targets with IGFBP2). (b) Correlation of expression of proteins in the TCGA RPPA data with IGFBP2 (x axis) and pSTAT3(Y705) (y axis). Each dot represents a protein. Proteins with correlation coefficients >0.2 are highlighted in orange. (c) Correlation of the seven proteins with the highest correlation coefficients with both IGFBP2 and STAT3. Also shown is the relationship of each protein with STAT3 (‘target’=STAT3 transcriptional target; ‘regulator’=STAT3 upstream regulator). Y=yes, a known target or upstream regulator of STAT3; N=not a known target or upstream regulator of STAT3.
Next we postulated that the most functionally important of the correlated genes would likely be associated with STAT3 activity (as measured by phosphorylation) in the reverse-phase protein array (RPPA) data of the same TCGA cohort. In this proteomic analysis, we identified the seven proteins (Figures 2b and c) that were most significantly and strongly correlated with both IGFBP2 and pSTAT3(Y705) (correlation coefficients >0.2). Of these seven strongly correlated proteins, five are closely related to the STAT3 signaling pathway, namely plasminogen activator inhibitor-1, fibronectin, cyclin B1, pHER2(Y1248) and, notably, pEGFR(Y1068). HER2 is a member of the EGFR family and an upstream regulator of STAT3; however, it has not been shown to have clinical significance in glioma.55, 56, 57, 58, 59 Thus these results from patient samples are consistent with the results of our in vitro cell line–based studies, and together these results illustrate the potential importance of the IGFBP2–EGFR–STAT3 signaling axis in glioma.
IGFBP2 co-precipitates and co-localizes with EGFR
To further evaluate the functional relationship between IGFBP2 and EGFR, we performed reciprocal immunoprecipitation (IP) studies followed by immunoblotting comparing IGFBP2-overexpressing SNB19 cells and empty vector control cells. Co-IP experiments revealed co-precipitation of IGFBP2 and EGFR (Figure 3a). We next treated U87 cells, which had been serum-starved overnight, with two different doses of exogenous IGFBP2 followed by IP analysis and immunoblotting. The results showed a dose-dependent increase of IGFBP2 co-precipitated with EGFR (Figure 3b). Confocal imaging analysis of SNB19.BP2 cells demonstrated clear co-localization of IGFBP2 and EGFR proteins on the cell membrane and in the cytoplasm and nucleus (Figure 3c). Co-localization of IGFBP2 and EGFR provides further evidence of a complex containing IGFBP2 and EGFR.

IGFBP2 co-precipitates and co-localizes with EGFR. (a) Co-IP of IGFBP2 and EGFR in SNB19.EV control cells versus SNB19.BP2 cells analyzed by immunoblotting (IB). (b) IP of IGFBP2 in U87 cells starved of serum overnight and then stimulated with two different doses of IGFBP2 for 30 min, analyzed by IB. (c) Confocal microscopic images of immunofluorescence staining for IGFBP2 (green), EGFR (red) and DAPI (blue) in SNB19.BP2 cells show IGFBP2 and EGFR co-localization; blue arrow=cell membrane; purple arrow=cytoplasm; white arrow=nucleus.
IGFBP2 facilitates EGFR nuclear accumulation
Because we observed IGFBP2 and EGFR co-localization in the cytoplasm and nucleus, we investigated whether nuclear IGFBP2 is closely linked to nuclear EGFR and whether this complex augments STAT3 transcriptional activation. We first fractionated SNB19.BP2 and SNB19.EV cells into cytoplasmic and nuclear fractions and performed immunoblotting to detect IGFBP2, EGFR and STAT3. Our results revealed that a substantial proportion of IGFBP2 and EGFR localized to the nucleus in SNB19.BP2 cells (Figure 4a). We then determined the ratio of nuclear to cytoplasmic EGFR via densitometric analysis and found that SNB19.BP2 cells had more than twice as much nuclear EGFR as SNB19.EV cells.

IGFBP2 drives EGFR nuclear accumulation. (a) Immunoblot analysis of cytoplasmic (cyt) and nuclear (nuc) fractions of SNB19.EV and SNB19.BP2 cells. Beta-tubulin represents a loading control for the cytoplasmic fraction, and PARP represents a loading control for the nuclear fraction. Densitometric analysis represented by the bar graph demonstrates the percentage of cytoplasmic or nuclear EGFR. (b) Confocal images of SNB19 parental cells and SNB19 parental cells stimulated with exogenous IGFBP2 protein (250 ng/ml for 30 min). Cells were stained for EGFR (red) and the nuclei were stained with DAPI (blue). (c) Immunoblot analysis of cytoplasmic and nuclear fractions of SNB19 parental cells stimulated with exogenous IGFBP2 (250 ng/ml for the indicated times). The graph represents fold-change of cytoplasmic or nuclear IGFBP2 and EGFR calculated from densitometric analysis of the immunoblot bands. (d) Immunoblot analysis comparing cytoplasmic and nuclear fractions of SNB19.BP2 cells depleted of IGFBP2 via two independent pools of IGFBP2 siRNA (BP2 siR #1, #2) to cells transfected with scrambled negative control siRNA (ctrl siR). Densitometric analysis represented by the bar graph demonstrates the percentage of cytoplasmic or nuclear EGFR. (e) Immunoblot analysis of cytoplasmic and nuclear fractions of transiently transfected SNB19.EV, SNB19.BP2 wild type (BP2 WT) and SNB19 with a mutated IGFBP2 nuclear localization signal (BP2ΔNLS). Densitometric analysis represented by the bar graph demonstrates the percentage of cytoplasmic or nuclear EGFR. (f) Immunoblot analysis of cytoplasmic and nuclear proteins in stable SNB19.EV, SNB19.BP2 WT and SNB19.BP2ΔNLS cells.
To investigate whether IGFBP2 facilitates EGFR nuclear accumulation, we stimulated SNB19.par cells, which had been serum-starved overnight with exogenous IGFBP2 protein and then visualized EGFR protein localization by confocal imaging. IGFBP2 stimulation of SNB19.par cells resulted in EGFR accumulation in the nucleus (Figure 4b). A time-course study with the same cells demonstrated that IGFBP2 nuclear accumulation paralleled EGFR nuclear accumulation in a time-dependent manner (Figure 4c). To validate that EGFR nuclear accumulation is mediated through IGFBP2, we knocked down IGFBP2 using two different pools of siRNA in SNB19.BP2 cells and performed immunoblotting analysis on the fractionated cells. IGFBP2 depletion led to impaired EGFR nuclear localization with coordinate cytoplasmic accumulation of EGFR, whereas control knockdown did not affect EGFR nuclear accumulation (Figure 4d). These results suggest that IGFBP2 has a role in promoting EGFR nuclear accumulation.
Nuclear translocation of IGFBP2 is required for IGFBP2-mediated EGFR nuclear accumulation
To better understand the mechanism of nuclear IGFBP2–mediated EGFR nuclear accumulation, we generated an IGFBP2 construct with a mutant nuclear localization signal60 (BP2ΔNLS; Supplementary Figure S4A). Transient transfection of BP2ΔNLS plasmid into SNB19.par cells resulted in the expected compromise of IGFBP2 nuclear entry and also impaired EGFR nuclear accumulation (Figure 4e). Next, we created a stable BP2ΔNLS-overexpressing cell line (SNB19.BP2ΔNLS). Impaired EGFR nuclear accumulation in fractionated stable SNB19.BP2ΔNLS cells, compared with SNB19.BP2 WT (wild-type IGFBP2), resulted in decreased nuclear expression of COX2 and c-MYC, which are known downstream targets of nuclear EGFR/STAT3 complex (Figure 4f). These results were replicated in another glioma cell line, T98G (Supplementary Figures S5A and B). To determine whether BP2ΔNLS can bind to EGFR, we transiently transfected U87 cells with BP2 WT or BP2ΔNLS plasmid and performed IP followed by immunoblotting (Supplementary Figure S6). The results showed that mutation of IGFBP2 NLS does not affect binding to EGFR, demonstrating that nuclear translocation of IGFBP2 is important for mediating EGFR nuclear accumulation. Because IGFBP2 is involved in glioma cell migration and invasion,15, 51 we then performed migration and invasion assays using the SNB19.EV, SNB19.BP2 WT and SNB19.BP2ΔNLS cell lines. Migration and invasion potential were significantly impaired in the SNB19.BP2ΔNLS cells compared with SNB19.BP2 WT (Supplementary Figures S4B and C), indicating that nuclear IGFBP2 is important for the invasive phenotype of glioma cells, plausibly through regulation of nuclear EGFR–STAT3 activity.
Levels of nuclear EGFR, nuclear IGFBP2 and pSTAT3 are significantly correlated in glioma
The RPPA LGG data from TCGA revealed a close relationship between IGFBP2, activated EGFR and activated STAT3 but did not provide spatial information. To further investigate localization of these proteins, we performed immunohistochemical analysis to determine the association between IGFBP2, EGFR and pSTAT3(Y705) in a clinical glioma tissue microarray (TMA) comprising 222 samples of grade 2–4 gliomas. We observed both cytosolic and nuclear localization of IGFBP2, both of which were strongly associated with STAT3 phosphorylation in these gliomas (Figures 5a and b, Supplementary Table S1). Both cytosolic and nuclear IGFBP2 expression positively correlated with increased fraction and degree of phosphorylation of STAT3 (P=0.023 and P=0.018, respectively), suggesting a functional link between IGFBP2 expression and STAT3 phosphorylation.

IGFBP2 correlates with STAT3 activation and nuclear EGFR localization in clinical samples. Expression and localization of IGFBP2, pSTAT3(Y705) and EGFR were detected with immunohistochemistry from a TMA that included 222 human grade 2–4 gliomas. (a) TMA immunostaining images (magnification × 40) representing weak and strong staining of IGFBP2 and pSTAT3(Y705). (b) Cytosolic and nuclear IGFBP2 expression associated with the percentage of cells positive for pSTAT3 and with pSTAT3 staining intensity. Bar graphs illustrate the increasing fractions of pSTAT3-positive cells and pSTAT3 intensity upon increasing IGFBP2 intensity or nuclear accumulation. (c) TMA immunostaining images (magnification × 40) representing low and high nuclear localization of IGFBP2 and EGFR. (d) Nuclear IGFBP2 associated with nuclear EGFR. The bar graph illustrates the fraction of samples with increasing nuclear EGFR localization upon increasing nuclear accumulation of IGFBP2. (e) Nuclear co-localization of IGFBP2, EGFR and phosphorylated STAT3 predicted poor survival among patients with human grade 2–4 gliomas. Patients were stratified into two cohorts based on the nuclear staining of all three proteins: triple positives (⩾1% of cells with nuclear expression, n=51, red line) and all other cases (n=83, blue line). Survival rates were visualized by using a Kaplan–Meier survival plot (P=0.0086).
We observed nuclear co-localization of IGFBP2 and EGFR in the clinical samples (Figures 5c and d, and Supplementary Table S1). Cytosolic IGFBP2 did not correlate with nuclear EGFR, and nuclear IGFBP2 did not correlate with cytosolic EGFR. However, nuclear IGFBP2 positively associated with nuclear EGFR localization (P=0.011). Furthermore, clinical samples that were triply positive for nuclear accumulation of IGFBP2, phosphorylated STAT3 and EGFR were strongly associated with poor survival (Figure 5e).
Discussion
Conventionally, proteins are categorized as secreted, membrane-bound or intracellular. However, accumulating evidence demonstrates that signaling molecules are spatiotemporally dynamic;61, 62, 63, 64, 65, 66 in addition to cell surface–initiated signaling, upon internalization, EGFR can mediate signaling in the endosome,67, 68, 69, 70 nucleus71 or mitochondria.72, 73 EGFR crosstalks with STAT3 through two levels: tyrosine kinase–mediated activation of STAT342, 43, 74 and nuclear cooperation as transcriptional cofactors.44, 45, 46 Our results significantly expand our understanding of this network by demonstrating that IGFBP2 stimulation or overexpression contributes to activation of EGFR–STAT3 and downstream pathways. IGFBP2 forms a complex containing EGFR and facilitates the nuclear accumulation of EGFR to potentiate nuclear EGFR–STAT3 activity. Thus IGFBP2 controls two fundamental functions of EGFR, cytoplasmic signal transduction and nuclear accumulation.
These results also elucidate newly identified oncogenic functions for nuclear IGFBP2. By facilitating nuclear EGFR accumulation, nuclear IGFBP2 activates an EGFR–STAT3-mediated transcriptional program, which promotes transcription of iNOS, c-MYC and COX2.44, 45, 46 The resulting aberrant activation of these genes leads to uncontrolled cell proliferation, survival and metastasis and hence poor prognosis. COX2 is upregulated and correlated with poor survival in glioma,75, 76, 77 iNOS is critical for glioma stem cell survival and tumorigenesis78, 79, 80, 81 and c-MYC is correlated with high-grade glioma and important for glioma stem cell maintenance.82, 83, 84 We also demonstrated a role for nuclear IGFBP2 in promoting cancer cell migration and invasion, a key hallmark of cancer and a major aspect of glioma aggressiveness and treatment response.
The dynamic nature of these oncogenic signaling molecules may contribute to the ineffectiveness of EGFR-targeted therapy in glioma, perhaps because EGFR is being actively translocated into the nucleus by IGFBP2, rendering the cells resistant to therapies targeting membrane EGFR. Thus IGFBP2 may serve as an escape mechanism for glioma cells from EGFR-targeted therapy. EGFR can be activated by eight different ligands,85, 86 including EGF, transforming growth factor α and heparin-binding EGF. EGF, transforming growth factor α and heparin-binding EGF are constitutively expressed in the developing and normal adult brain.87, 88, 89, 90 Therefore, therapeutic targeting of these ligands is not feasible. Conversely, IGFBP2 is highly expressed in the fetal brain and glioma but not in the normal adult brain,26, 27, 30, 91, 92 making it a better therapeutic target than the known EGF ligands. EGFR amplification and activation are among the most common oncogenic events in human cancer,93, 94 and IGFBP2 overexpression is also a frequent event in glioma and other cancers. Our analysis of three independent clinical data sets revealed strong correlation between IGFBP2 and EGFR in terms of both gene expression and protein localization in the cell. The development of therapeutic strategies that target both molecules may represent a rational approach for cancer therapeutics.
Materials and methods
Cell culture, treatments, plasmids and transfections
SNB19, U87 and T98G cells were obtained from ATCC (Manassas, VA, USA). Cells were cultured in Dulbecco modified essential/F12 50:50 medium supplemented with 10% fetal bovine serum and 5% penicillin/streptomycin in an incubator with 5% CO2 at 37 °C. SNB19.EV (empty vector) and SNB19.BP2 WT (IGFBP2 wild type) cells were created as previously described.51 To generate BP2ΔNLS (IGFBP2 mutation at the nuclear localization signal), amino-acid residues 179PKKLRPP185 of the IGFBP2 nuclear localization signal were mutated to 179PNNLAPP185 using the Quikchange Lightning site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer’s protocol. A stable SNB19.BP2ΔNLS cell line was created by transfection of pcDNA3.1.IGFBP2ΔNLS plasmid via FuGENE HD (Promega, Fitchburg, WI, USA) according to the manufacturer’s protocol, followed by G418 selection for 3 weeks.
IGFBP2 stimulation experiments were performed by using recombinant IGFBP2 (ab63223; Abcam, Cambridge, MA, USA) with cells starved of serum overnight. Depletion of IGFBP2 and EGFR was achieved via transfection of Lipofectamine RNAiMAX (Life Technologies, Grand Island, NY, USA) according to the manufacturer’s protocol with two different pools of siRNA from Mission siRNA (Sigma, St Louis, MO, USA) for 48 h. Some cells were treated with a broad-spectrum ADAM inhibitor, TAPI-2 (no. 14695; Cayman Chemical, Ann Arbor, MI, USA) or marimatstat (no. M2699; Sigma) at 20 μM for 2 h. Depletion of ADAM17 siRNA was achieved via transfection of Lipofectamine RNAiMAX according to the manufacturer’s protocol with two different pools of siRNA from Life Technologies (nos. s13718 and s13719).
Gene set enrichment analysis
A total of 268 LGG samples obtained from the TCGA data portal (https://tcga-data.nci.nih.gov/tcga/) were subjected to RNA sequencing. The gene expression data were median-centered and then transformed to log2 space. We calculated the correlation of IGFBP2 gene expression with all other genes in the genome and ranked the genes in descending order based on the correlation coefficients. Using the gene expression correlation as the ranking metric, GSEA was then used to calculate the score for the degree of enrichment of the genes with higher correlation coefficients among genes involved in the STAT3 signaling pathway.95
In a similar manner, the correlation of IGFBP2 or STAT3 protein expression with proteins in the TCGA TMA was calculated for 257 LGG samples for which RPPA data were available. Proteins that had higher correlation coefficients with both IGFBP2 and STAT3 proteins were considered the most likely candidates to represent molecular mechanisms underlying the association of IGFBP2 and the STAT3 signaling pathway.
Ingenuity Pathway Analysis
The interaction network feature of Ingenuity Pathway Analysis was used to determine direct downstream targets of STAT3. Interactions were filtered on the basis of their confidence level so that only interactions experimentally observed in humans were included in the table of results. Interactions were also filtered by relationship type so that only interactions of type ‘expression’ or ‘transcription’ were included.
IP, immunoblotting and cellular fractionation
For IP, cells were subjected to lysis in NP-40 buffer with 0.1% phosphatase inhibitor cocktail (Pierce Biotechnology, Thermo Fisher Scientific, Waltham, MA, USA). After preclearing for 1 h at 4 °C with Protein G agarose beads (SC no. 2002; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and appropriate species normal immnuoglobulin G (IgG), lysates were immunoprecipitated overnight at 4 °C with Protein G agarose beads using antibodies to IGFBP2 (no. SC-6001; Santa Cruz Biotechnology; 1:100) and EGFR (no. 2256; Cell Signaling Technology, Beverly, MA, USA; 1:100). Beads were washed with NP-40 buffer three times and boiled in Laemmli buffer. Proteins from the IP experiment or extracted from cell lysates were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (10%) in running buffer and transferred onto an Immobilon TM-PVDF membrane (Millipore, Billerica, MA, USA) for 1 h at 100 V in transfer buffer (24 mM Tris base, 191 mM glycine and 20% (v/v) methanol). Membranes were blocked for 1 h at room temperature with 5% (w/v) non-fat milk powder in phosphate-buffered saline solution (PBS) with 0.1% Tween-20 (PBST) and incubated overnight at 4 °C with primary antibody: IGFBP2 (no. SC-6001; 1:500); EGFR (no. 4267; Cell Signaling Technology; 1:1000), EGFR-Y1068 (no. 3777; Cell Signaling Technology; 1:1000), beta-tubulin (no. 2128; Cell Signaling Technology; 1:1000), PARP (poly ADP-ribose polymerase; no. 9542; Cell Signaling Technology; 1:1000), STAT3 (no. 9139; Cell Signaling Technology; 1:1000), STAT3-Y705 (no. 9145; Cell Signaling Technology; 1:1000), Bcl-xL (no. 2764; Cell Signaling Technology; 1:1000), cyclin D1 (no. 2978; Cell Signaling Technology; 1:1000), c-MYC (no. SC-40; Santa Cruz Biotechnology; 1:1000), COX-2 (no. 160112; Cayman Chemical; 1:250), or ADAM17 (no. T5442; Sigma; 1:500) in blocking solution. After washing in PBST, blots were incubated for 1 h at room temperature in PBST with secondary antibodies (anti-goat IgG, anti-rabbit IgG, or anti-mouse IgG; Santa Cruz Biotechnology; 1:5000) coupled to horseradish peroxidase (HRP). Immunoblots were incubated with enhanced chemiluminescence SuperSignal West Pico or Femto solution (Pierce Biotechnology). Cellular fractionation was performed by using the NE-PER Nuclear and Cytoplasmic Kit (Pierce Biotechnology) according to the manufacturer’s protocol. Densitometric analysis of immunoblot bands were quantified using the ImageJ software (US National Institutes of Health, Bethesda, MD, USA).
Confocal imaging
Cell on chamber slides were fixed in 4% paraformaldehyde, permeabilized with 0.5% Triton X-100 and incubated with primary antibody to EGFR (no. 4267; 1:100) and IGFBP2 (no. SC-6001; 1:100) at 4 °C overnight. They were then incubated with secondary antibody (Life Technologies (Alexa Fluor); 1:500) for 1 h at room temperature in 1% bovine serum albumin/PBS buffer. They were mounted in Vectashield (Vector Laboratories, Burlingame, CA, USA), and nuclei were counterstained with DAPI (4′,6-diamidino-2-phenylindole, dihydrochloride). Immunofluorescence images were acquired by using an Olympus FV1000 Laser Confocal Microscope at × 40/NA 1.3 objective (stacking from basement membrane to apical site at 1-μM intervals).
TMA construction and immunohistochemical analysis
Tumor samples were collected, and the TMA comprising formalin-fixed, paraffin-embedded astrocytoma tissues was processed at Tampere University Hospital as described previously.96 Briefly, histologically representative tumor regions were selected by a neuropathologist (HH), and samples from these areas were placed in TMA blocks using a custom-built instrument (Beecher Instruments, Sun Prairie, WI, USA). The diameter of the tissue cores in the microarray block was 1 mm. Altogether, 222 diffusely infiltrating astrocytomas (167 glioblastomas, 17 grade 3 astrocytomas and 38 grade 2 astrocytomas) were included in the immunohistochemical analysis. For staining, 5-μm sections from TMA blocks were deparaffinized in xylene or hexane and rehydrated through an ethanol dilution series. Immunohistochemical staining was performed with goat antibodies against human IGFBP2 (no. SC-6001; 1:300), phosphorylated STAT3 (no. 9145; 1:100) and EGFR (GR-01, Calbiochem, San Diego, CA, USA; 1:50), together with the HRP-diaminobenzidine (DAB)–based Cell and Tissue Staining Kit (R&D Systems, Minneapolis, MN, USA) or the Envision+System HRP-DAB kit (Dako, Carpenteria, CA, USA).
Intensity of cytosolic expression levels of the proteins in tumor cells was manually quantified by using a scoring system from 0 to 3 (0=no signal, 1=weak signal, 2=moderate signal and 3=strong signal). The proportion of the cells with nuclear protein localization was manually classified into four categories: 0, <10, 10–30 and ⩾30%. Intensity of nuclear expression levels in tumor cells was manually quantified by using a scoring system from 0 to 2 (0=no signal, 1=weak signal, 2=strong signal). The TMA samples were examined and scored by two neuropathologists who were blinded to the clinical data. A survival association analysis of the patients from whom these samples were taken compared survival in patients with nuclear co-localization of all three proteins—IGFBP2, EGFR and phosphorylated STAT3 (⩾1% cells with nuclear staining)—with survival of all the other patients. The survival data were analyzed by the log-rank test and visualized with a Kaplan–Meier plot. Statistical analyses were run with the SPSS 20.0 software for Windows (SPSS Inc., Chicago, IL, USA). The statistical significance of associations was evaluated by using the Pearson chi-square test.
Invasion and migration assays
The cell invasion assay was performed in triplicate in Matrigel-coated transwell chambers (8-μm pore size; (BD Biosciences, San Jose, CA, USA). The cells were plated in 500 μl of serum-free medium (4 × 104 cells per transwell) and allowed to invade toward a medium containing 10% fetal bovine serum for 16 h. Cells that invaded into the underside of the filter were fixed and stained with HEMA-DIFF solution (Thermo Fisher Scientific). The numbers of invaded cells from five randomly chosen fields from each membrane were counted. The cell migration assay was performed the same way as the invasion assay, using transwell chambers (8-μm pore size, BD Biosciences), and the cells were allowed to migrate for 4 h. The data were expressed as means±s.e.m. and analyzed by Student's t-test for difference between the two groups.
Statistical analysis
Experiments were performed at least three times. GraphPrism 6 (GraphPad, La Jolla, CA, USA) and SPSS 20.0 software for Windows (SPSS Inc.) were used for statistical analysis and graphing. The Spearman correlation test was used to examine correlation between protein or phosphoprotein expression in the TCGA RPPA data set. The survival data were analyzed by the log-rank test and visualized with a Kaplan–Meier plot. The statistical significance of protein associations in the TMA data set was evaluated by using the Pearson chi-square test. Statistical test on GSEA was estimated as previously described.95 Student's t-tests were used for paired comparisons where variances were estimated to be similar. Except for one-side test for the GSEA analysis,95 all other tests were two-sided, with P<0.05 as the threshold for statistical significance in all the tests. Indicated annotations correspond to the following P-values: *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001.
References
Dobrowolski R, De Robertis EM . Endocytic control of growth factor signalling: multivesicular bodies as signalling organelles. Nat Rev Mol Cell Biol 2012; 13: 53–60.
Sorkin A, Von Zastrow M . Signal transduction and endocytosis: close encounters of many kinds. Nat Rev Mol Cell Biol 2002; 3: 600–614.
Sorkin A, von Zastrow M . Endocytosis and signalling: intertwining molecular networks. Nat Rev Mol Cell Biol 2009; 10: 609–622.
McMahon HT, Boucrot E . Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol 2011; 12: 517–533.
Baxter RC . Insulin-like growth factor binding protein-3 (IGFBP-3): Novel ligands mediate unexpected functions. J Cell Commun Signal 2013; 7: 179–189.
Joy A, Moffett J, Neary K, Mordechai E, Stachowiak EK, Coons S et al. Nuclear accumulation of FGF-2 is associated with proliferation of human astrocytes and glioma cells. Oncogene 1997; 14: 171–183.
Stachowiak MK, Fang X, Myers JM, Dunham SM, Berezney R, Maher PA et al. Integrative nuclear FGFR1 signaling (INFS) as a part of a universal "feed-forward-and-gate" signaling module that controls cell growth and differentiation. J Cell Biochem 2003; 90: 662–691.
Arese M, Chen Y, Florkiewicz RZ, Gualandris A, Shen B, Rifkin DB . Nuclear activities of basic fibroblast growth factor: potentiation of low-serum growth mediated by natural or chimeric nuclear localization signals. Mol Biol Cell 1999; 10: 1429–1444.
Lin SY, Makino K, Xia W, Matin A, Wen Y, Kwong KY et al. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol 2001; 3: 802–808.
Sarfstein R, Werner H . Minireview: nuclear insulin and insulin-like growth factor-1 receptors: a novel paradigm in signal transduction. Endocrinology 2013; 154: 1672–1679.
Rakowicz-Szulczynska EM, Rodeck U, Herlyn M, Koprowski H . Chromatin binding of epidermal growth factor, nerve growth factor, and platelet-derived growth factor in cells bearing the appropriate surface receptors. Proc Natl Acad Sci USA 1986; 83: 3728–3732.
Baxter RC . Circulating binding proteins for the insulinlike growth factors. Trends Endocrinol Metab 1993; 4: 91–96.
Clemmons DR . Insulinlike growth factor binding proteins. Trends Endocrinol Metab 1990; 1: 412–417.
Scharf J, Ramadori G, Braulke T, Hartmann H . Synthesis of insulinlike growth factor binding proteins and of the acid-labile subunit in primary cultures of rat hepatocytes, of Kupffer cells, and in cocultures: regulation by insulin, insulinlike growth factor, and growth hormone. Hepatology 1996; 23: 818–827.
Holmes KM, Annala M, Chua CY, Dunlap SM, Liu Y, Hugen N et al. Insulin-like growth factor-binding protein 2-driven glioma progression is prevented by blocking a clinically significant integrin, integrin-linked kinase, and NF-kappaB network. Proc Natl Acad Sci USA 2012; 109: 3475–3480.
Wang GK, Hu L, Fuller GN, Zhang W . An interaction between insulin-like growth factor-binding protein 2 (IGFBP2) and integrin alpha5 is essential for IGFBP2-induced cell mobility. J Biol Chem 2006; 281: 14085–14091.
Pereira JJ, Meyer T, Docherty SE, Reid HH, Marshall J, Thompson EW et al. Bimolecular interaction of insulin-like growth factor (IGF) binding protein-2 with alphavbeta3 negatively modulates IGF-I-mediated migration and tumor growth. Cancer Res 2004; 64: 977–984.
Mehrian-Shai R, Chen CD, Shi T, Horvath S, Nelson SF, Reichardt JK et al. Insulin growth factor-binding protein 2 is a candidate biomarker for PTEN status and PI3K/Akt pathway activation in glioblastoma and prostate cancer. Proc Natl Acad Sci USA 2007; 104: 5563–5568.
Han S, Li Z, Master LM, Master ZW, Wu A . Exogenous IGFBP-2 promotes proliferation, invasion, and chemoresistance to temozolomide in glioma cells via the integrin beta1-ERK pathway. Br J Cancer 2014; 111: 1400–1409.
Azar WJ, Azar SH, Higgins S, Hu JF, Hoffman AR, Newgreen DF et al. IGFBP-2 enhances VEGF gene promoter activity and consequent promotion of angiogenesis by neuroblastoma cells. Endocrinology 2011; 152: 3332–3342.
Terrien X, Bonvin E, Corroyer S, Tabary O, Clement A, Henrion Caude A . Intracellular colocalization and interaction of IGF-binding protein-2 with the cyclin-dependent kinase inhibitor p21CIP1/WAF1 during growth inhibition. Biochem J 2005; 392: 457–465.
Miyako K, Cobb LJ, Francis M, Huang A, Peng B, Pintar JE et al. PAPA-1 Is a nuclear binding partner of IGFBP-2 and modulates its growth-promoting actions. Mol Endocrinol 2009; 23: 169–175.
Hoeflich A, Reisinger R, Schuett BS, Elmlinger MW, Russo VC, Vargas GA et al. Peri/nuclear localization of intact insulin-like growth factor binding protein-2 and a distinct carboxyl-terminal IGFBP-2 fragment in vivo. Biochem Biophys Res Commun 2004; 324: 705–710.
Besnard V, Corroyer S, Trugnan G, Chadelat K, Nabeyrat E, Cazals V et al. Distinct patterns of insulin-like growth factor binding protein (IGFBP)-2 and IGFBP-3 expression in oxidant exposed lung epithelial cells. Biochim Biophys Acta 2001; 1538: 47–58.
Gerrard DE, Okamura CS, Grant AL . Expression and location of IGF binding proteins-2, -4, and -5 in developing fetal tissues. J Anim Sci 1999; 77: 1431–1441.
Green BN, Jones SB, Streck RD, Wood TL, Rotwein P, Pintar JE . Distinct expression patterns of insulin-like growth factor binding proteins 2 and 5 during fetal and postnatal development. Endocrinology 1994; 134: 954–962.
Lee WH, Michels KM, Bondy CA . Localization of insulin-like growth factor binding protein-2 messenger RNA during postnatal brain development: correlation with insulin-like growth factors I and II. Neuroscience 1993; 53: 251–265.
Wood TL, Streck RD, Pintar JE . Expression of the IGFBP-2 gene in post-implantation rat embryos. Development 1992; 114: 59–66.
Huynh H, Zheng J, Umikawa M, Zhang C, Silvany R, Iizuka S et al. IGF binding protein 2 supports the survival and cycling of hematopoietic stem cells. Blood 2011; 118: 3236–3243.
Fuller GN, Rhee CH, Hess KR, Caskey LS, Wang R, Bruner JM et al. Reactivation of insulin-like growth factor binding protein 2 expression in glioblastoma multiforme: a revelation by parallel gene expression profiling. Cancer Res 1999; 59: 4228–4232.
Busund LT, Richardsen E, Busund R, Ukkonen T, Bjornsen T, Busch C et al. Significant expression of IGFBP2 in breast cancer compared with benign lesions. J Clin Pathol 2005; 58: 361–366.
Yazawa T, Sato H, Shimoyamada H, Okudela K, Woo T, Tajiri M et al. Neuroendocrine cancer-specific up-regulating mechanism of insulin-like growth factor binding protein-2 in small cell lung cancer. Am J Pathol 2009; 175: 976–987.
Dunlap SM, Celestino J, Wang H, Jiang R, Holland EC, Fuller GN et al. Insulin-like growth factor binding protein 2 promotes glioma development and progression. Proc Natl Acad Sci USA 2007; 104: 11736–11741.
Colman H, Zhang L, Sulman EP, McDonald JM, Shooshtari NL, Rivera A et al. A multigene predictor of outcome in glioblastoma. Neuro Oncol 2010; 12: 49–57.
Hsieh D, Hsieh A, Stea B, Ellsworth R . IGFBP2 promotes glioma tumor stem cell expansion and survival. Biochem Biophys Res Commun 2010; 397: 367–372.
Li X, Liu Y, Granberg KJ, Wang Q, Moore LM, Ji P et al. Two mature products of MIR-491 coordinate to suppress key cancer hallmarks in glioblastoma. Oncogene 2014; 34: 1619–1628.
Lo HW, Cao X, Zhu H, Ali-Osman F . Constitutively activated STAT3 frequently coexpresses with epidermal growth factor receptor in high-grade gliomas and targeting STAT3 sensitizes them to Iressa and alkylators. Clin Cancer Res 2008; 14: 6042–6054.
Scrideli CA, Carlotti Jr CG, Mata JF, Neder L, Machado HR, Oba-Sinjo SM et al. Prognostic significance of co-overexpression of the EGFR/IGFBP-2/HIF-2A genes in astrocytomas. J Neurooncol 2007; 83: 233–239.
Nishikawa R, Ji XD, Harmon RC, Lazar CS, Gill GN, Cavenee WK et al. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci USA 1994; 91: 7727–7731.
Ekstrand AJ, Longo N, Hamid ML, Olson JJ, Liu L, Collins VP et al. Functional characterization of an EGF receptor with a truncated extracellular domain expressed in glioblastomas with EGFR gene amplification. Oncogene 1994; 9: 2313–2320.
Heimberger AB, Hlatky R, Suki D, Yang D, Weinberg J, Gilbert M et al. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin Cancer Res 2005; 11: 1462–1466.
Shao H, Cheng HY, Cook RG, Tweardy DJ . Identification and characterization of signal transducer and activator of transcription 3 recruitment sites within the epidermal growth factor receptor. Cancer Res 2003; 63: 3923–3930.
Park OK, Schaefer TS, Nathans D . In vitro activation of Stat3 by epidermal growth factor receptor kinase. Proc Natl Acad Sci USA 1996; 93: 13704–13708.
Lo HW, Cao X, Zhu H, Ali-Osman F . Cyclooxygenase-2 is a novel transcriptional target of the nuclear EGFR-STAT3 and EGFRvIII-STAT3 signaling axes. Mol Cancer Res 2010; 8: 232–245.
Lo HW, Hsu SC, Ali-Seyed M, Gunduz M, Xia W, Wei Y et al. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell 2005; 7: 575–589.
Jaganathan S, Yue P, Paladino DC, Bogdanovic J, Huo Q, Turkson J . A functional nuclear epidermal growth factor receptor, SRC and Stat3 heteromeric complex in pancreatic cancer cells. PLoS One 2011; 6: e19605.
Lo HW, Xia W, Wei Y, Ali-Seyed M, Huang SF, Hung MC . Novel prognostic value of nuclear epidermal growth factor receptor in breast cancer. Cancer Res 2005; 65: 338–348.
Hoshino M, Fukui H, Ono Y, Sekikawa A, Ichikawa K, Tomita S et al. Nuclear expression of phosphorylated EGFR is associated with poor prognosis of patients with esophageal squamous cell carcinoma. Pathobiology 2007; 74: 15–21.
Xia W, Wei Y, Du Y, Liu J, Chang B, Yu YL et al. Nuclear expression of epidermal growth factor receptor is a novel prognostic value in patients with ovarian cancer. Mol Carcinog 2009; 48: 610–617.
Lo HW, Hung MC . Nuclear EGFR signalling network in cancers: linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br J Cancer 2006; 94: 184–188.
Wang H, Wang H, Shen W, Huang H, Hu L, Ramdas L et al. Insulin-like growth factor binding protein 2 enhances glioblastoma invasion by activating invasion-enhancing genes. Cancer Res 2003; 63: 4315–4321.
Blobel CP . ADAMs: key components in EGFR signalling and development. Nat Rev Mol Cell Biol 2005; 6: 32–43.
Zhou BB, Peyton M, He B, Liu C, Girard L, Caudler E et al. Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer Cell 2006; 10: 39–50.
Zheng X, Jiang F, Katakowski M, Kalkanis SN, Hong X, Zhang X et al. Inhibition of ADAM17 reduces hypoxia-induced brain tumor cell invasiveness. Cancer Sci 2007; 98: 674–684.
Haynik DM, Roma AA, Prayson RA . HER-2/neu expression in glioblastoma multiforme. Appl Immunohistochem Mol Morphol 2007; 15: 56–58.
Waage IS, Vreim I, Torp SH . C-erbB2/HER2 in human gliomas, medulloblastomas, and meningiomas: a minireview. Int J Surg Pathol 2013; 21: 573–582.
Torp SH, Helseth E, Unsgaard G, Dalen A . C-erbB-2/HER-2 protein in human intracranial tumours. Eur J Cancer 1993; 29A: 1604–1606.
Tuzi NL, Venter DJ, Kumar S, Staddon SL, Lemoine NR, Gullick WJ . Expression of growth factor receptors in human brain tumours. Br J Cancer 1991; 63: 227–233.
Haapasalo H, Hyytinen E, Sallinen P, Helin H, Kallioniemi OP, Isola J . c-erbB-2 in astrocytomas: infrequent overexpression by immunohistochemistry and absence of gene amplification by fluorescence in situ hybridization. Br J Cancer 1996; 73: 620–623.
Azar WJ, Zivkovic S, Werther GA, Russo VC . IGFBP-2 nuclear translocation is mediated by a functional NLS sequence and is essential for its pro-tumorigenic actions in cancer cells. Oncogene 2014; 33: 578–588.
Smalheiser NR . Proteins in unexpected locations. Mol Biol Cell 1996; 7: 1003–1014.
Ganesan A, Zhang J . How cells process information: quantification of spatiotemporal signaling dynamics. Protein Sci 2012; 21: 918–928.
Kholodenko BN . Cell-signalling dynamics in time and space. Nat Rev Mol Cell Biol 2006; 7: 165–176.
Housden BE, Perrimon N . Spatial and temporal organization of signaling pathways. Trends Biochem Sci 2014; 39: 457–464.
Wiley HS . Trafficking of the ErbB receptors and its influence on signaling. Exp Cell Res 2003; 284: 78–88.
Tomas A, Futter CE, Eden ER . EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol 2014; 24: 26–34.
Wang Y, Pennock S, Chen X, Wang Z . Endosomal signaling of epidermal growth factor receptor stimulates signal transduction pathways leading to cell survival. Mol Cell Biol 2002; 22: 7279–7290.
Pennock S, Wang Z . Stimulation of cell proliferation by endosomal epidermal growth factor receptor as revealed through two distinct phases of signaling Mol Cell Biol 2003; 23: 5803–5815.
Miaczynska M, Pelkmans L, Zerial M . Not just a sink: endosomes in control of signal transduction. Curr Opin Cell Biol 2004; 16: 400–406.
Burke P, Schooler K, Wiley HS . Regulation of epidermal growth factor receptor signaling by endocytosis and intracellular trafficking. Mol Biol Cell 2001; 12: 1897–1910.
Brand TM, Iida M, Li C, Wheeler DL . The nuclear epidermal growth factor receptor signaling network and its role in cancer. Discov Med 2011; 12: 419–432.
Boerner JL, Demory ML, Silva C, Parsons SJ . Phosphorylation of Y845 on the epidermal growth factor receptor mediates binding to the mitochondrial protein cytochrome c oxidase subunit II. Mol Cell Biol 2004; 24: 7059–7071.
Demory ML, Boerner JL, Davidson R, Faust W, Miyake T, Lee I et al. Epidermal growth factor receptor translocation to the mitochondria: regulation and effect. J Biol Chem 2009; 284: 36592–36604.
Coffer PJ, Kruijer W . EGF receptor deletions define a region specifically mediating STAT transcription factor activation. Biochem Biophys Res Commun 1995; 210: 74–81.
Shono T, Tofilon PJ, Bruner JM, Owolabi O, Lang FF . Cyclooxygenase-2 expression in human gliomas: prognostic significance and molecular correlations. Cancer Res 2001; 61: 4375–4381.
Joki T, Heese O, Nikas DC, Bello L, Zhang J, Kraeft SK et al. Expression of cyclooxygenase 2 (COX-2) in human glioma and in vitro inhibition by a specific COX-2 inhibitor, NS-398. Cancer Res 2000; 60: 4926–4931.
Xu K, Wang L, Shu HK . COX-2 overexpression increases malignant potential of human glioma cells through Id1. Oncotarget 2014; 5: 1241–1252.
Yang DI, Yin JH, Mishra S, Mishra R, Hsu CY . NO-mediated chemoresistance in C6 glioma cells. Ann NY Acad Sci 2002; 962: 8–17.
Giannopoulou E, Ravazoula P, Kalofonos H, Makatsoris T, Kardamakis D . Expression of HIF-1alpha and iNOS in astrocytic gliomas: a clinicopathological study. In Vivo 2006; 20: 421–425.
Hara A, Okayasu I . Cyclooxygenase-2 and inducible nitric oxide synthase expression in human astrocytic gliomas: correlation with angiogenesis and prognostic significance. Acta Neuropathol 2004; 108: 43–48.
Jahani-Asl A, Bonni A . iNOS: a potential therapeutic target for malignant glioma. Curr Mol Med 2013; 13: 1241–1249.
Wang J, Wang H, Li Z, Wu Q, Lathia JD, McLendon RE et al. c-Myc is required for maintenance of glioma cancer stem cells. PLoS One 2008; 3: e3769.
Orian JM, Vasilopoulos K, Yoshida S, Kaye AH, Chow CW, Gonzales MF . Overexpression of multiple oncogenes related to histological grade of astrocytic glioma. Br J Cancer 1992; 66: 106–112.
Herms JW, von Loewenich FD, Behnke J, Markakis E, Kretzschmar HA . c-myc oncogene family expression in glioblastoma and survival. Surg Neurol 1999; 51: 536–542.
Schneider MR, Wolf E . The epidermal growth factor receptor ligands at a glance. J Cell Physiol 2009; 218: 460–466.
Swindle CS, Tran KT, Johnson TD, Banerjee P, Mayes AM, Griffith L et al. Epidermal growth factor (EGF)-like repeats of human tenascin-C as ligands for EGF receptor. J Cell Biol 2001; 154: 459–468.
Kaser MR, Lakshmanan J, Fisher DA . Comparison between epidermal growth factor, transforming growth factor-alpha and EGF receptor levels in regions of adult rat brain. Brain Res Mol Brain Res 1992; 16: 316–322.
Kudlow JE, Leung AW, Kobrin MS, Paterson AJ, Asa SL . Transforming growth factor-alpha in the mammalian brain. Immunohistochemical detection in neurons and characterization of its mRNA. J Biol Chem 1989; 264: 3880–3883.
Ferrer I, Blanco R, Carulla M, Condom M, Alcantara S, Olive M et al. Transforming growth factor-alpha immunoreactivity in the developing and adult brain. Neuroscience 1995; 66: 189–199.
Lazar LM, Blum M . Regional distribution and developmental expression of epidermal growth factor and transforming growth factor-alpha mRNA in mouse brain by a quantitative nuclease protection assay. J Neurosci 1992; 12: 1688–1697.
Becher OJ, Peterson KM, Khatua S, Santi MR, MacDonald TJ . IGFBP2 is overexpressed by pediatric malignant astrocytomas and induces the repair enzyme DNA-PK. J Child Neurol 2008; 23: 1205–1213.
McDonald KL, O'Sullivan MG, Parkinson JF, Shaw JM, Payne CA, Brewer JM et al. IQGAP1 and IGFBP2: valuable biomarkers for determining prognosis in glioma patients. J Neuropathol Exp Neurol 2007; 66: 405–417.
Santarius T, Shipley J, Brewer D, Stratton MR, Cooper CS . A census of amplified and overexpressed human cancer genes. Nat Rev Cancer 2010; 10: 59–64.
Salomon DS, Brandt R, Ciardiello F, Normanno N . Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol 1995; 19: 183–232.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005; 102: 15545–15550.
Sallinen SL, Sallinen PK, Haapasalo HK, Helin HJ, Helen PT, Schraml P et al. Identification of differentially expressed genes in human gliomas by DNA microarray and tissue chip techniques. Cancer Res 2000; 60: 6617–6622.
Acknowledgements
We thank Dr Oliver Bogler, Dr Zhimin Lu, Dr Frederick Lang, Dr Paul Chiao and Dr Tapio Visakorpi for their helpful comments and discussions and Kathryn L Hale, Department of Scientific Publications at MD Anderson Cancer Center, for editing the manuscript. We thank Ville Kytölä for contributions to the immunohistochemical association analyses. This work was partially supported by grants from the US National Institutes of Health (CA098503, CA141432 and CA143835 to WZ and GNF and U24 CA143835 to WZ), by funding for the Cancer Systems Informatics Center from the National Foundation for Cancer Research (to WZ), by NIH/NCI grant P30CA016672 to MD Anderson Cancer Center supporting the Flow Cytometry and Cellular Imaging Core Facility and by the Finnish Funding Agency for Technology and Innovation Finland Distinguished Professor program and the Academy of Finland (grant 259038 to KG). WKC is a Fellow of the National Foundation for Cancer Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Chua, C., Liu, Y., Granberg, K. et al. IGFBP2 potentiates nuclear EGFR–STAT3 signaling. Oncogene 35, 738–747 (2016). https://doi.org/10.1038/onc.2015.131
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2015.131
- Springer Nature Limited
This article is cited by
-
IGFBP2 promotes proliferation and cell migration through STAT3 signaling in Sonic hedgehog medulloblastoma
Acta Neuropathologica Communications (2023)
-
STAT3 potentiates RNA polymerase I-directed transcription and tumor growth by activating RPA34 expression
British Journal of Cancer (2023)
-
Deficiency of N-glycanase 1 perturbs neurogenesis and cerebral development modeled by human organoids
Cell Death & Disease (2022)
-
EGFR-upregulated LIFR promotes SUCLG2-dependent castration resistance and neuroendocrine differentiation of prostate cancer
Oncogene (2020)
-
IGFBP2: integrative hub of developmental and oncogenic signaling network
Oncogene (2020)