
Abstract
Patients with chronic psychotic disorders (CPD) exhibit deficient sensorimotor gating (measured by prepulse inhibition (PPI) of startle) and mismatch negativity (MMN). In healthy subjects (HS), N-methyl-D-aspartate (NMDA) antagonists like memantine and ketamine increase PPI, and under some conditions, memantine enhances MMN; these findings present a challenge to understanding the basis for deficient PPI and MMN in psychotic disorders, as reduced NMDA activity is implicated in the pathogenesis of these disorders. Here we assessed for the first time the effects of memantine on PPI and MMN in CPD subjects. Baseline PPI was measured in HS and patients with a diagnosis of schizophrenia or schizoaffective disorder, depressed type. Subjects (total n=84) were then tested twice, in a double-blind crossover design, comparing either: (1) placebo vs 10 mg of memantine or (2) placebo vs 20 mg memantine. Tests included measures of acoustic startle magnitude and habituation, PPI, MMN, autonomic indices, and subjective self-rating scales. Memantine (20 mg) significantly enhanced PPI in CPD subjects, and enhanced MMN across subject groups. These effects on PPI were age dependent and most evident in older CPD patients, whereas those on MMN were most evident in younger subjects. The lower dose (10 mg) either had no detectable effect or tended to degrade these measures. The NMDA antagonist, memantine, has dose-dependent effects on preconscious, automatic measures of sensorimotor gating and auditory sensory processing that are associated with enhanced cognition and function in CPD patients. Ongoing studies will determine whether these memantine-induced changes predict acute pro-cognitive or otherwise clinically beneficial effects in CPD patients.
Similar content being viewed by others

Introduction
Individuals with chronic psychotic disorders (CPD) exhibit deficits in measures of automatic, ‘preconscious’ information processing. In one such measure, prepulse inhibition (PPI), startle is reduced when the startling stimulus is preceded by a weak nonstartling sensory event or ‘prepulse’ (Graham 1975). PPI is impaired in schizophrenia patients (Braff et al, 1978; Swerdlow et al, 2013) and this effect is diminished or eliminated by second-generation antipsychotics (APs; Kumari et al, 1999; see, for example, Swerdlow et al, 2006a). Mismatch negativity (MMN) is a negative-going deflection in the event-related potential (ERP), evoked when a sequence of repetitive ‘standard’ stimuli is interrupted by infrequent oddball or ‘deviant’ stimuli that differ in some physical dimension such as duration or pitch. Like PPI, MMN is presumed to reflect a predominantly automatic, pre-conscious process—in this case, one of detecting a ‘mismatch’ between the deviant stimulus and a sensory–memory trace (Näätänen et al, 1989). MMN amplitude reduction in schizophrenia was reported over 20 years ago (Shelley et al, 1991) with subsequent studies consistently identifying deficits in chronic and unmedicated patients (Rissling et al, 2012; Bodatsch et al, 2011; Brockhaus-Dumke et al, 2005; Light & Braff, 2005; Kirino & Inoue 1999; Catts et al, 1995).
Both PPI and MMN reflect a neural response to simple, discrete, weak stimuli, and they both are regulated by distributed neural circuits, involving both frontal and temporal cortex, and in the case of PPI, subcortical regions including the ventral striatum, pallidum, and pontine tegmentum (Takahashi et al, 2012; see, for example, Swerdlow et al, 2008). Deficient forebrain N-methyl-D-aspartate (NMDA) activity has been proposed as one mechanism contributing to the pathogenesis of schizophrenia and its associated deficits in information processing (Javitt et al, 2012). Interestingly, whereas NMDA systems have been implicated in the regulation of both PPI and MMN, the evidence linking reduced NMDA activity to deficits in PPI and/or MMN is not entirely straightforward. Thus, PPI is actually increased in healthy subjects (HS) by NMDA antagonists such as ketamine (Duncan et al, 2001; Abel et al, 2003), the low- to moderate-affinity NMDA-receptor antagonist, memantine (Swerdlow et al, 2009), and by the mixed NMDA antagonist/dopamine agonist, amantadine (Swerdlow et al, 2002b). Although MMN has been reported to be reduced in HS by ketamine (Umbricht et al, 2000; Umbricht et al, 2002), it has also been shown to be increased by memantine (Korostenskaja et al, 2007).
There are reasons to think that memantine-induced increases in PPI and/or MMN are signals of ‘target’ circuitries being engaged in a manner that might benefit CPD patients. Increases in both PPI and MMN have been associated with enhanced neurocognition, function, and treatment responsivity (Giakoumaki et al, 2006; Light et al, 2007; Swerdlow et al, 2006a). There may also be a rational basis for suggesting therapeutic value of memantine under conditions requiring improved frontal function. For example, 20 mg memantine acutely enhances cortical metabolic efficiency (Kim et al, 2010) and improves neurocognition after brain injury, and these effects are strongly associated with increased left frontal and parietal lobe glucose metabolism (Willenborg et al, 2011). We might predict that enhanced frontal cortical metabolic efficiency should have positive effects on frontal-regulated functions, such as sensorimotor gating and MMN, and that this effect might be particularly beneficial to patients whose frontal lobe efficiency is compromised by a disease process. Previous studies have focused on the impact of memantine on PPI and MMN in HS, which is a rational strategy for identifying evidence of ‘target engagement’ and bioactivity within relevant neural systems. The present study was designed to take the ‘next step,’ that is, to determine the impact of memantine on PPI and MMN in schizophrenia patients.
Memantine was used in these studies rather than ketamine because of its superior safety profile—particularly in CPD subjects—its linear kinetics, 100% bioavailability after oral administration and relative NMDA-receptor selectivity (Wilcock et al, 2002; Aerosa et al, 2005). Memantine does not cause the sensory or dissociative symptoms produced by ketamine (Duncan et al, 2001; Abel et al, 2003; Umbricht et al, 2002), which might complicate the interpretation of changes in PPI and MMN. Doses (10 and 20 mg, po) were selected based on published reports in HS, which noted dizziness and other mild adverse reactions at higher doses (Swerdlow et al, 2009).
Materials and Methods
Methods were approved by the UCSD Human Subject Institutional Review Board. Healthy 18–50-year-old adult subjects (HS) and patients with a clinically stable CPD participated in this study. The enrollment target, based on power considerations for the major dependent measures, was 40 HS and 40 CPD subjects (20 per dose per group); enrollment continued until the target was reached (total screened and participated in at least some startle testing, n=84). On the basis of measure-specific exclusion criteria (Table 1), effective ‘n’s’ were n=70 for PPI and n=69 for MMN.
Subjects who passed a phone checklist were invited for an in-person screening session that included a urine toxicology, hearing test (Saico Audiometer, Assens Denmark), and startle reflex tests and questionnaires related to demographic and clinical information (Table 2). Diagnoses in CPD patients were confirmed by the MINI International Neuropsychiatric Interview (6.0), and symptom severity was assessed with the Positive and Negative Syndrome Scale (Kay et al, 1987; R’s for subscales 0.903–0.947). Among HS, personality traits were quantified using the Tridimensional Personality Questionnaire (Cloninger, 1991), the Sensation Seeking Scale (Zuckerman et al, 1974), and the Eysenck Personality Questionnaire (Eysenck & Eysenck, 1975).
Startle was measured as previously described (eg, Swerdlow et al, 2006a; see Supplementary Methods for details of signal acquisition and processing). Broadband noise (70 dB(A)) preceded active stimuli by 3 min and persisted as a background noise during the test. During acclimation, blinks were counted remotely by trained staff as previously described (interobserver R=0.97 (Swerdlow et al, 2002a)). The session consisted of 42 trials, with six conditions: a 118-dB(A) 40-ms noise burst (pulse alone) and the same burst preceded 10, 20, 30, 60, and 120 ms by a prepulse of 16 dB above background; using 16-dB prepulses with this startle system, prepulse-associated EMG activity is <0.5% of startle stimulus-induced levels (Swerdlow et al, 2006a). To measure startle habituation, three pulse-alone trials were presented at the session beginning (Block 1) and end (Block 3).
Participants who met the study inclusion criteria returned for testing after 6–10 days, and for a second test 7 days later. Testing followed a double-blind, placebo-controlled, pseudo-randomized balanced drug order design. Before the test days, subjects were instructed to maintain their normal patterns of caffeine intake, due to the effects of caffeine withdrawal on PPI and subjective rating scales (Swerdlow et al, 2000). CPD subjects were asked to maintain their medication doses or to inform us of any medication changes during the study. Subjects arrived at ~0830 hours on the test day, ate a standardized breakfast, and completed a urine toxicology test and a hearing test. At 0900 hours, either memantine HCl (10 or 20 mg) or placebo was administered (po). Memantine pharmacokinetics are linear for single oral doses of 5–40 mg; Tmax for 10 and 20 mg single-dose administration are ~6.0 and 6.89 h for 10 and 20 mg po, and elimination T(1/2) is 60–80 h (Liu et al, 2008). For the first 40 subjects (20 from each group), the active dose of memantine HCl was 10 mg; for the next 40 subjects, the active dose was 20 mg. On the basis of subject attrition, four additional subjects were tested, bringing the total sample screened and enrolled to 84.
Startle testing began 210 min after pill administration, based on memantine pharmacokinetics in HS (Sonkusare et al, 2005), using a test session identical to that administered on the screening-day. During active and placebo drug tests, startle ‘nonresponsiveness’ was defined by a mean startle magnitude <10 units on pulse alone trials (Swerdlow et al, 2002a, b, 2006a). %PPI was defined as (100−(100 × magnitude on prepulse trial/magnitude on pulse alone trials)), and used as the major dependent variable for this measure.
At minute 345 post pill, MMN testing commenced. Auditory stimuli were presented at 85 dB SPL via Etymotic ER3-A insert earphones. A four-tone auditory oddball paradigm comprised of 82% standards (50-ms, 1000-Hz) and 18% deviant stimuli (6% per deviant type) that differed from the standard in pitch (50-ms tones at 1100-Hz), duration (125-ms tones at 1000-Hz), or both pitch and duration (125-ms tones at 1100-Hz). A pseudorandomized sequence ensured that a minimum of three standard tones were presented between each deviant stimulus. All tones had 5-ms rise/fall times and were presented with a fixed 500-ms stimulus onset asynchrony. Total recording time was ~45 min per session. Subjects were instructed to ignore auditory stimuli while viewing a silent movie.
EEG data were continuously recorded at a sampling rate of 2048-Hz from 64 channels using a BioSemi ActiveTwo system (www.biosemi.com) and downsampled to 512-Hz (for details of electrode configuration and offline processing, see Supplementary Methods). On average, 4127.09 standards, 244.93 pitch, 245.91 duration, and 247.52 pitch plus duration trials were used for ERP averages. Deviant-minus-standard difference waves were generated for each deviant type and low-pass filtered (20-Hz zerophase shift, 24 dB/octave rolloff). MMN was computed as the mean amplitude across 135–205 ms range for each deviant type in the difference waveforms at electrode Fz.
Symptom rating scales (SRS) were assessed 30, 90, 150, 200, 230, 335, 395, and 425 min after pill consumption, as previously described (eg, Swerdlow et al, 2009; see Supplementary Methods). Our previous study in HS detected an increase in SRS scores for ‘happy’ after administration of 20 mg of memantine (Swerdlow et al, 2009). At 1130 h, the subjects received a standardized lunch.
Data were analyzed by RM-ANOVA, with diagnosis and dose group (10 mg or 20 mg) as between-subject factors, and drug condition (placebo vs active) as a within-subject factor. For PPI, prepulse interval was also a within-subject factor, and for MMN, oddball type was a within-subject factor. A specific prediction was possible for the 20-mg memantine dose effect on PPI, based on past studies (Swerdlow et al, 2009), but this was our first experience with the 10-mg dose. Planned comparisons assessed the relationship of baseline (screening and placebo) PPI levels to memantine PPI effects, and the relationship of memantine effects on PPI to those on MMN. These analyses utilized categorical factors (‘low’ vs ‘high’ based on median splits) within ANOVAs to permit assessments of embedded factors (eg, prepulse intervals and MMN deviant types), and continuous comparisons within regression analyses with a ‘difference score’ calculated as (mean PPI (or MMN) after memantine minus mean PPI (or MMN) after placebo). Baseline SRS and autonomic measures (before pill ingestion) and immediately before startle testing were continuous variables and analyzed by ANOVA as ‘change scores’ (difference from baseline (Swerdlow et al, 2002a). Because most CPD subjects were receiving atypical (and in many cases, typical) AP medications at the time of testing, and these medications are known to ‘normalize’ PPI in CPD patients (Kumari et al, 1999; see, for example, Swerdlow et al, 2006a), there was no clear prediction for the effects of diagnosis on PPI per se.
Study recruitment did not balance groups for sex, age, or medication status, but where possible, these variables were examined post hoc to determine their potential impact on memantine effects. Drug order (placebo test 1 vs active test 1) was also assessed as a potential moderator of drug effects. For all statistical comparisons, alpha was 0.05 for two-tailed contrasts, and 0.10 for one-tailed contrasts. For post hoc comparisons with smaller cell sizes, effect sizes (Cohen’s d) are reported.
Results
Subject Characteristics
Demographic and clinical characteristics of the final cohorts in the 10 mg and 20 mg studies are shown in Table 2. Two CPD patients carried a diagnosis of schizoaffective disorder, depressed type; all others were diagnosed with schizophrenia. Most (77/84) patients were taking atypical APs at the time of testing.
Subjective and Physiological Measures
Among the 84 subjects who participated in startle testing, resting blink rate was elevated among CPD patients vs HS (F=5.03, df(1,80), P<0.03), but there were no significant effects of memantine, differences across dose groups or any significant two- or three-way interactions. Analyses of autonomic measures (Supplementary Figure S1) revealed no significant main or interaction effects of diagnosis, memantine, or dose group on change in heart rate or diastolic blood pressure; for systolic blood pressure, a statistically significant interaction of diagnosis × memantine × time (P=0.02) was most easily attributable to small HS vs CPD differences in placebo-level SBP rather than drug effects per se.
SRS data generally yielded no meaningful main or interaction effects; however, inspection of the data revealed a non-normal distribution, with a substantial number of subjects providing maximum scores of 100 for measures of ‘happy,’ and scores of 0 for measures of ‘drowsy’. Distributions restricted to subjects with at least 5 units of range at either tail suggested that compared to placebo, 10 mg memantine was associated with higher ‘happy’ ratings in CPD patients (see Supplementary Results).
Habituation and Startle Magnitude
Seventy subjects completed two full test days with mean startle values ≥10 units (‘responders’). ANOVA of startle during Blocks 1 and 3 (to assess reflex habituation) revealed significantly reduced startle magnitude among CPD patients compared with HS (Table 3), but no significant effects of memantine, dose group or any two- or three-way interactions. The lack of significant interactions with trial block reflects no impact of any factor on reflex habituation. ANOVA of startle magnitude during PPI testing (Block 2) also revealed significantly reduced startle magnitude among CPD patients, but no significant effects of memantine, dose group or any two- or three-way interactions (Table 3).
PPI
ANOVA of PPI (Figure 1) revealed no significant main effects of diagnosis, memantine, or dose group, but a significant interaction of diagnosis × memantine × dose group (F=6.87, df(1,66), P=0.01). There was the expected main effect of prepulse interval (P<0.0001), but not other significant main or interaction effects. This three-way interaction remained significant (F=7.25, df(1,62), P=0.009) when drug order (placebo first vs active first) was included as a factor; ANOVA using test day (test 1 vs test 2) as a grouping factor revealed no significant main or interaction effects of this variable. To understand this three-way interaction, separate analyses were conducted on each dose of memantine.

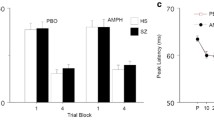
Memantine effects on prepulse inhibition: %PPI in HS and CPD patients in low dose (placebo vs 10 mg memantine; left) and high dose (placebo vs 20 mg memantine; right) groups. PPI in CPD patients was reduced by 10 mg of memantine and significantly increased by 20 mg memantine (*). CPD, chronic psychotic disorder; HS, healthy subjects.
Analysis of the 10-mg dose group revealed no significant main effects of diagnosis or memantine, but a near-significant interaction of diagnosis × memantine (F=3.52, df(1,32), P<0.07), reflecting a trend toward a memantine-induced reduction of PPI in CPD patients, seen arithmetically at each of the five prepulse intervals, with maximum reduction at 60 ms (d=0.71). This pattern was recapitulated in analyses limited to men (n=16: maximal reduction at the 60 ms interval, d=0.72) and women (n=4; maximum reduction at the 60 ms interval, d=0.48), and in patients taking atypical APs (n=16; maximal reduction at the 60 ms interval, d=0.66). When AP status (with (n=16) or without (n=4)) was used as a grouping factor in CPD patients, ANOVA again showed a trend towards PPI-reducing effects of memantine (F=3.97, df(1,18), P<0.065), with no interaction of memantine × AP status (F<1), though the small number of unmedicated subjects makes this analysis less compelling. Effect size calculations suggest that the impact of memantine across prepulse intervals may be blunted in AP-medicated compared with unmedicated patients (d=0.21 vs 0.70, respectively).
In contrast, analysis of the 20-mg dose group revealed no significant effect of diagnosis, a significant effect of memantine (F=4.34, df(1,34), P<0.05), and a near-significant interaction of diagnosis × memantine (F=3.39, df(1,34), P=0.07); in this case, the main and interaction effects reflected a memantine-induced potentiation of PPI in CPD patients (F=5.04, df(1,17), P<0.04). This PPI-potentiating effect of memantine was not statistically significant in HS across the five prepulse intervals, though a moderate effect size increase was evident at 60 ms intervals (d=0.46) where we previously detected maximum effects in HS (Swerdlow et al, 2009). Sex distributions among CPD patients in the 20-mg dose group (M:F=10:8) permitted a meaningful contrast; with sex added as a grouping factor, the PPI-enhancing effects of memantine in this group persisted (F=5.12, df(1,16), P<0.04), with no significant sex × memantine interaction (F<1). PPI-potentiating effects of memantine were also significant among the subgroup of 18 CPD patients taking atypical APs (F=5.04, df(1,17), P<0.04), and among two patients taking only haloperidol (d=0.94).
We previously reported that the PPI-enhancing effects of memantine were most robust among HS with the lowest baseline PPI levels, so-called ‘low-gaters’ (Swerdlow et al, 2009). Similar baseline-dependent effects have been reported with atypical APs (Swerdlow et al, 2006b; Vollen-weider et al, 2006). For the current data, the overall correlation between baseline PPI (screening session) and the magnitude of memantine-enhanced PPI missed significance (r=−0.22, P<0.07), whereas the correlation between placebo-level PPI and the magnitude of memantine-enhanced PPI was quite robust (r=−0.60, P<0.0001), particularly among all 20 mg dose subjects (r=−0.70, P<0.0001) and specifically among patients in the 20-mg dose group (r=−0.85, P<0.0001). A conservative view of the data (shown in Figure 2 with a median split of baseline PPI values, and median split of placebo PPI values) suggests that future analyses might consider whether low baseline PPI can be used to identify patients who are most sensitive to the PPI-enhancing effects of memantine.

Memantine (20 mg) effect on PPI—relationship to baseline and placebo PPI levels: based on our previous findings (Swerdlow et al, 2009), we assessed the relationship between memantine-enhanced PPI and baseline PPI levels, in two ways. (a) Relationship for HS (left) and CPD patients (right) divided based on their level of PPI exhibited during the screening session, about 1 week before drug testing. (b) Relationship in the same manner, except that the groups are divided based on their level of PPI exhibited in the placebo condition. In both cases, memantine enhanced PPI only among ‘low-gating’ HS and CPD patients. (*) significant main effect of memantine in ‘low-gating’ CPD patients; (#) significant PPI-enhancing effect of memantine in ‘low gating’ HS at 30–60 ms intervals (P=0.025) after significant drug × interval interaction (P<0.005). CPD, chronic psychotic disorder; HS, healthy subjects; PPI, prepulse inhibition.
Analyses of demographic and clinical variables revealed that age correlated positively with the magnitude of the 20 mg memantine effect (r=0.50, P=0.002; Figure 5a); this relationship was roughly comparable among subgroups limited to CPD patients (r=0.44) and HS (r=0.36). In contrast, in this dose group, age did not correlate significantly with either baseline PPI or placebo-level PPI (r’s=0.26 and −0.11, respectively, both NS). As seen in Figure 3, memantine significantly enhanced PPI only among older CPD patients (memantine effect in younger vs older CPD patients: F=9.70, df(1,16), P<0.007); this effect remained when screening or placebo levels of PPI were used as covariates (P=0.01 and P<0.02, respectively). A similar, though weaker, pattern was evident among the HS group, which was significantly younger than the CPD group. Supporting the role of age rather than diagnosis per se in this memantine sensitivity, a repeated measures ANCOVA of PPI with age as a covariate revealed significant PPI-enhancing effects of memantine (P<0.05), no significant main effects of diagnosis or age, and a significant interaction of memantine × age (P<0.02), reflecting the age-related patterns of memantine sensitivity described above. On the basis of this age effect on sensitivity to 20 mg memantine, we examined the effect of age on sensitivity to memantine among patients in the 10-mg group. Only older CPD patients were sensitive to the PPI-disruptive effects of this lower dose of memantine (younger vs older CPD patients: F=5.46, df(1,18), P<0.035). Thus, regardless of the ‘direction’ of the memantine effect on PPI, among CPD patients, age strongly predicted memantine sensitivity.

Memantine (20 mg) effect on PPI—relationship to age: %PPI in HS (left) and CPD patients (right) after placebo or 20 mg memantine. Memantine significantly enhanced PPI only among the older CPD patients (*). A similar, though weaker, pattern was evident among the HS group, which was significantly younger than the CPD group (‘older’ HS age (range) vs ‘older’ CPD patients=33.0 (30–36) vs 42.4 (39–48) years). CPD, chronic psychotic disorder; HS, healthy subjects; PPI, prepulse inhibition.
MMN
Sixty-nine subjects completed ERP testing. ANOVA of MMN revealed a significant effect of diagnosis (F=10.13, df(1,65), P<0.003) and diagnosis × oddball type (pitch (pMMN), duration (dMMN), and combined (cMMN)) interaction (F=6.63, df(2,130), P=0.02). Despite this interaction, separate ANOVAs revealed significant HS>CPD group MMN for pMMN (P<0.01), dMMN (P<0.002) and cMMN (P<0.009). There was no significant effect of memantine or dose group, but there was a significant interaction of memantine × dose group (F=5.47, df(1,65), P<0.025) (Figure 4a).

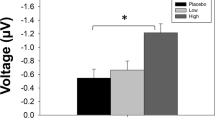
Memantine effect on mismatch negativity. (a) MMN (μV) in response to three different ‘oddball’ stimuli, differing from standards in duration (D), pitch (P), and both duration and pitch (C (combined)). Data are shown for HS and CPD in both dose groups. Although a significant effect of diagnosis was detected for both dose groups (*), memantine effects on MMN were evident only for the 20-mg dose group (#; see text). (b) ERP waveform averaged across oddball types for placebo and 20 mg memantine in age-matched subgroups of HS and CPD patients. Note comparable memantine-induced increases in MMN in both groups (main effect of memantine: P<0.04; memantine × diagnosis interaction: NS). Analyses of P3a amplitude consistently detected no significant main effects of memantine or significant two- or three-way interactions (data not shown). CPD, chronic psychotic disorder; ERP, event-related potential; HS, healthy subjects.
Analysis of the 10-mg dose group revealed no significant effect of memantine, or significant interactions of memantine × diagnosis or memantine × oddball type, or three-way interaction. Collapsed across diagnoses and oddball types, 10 mg memantine weakly reduced MMN (d=0.09); this tendency was roughly comparable in HS and CPD patients, and was not impacted by variables of sex or AP status (data not shown).
In contrast, analysis of the 20-mg dose group revealed significant MMN-enhancing effects of memantine (F=6.92, df(1,31), P<0.015), with no significant interaction of memantine × diagnosis or memantine × oddball type, or three-way interaction. Collapsed across diagnoses and oddball types, this MMN-enhancing effect of 20 mg memantine was small (d=0.19), and was arithmetically larger in HS vs CPD patients (d=0.30 vs 0.11). As with PPI, the significant MMN-enhancing effects of this 20 mg dose were independent of drug order, and were evident when the samples were limited to males only, or to patients taking atypical APs (data not shown).
Althugh baseline (screening day) MMN was not measured, analyses suggested that MMN-enhancing effects of 20 mg memantine were most robust among patients with low placebo levels of MMN. ANOVA using a median split of placebo MMN levels among CPD patients revealed a significant interaction of memantine × placebo level (F=6.42, df(1,18), P=0.02; main effect of memantine among low placebo-level patients: F=4.90, df(1,9), P=0.05).
As with PPI, analyses of demographic or clinical variables revealed that only age had a significant impact on the magnitude of the 20-mg memantine effect on MMN, but compared with PPI, this MMN relationship with age was inverted. Thus, age correlated positively with the magnitude of the 20-mg memantine effect (ie, r=0.40, P<0.025; Figure 5b), ie, younger subjects exhibited greater negativity after memantine; this relationship was comparable among the subgroup limited to CPD patients (r=0.44) but not HS (r=0.10). In contrast, in this dose group, age did not correlate significantly with placebo-level MMN in either HS or CPD patients (r’s=0.32 and 0.03, respectively; both NS). Memantine 20 mg had weak MMN-enhancing effects in younger (d=0.25) but not in older CPD patients (d=−0.03). When CPD patients were age matched to the younger HS subjects, 20 mg memantine significantly enhanced MMN to a comparable degree across groups (main effect of memantine: F=4.74, df(1,28), P<0.04; memantine × diagnosis interaction: F=1.28, df 1,28, NS (Figure 4b)). Among subjects in the 10-mg memantine group, no moderating effects of age were evident among either HS or CPD patients. Thus, in contrast to what was observed for PPI, sensitivity to the MMN-enhancing effects of 20 mg memantine was only evident among younger CPD patients. In comparing drug effects across measures, it should be noted that MMN testing occurred substantially later in the test day than did PPI.

Magnitude of memantine effects across measures and age: memantine-enhanced PPI and MMN appear to reflect effects on distinct CPD subgroups. (a) Older CPD patients exhibit greater PPI-enhancing effects of 20 mg memantine. (b) Younger CPD patients exhibit greater MMN-enhancing effects of 20 mg memantine. (c) Significant negative correlation of 20 mg memantine-induced changes in PPI and MMN among 17 subjects tested on both measures. CPD, chronic psychotic disorder; MMN, mismatch negativity; PPI, prepulse inhibition.
Correlated Memantine Effects on PPI and MMN
Fifty-eight subjects had valid PPI and MMN data for both test days. Using absolute values of MMN amplitude, so that changes toward ‘greater negativity’ (ie, a more ‘normal’ score) were positive values, the PPI- and MMN-enhancing effects of 20 mg memantine were significantly negatively correlated. Simple regression of the ‘20-mg memantine effect’ (20 mg dose minus placebo) collapsed across PPI intervals and MMN conditions revealed a significant negative correlation (r=−0.44, P<0.02) across all subjects, and when the analysis was limited to CPD patients (r=−0.54, P<0.03) (Figure 5c).
Discussion
The low- to moderate-potency NMDA antagonist, memantine, exhibited dose-dependent effects on two distinct measures of automatic, preconscious information processing in CPD patients. In each case, 20 mg of memantine changed performance in a direction associated with enhanced neurocognition and function (Bitsios et al, 2006; Swerdlow et al, 2006a; Kawakubo et al, 2007). The ability of this acute drug challenge to enhance PPI and MMN in CPD patients has several implications.
First, these findings suggest malleability within systems that regulate pre-attentive processing, even in patients with disorders characterized by severe pathological changes within those regulatory systems. In other words, the circuitry responsible for sensorimotor gating and sensory discrimination remain sufficiently intact and dynamic in CPD patients to permit an increase in these processes in response to an acute drug challenge. Conceivably, this plasticity may represent a neural resource that could be engaged in a therapeutic capacity, which is a core tenet of the ‘PACT’ strategy (‘pharmacologic augmentation of cognitive therapy’ (Swerdlow, 2011a,b,). This is not to say that a single dose of memantine would be expected to have therapeutic effects in CPD patients; however, the neural signal elicited by this drug challenge provides some evidence that mechanisms can be accessed that lead to neurobehavioral and electrophysiological evidence of enhanced sensorimotor gating and sensory discrimination, respectively. Memantine engaged the ‘target’ circuitry regulating PPI and MMN, and the resulting signal provides a metric of specific available neural resources within any given individual.
Second, to the degree that these acute effects of memantine reflect reduced NMDA neurotransmission, the present findings raise the paradox that reduced NMDA neurotransmission—implicated in the pathogenesis of CPDs (Javitt, 2012; Javitt et al, 2012)—generated signals in these patients that are associated with more normalized function, rather than greater deficits. In previous reports in HS, acute exposure to NMDA antagonists led to greater levels of both PPI (Duncan et al, 2001; Abel et al, 2003; Swerdlow et al, 2002b, 2009) and—in the case of memantine—MMN (Korostenskaja et al, 2007), but the present findings provide the first evidence for such effects in CPD patients. Conceivably, NMDA activity might be deficient within substrates responsible for core CPD symptoms, but be normal or even elevated in other brain regions regulating PPI and MMN. Alternatively, the effects of acute pharmacologic disruption on PPI and MMN might not reproduce the effects of sustained NMDA deficits on these measures in CPD patients. Hypotheses regarding the location and/or chronicity of NMDA blockade notwithstanding, the empirical evidence is that neither the present findings, nor those cited above for ketamine, memantine or amantadine in HS, are easily reconcilable with the simplest version of the ‘NMDA hypothesis’ for CPDs.
Although it is clear that memantine has a very distinct clinical profile compared with the dissociative and cognitively impairing properties of NMDA antagonists such as ketamine, the precise neurochemical basis for these differences in not known. At the level of NMDA receptors, memantine has lower affinity and faster kinetics compared with ketamine, and the two drugs may act on different populations of NMDA receptors (see, for example, Johnson et al, 2015). At a systemic level, memantine does not stimulate cortisol release, whereas ketamine does (Hergovich et al, 2001). Either of these effects might contribute both to clinical differences and to the opposite effects of these two drugs on MMN, which is reduced under conditions of elevated cortisol (Elling et al, 2011). In contrast to these differential drug effects, memantine and ketamine both enhance sensorimotor gating in HS, and in the case of memantine, this effect is also seen in CPD patients. The simplest explanation for this pattern of findings is that the PPI-enhancing effects of these drugs reflect their shared chemical properties, whereas the divergent effects of these drugs on neurocognition and MMN reflects their distinct chemical properties. It is not clear what these shared and distinct properties of ketamine and memantine might say about the functional significance of studies using one drug vs the other; however, it is clear that a therapeutic algorithm requiring repeated exposures to ketamine in CPD patients would be much more problematic compared with a similar regimen with memantine.
Most CPD patients were medicated with second-generation AP medications; to the degree that this issue could be assessed among the small subgroups, atypical AP use was not required for, nor protective against, either the PPI-disruptive effects of 10 mg memantine, nor the PPI-enhancing effects of 20 mg memantine. PPI levels among AP-medicated CPD patients were comparable to their HS counterparts. Thus, memantine did not actually ‘normalize’ PPI in these patients, but rather elevated it to ‘supra’-normal levels. Because atypical APs ‘normalize’ PPI in CPD patients, the fact that there is still ‘room to move’, ie, for memantine to further elevate PPI in these patients, raises the possibility that AP- and memantine-induced effects are mediated by distinct mechanisms.
The present findings do not address the potential therapeutic value of memantine in daily dosing regimens in CPD (the evidence of which is mixed: de Lucena et al, 2009; Lieberman et al, 2009; Krivoy et al, 2007; Zdanys & Tampi, 2008), but do suggest the value for some CPD patients of adding an acute pharmacologic challenge with memantine to augment cognitive therapies that would benefit from gains in pre-attentive information processing (Swerdlow, 2011a, b,). Of course, memantine may not be the optimal drug for such a strategy: newer analogs of this compound have been developed, and our ongoing studies are assessing other drug classes (Bhakta et al, 2014; Swerdlow et al, 2014).
These findings confirm that oral memantine at these doses and time course is ‘neuro-active’, consistent with many reports (eg, Jocham et al, 2014); the observed increases in PPI and MMN also suggest that circuitry is ‘engaged’ that regulated pre-attentive information processing. The finding that 10 vs 20 mg doses of memantine appeared in some cases to have opposite effects on measures, particularly in CPD patients, might reflect the complex pharmacology of memantine, which has distinct effects on activity within NMDA, dopamine, and perhaps other neural substrates (eg, Seeman et al, 2008), each of which might have different levels of sensitivity and thus be preferentially impacted by lower vs higher drug doses.
The fact that the observed effects of 20 mg memantine on PPI and MMN segregated to separate populations distinguished by age was unexpected and post hoc, but might suggest that memantine-enhanced PPI and MMN reflect drug actions on two different substrates. Perhaps more importantly, this moderation of memantine effects by age suggests that this drug might have different neurocognitive benefits in younger vs older CPD patients, which might ultimately predict different clinical uses across the course of illness. Although ‘age’ effects among CPD patients might reflect an aggregate of related factors, eg, duration of illness and medication exposure, an explanation based on simple confounding effects of sustained illness is made less compelling by evidence that: (1) similar (albeit weaker) age effects were detected among the HS 20-mg group (despite their overall younger age); and (2) enhanced sensitivity is bidirectional (ie, greater disruption of PPI in the 10-mg dose group, and greater enhancement of PPI in the 20 mg dose group). Clearly, additional studies comparing memantine effects on PPI vs MMN at the same time post pill, and across a larger range of ages in HS and CPD patients, would help clarify the implications of these age-dependent drug effects.
It is conceivable that neurophysiological markers such as PPI or MMN might be used to identify CPD patients more likely to have memantine-sensitive pre-attentional mechanisms, and thus who might be prone to exhibit clinical benefits over the course of an extended therapeutic trial. Such a predictive biomarker might spare patients and families the substantial time, effort, and resources lost to hit-or-miss treatment strategies. The hope that laboratory-based biomarkers could predict treatment response is not a new one, but the application suggested by the present findings is distinct from many biomarker-based strategies because the presence of the biomarker is not expected to be informative about diagnosis per se, nor the varied causes and pathology associated with a CPD diagnosis: instead, the presence of memantine-enhanced PPI and/or MMN is simply evidence that the drug has engaged an existing potential for neuroplasticity within a target neurocognitive domain, which might be harnessed to meet the demands of a cognitive intervention. This domain-dependent approach may ultimately be more feasible than diagnosis-dependent biomarkers, because it is relatively agnostic to any one of the heterogeneous biological bases for the CPD clinical diagnosis.
In summary, memantine produces dose-dependent increases in measures of pre-attentive information processing in HS and CPD patients. These effects appear to be moderated by age and, in the case of PPI, baseline levels. We view these results to suggest a level of target engagement and plasticity within pre-attentive mechanisms in CPD patients that might be leveraged in the service of cognitive therapies that engage and require functionality within these pre-attentive mechanisms.
Funding and Disclosure
This work was supported by MH59803, MH94320, and MH93453 (NRS, PI). SB and GAL were also supported by the VISN 22 MIRECC, GAL by the Sidney R Baer Jr Foundation and Brain and Behavior Research Foundation, SB and H-HC by Young Investigator Awards from the Brain and Behavior Research Foundation, and HHC by the APF/Merck Early Academic Career Award. In the past 3 years, NRS has had support from Neurocrine, and GAL has received support from Astellas and Forum Pharmaceuticals. Neither NRS nor GAL receives or ever received support from companies that develop or market memantine. The remaining authors declare no conflict of interest.
References
Abel KM, Allin MP, Hemsley DR, Geyer MA (2003). Low doses of ketamine increases prepulse inhibition in healthy men. Neuropharmacology 44: 729–737.
Areosa SA, Sherriff F, McShane R (2005). Memantine for dementia. Cochrane Database Syst Rev CD 003154.pub3.
Bhakta SG, Talledo J, Lamb SN, Balvaneda B, Chou HH, Rana B et al (2014). Effects of tolcapone on neurocognitive and neurophysiological measures in healthy adults. Neuropsychopharmacology 39: S514.
Bitsios P, Giakoumaki SG, Theou K, Frangou S (2006). Increased prepulse inhibition of the acoustic startle response is associated with better strategy formation and execution times in healthy males. Neuropsychologia 44: 2494–2499.
Bodatsch M, Ruhrmann S, Wagner M, Muller R, Schultze-Lutter F, Frommann I et al (2011). Prediction of psychosis by mismatch negativity. Biol Psychiatry 69: 959–966.
Braff D, Stone C, Callaway E, Geyer M, Glick I, Bali L (1978). Prestimulus effects on human startle reflex in normals and schizophrenics. Psychophysiology 15: 339–343.
Brockhaus-Dumke A, Tendolkar I, Pukrop R, Schultze-Lutter F, Klosterkotter J, Ruhrmann S (2005). Impaired mismatch negativity generation in prodromal subjects and patients with schizophrenia. Schizophr Res 73: 297–310.
Catts SV, Shelley AM, Ward PB, Liebert B, McConaghy N, Andrews S et al (1995). Brain potential evidence for an auditory sensory memory deficit in schizophrenia. Am J Psychiatry 152: 213–219.
Cloninger CR, Przybeck TR, Svrakic DM (1991). The Tridimensional Personality Questionnaire: U.S. normative data. Psychol Rep 69: 1047–1057.
de Lucena D, Fernandes BS, Berk M, Dodd S, Medeiros DW, Pedrini M et al (2009). Improvement of negative and positive symptoms in treatment refractory schizophrenia: a double-blind, randomized, placebo-controlled trial with memantine as add-on therapy to clozapine. J Clin Psychiatry 70: 1416–1423.
Duncan EJ, Madonick SH, Parwani A, Angrist B, Rajan R, Chakravorty S et al (2001). Clinical and sensorimotor gating effects of ketamine in normals. Neuropsychopharmacology 25: 72–83.
Giakoumaki SG, Bitsios P, Frangou S (2006). The level of prepulse inhibition in healthy individuals may index cortical modulation of early information processing. Brain Res 1078: 168–170.
Elling L, Steinberg C, Bröckelmann AK, Dobel C, Bölte J, Junghofer M (2011). Acute stress alters auditory selective attention in humans independent of HPA: a study of evoked potentials. PLoS One 6: e18009.
Eysenck HJ, Eysenck SBG (1975) Manual of the Eysenck Personality Questionnaire. Hodder and Stoughton: London.
Graham FK (1975). The more or less startling effects of weak prestimulation. Psychopsysiology 12: 238–248.
Hergovich N, Singer E, Agneter E, Eichler HG, Graselli U, Simhandl C et al (2001). Comparison of the effects of ketamine and memantine on prolactin and cortisol release in men. a randomized, double-blind, placebo-controlled trial. Neuropsychopharmacology 24: 590–593.
Javitt DC, Zukin SR, Heresco-Levy U, Umbricht D (2012). Has an angel shown the way? Etiological and therapeutic implications of the PCP/NMDA model of schizophrenia. Schizophr Bull 38: 958–966.
Javitt DC (2012). Twenty-five years of glutamate in schizophrenia: are we there yet? Schizophr Bull 38: 911–913.
Jocham G, Klein TA, Ullsperger M (2014). Differential modulation of reinforcement learning by D2 dopamine and NMDA glutamate receptor antagonism. J Neurosci 34: 13151–13162.
Johnson JW, Glasgow NG, Povysheva NV (2015). Recent insights into the mode of action of memantine and ketamine. Curr Opin Pharmacol 20: 54–63.
Kawakubo Y, Kamio S, Nose T, Iwanami A, Nakagome K, Fukuda M et al (2007). Phonetic mismatch negativity predicts social skills acquisition in schizophrenia. Psychiatry Res 152: 261–265.
Kay SR, Fiszbein A, Opler LA (1987). The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr Bull 13: 261–276.
Kim YW, Shin JC, An YS (2010). Changes in cerebral glucose metabolism in patients with posttraumatic cognitive impairment after memantine therapy: a preliminary study. Ann Nucl Med 24: 363–369.
Kirino E, Inoue R (1999). The relationship of mismatch negativity to quantitative EEG and morphological findings in schizophrenia. J Psychiatr Res 33: 445–456.
Korostenskaja M, Nikulin VV, Kicić D, Nikulina AV, Kähkönen S (2007). Effects of NMDA receptor antagonist memantine on mismatch negativity. Brain Res Bull 72: 275–283.
Krivoy A, Weizman A, Laor L, Hellinger N, Zemishlany Z, Fischel T (2007). Addition of memantine to antipsychotic treatment in schizophrenia inpatients with residual symptoms: a preliminary study. Eur Neuropsychopharmacol 18: 117–121.
Kumari V, Soni W, Sharma T (1999). Normalization of information processing deficits in schizophrenia with clozapine. Am J Psychiatry 156: 1046–1051.
Lieberman JA, Papadakis K, Csernansky J, Litman R, Volavka J, Jia XD et alMEM-MD-29 Study Group (2009). A randomized, placebo-controlled study of memantine as adjunctive treatment in patients with schizophrenia. Neuropsychopharmacology 34: 1322–1329.
Light GA, Braff DL (2005). Stability of mismatch negativity deficits and their relationship to functional impairments in chronic schizophrenia. Am J Psychiatry 162: 1741–1743.
Light GA, Swerdlow NR, Braff DL (2007). Preattentive sensory processing is associated with cognitive and psychosocial functioning in healthy adults. J Cog Neurosci 19: 1624–1632.
Liu MY, Meng SN, Wu HZ, Wang S, Wei MJ (2008). Pharmacokinetics of single-dose and multiple-dose memantine in healthy chinese volunteers using an analytic method of liquid chromatography-tandem mass spectrometry. Clin Ther 30: 641–653.
Näätänen R, Paavilainen P, Reinikainen K (1989). Do event-related potentials to infrequent decrements in duration of auditory stimuli demonstrate a memory trace in man? Neurosci Lett 107: 347–352.
Rissling AJ, Braff DL, Swerdlow NR, Hellemann G, Rassovsky Y, Sprock J et al (2012). Disentangling early sensory information processing deficits in schizophrenia. Clin Neurophysiol 123: 1942–1949.
Seeman P, Caruso C, Lasaga M (2008). Memantine agonist action at dopamine D2High receptors. Synapse 62: 149–153.
Shelley AM, Ward PB, Catts SV, Michie PT, Andrews S, McConaghy N (1991). Mismatch negativity: an index of a preattentive processing deficit in schizophrenia. Biol Psychiatry 30: 1059–1062.
Sonkusare SK, Kaul CL, Ramarao P (2005). Dementia of Alzheimer’s disease and other neurodegenerative disorders—memantine, a new hope. Pharmacol Res 51: 1–17.
Swerdlow NR (2011a). Are we studying and treating schizophrenia correctly? Schizophr Res 130: 1–10.
Swerdlow NR (2011b). Beyond antipsychotics: Pharmacologically-augmented cognitive therapies (PACTs) for schizophrenia. Neuropsychopharmacology 37: 310–311.
Swerdlow N, Bhakta S, Chou HH, Lamb S, Balvaneda B, Rana B et al (2014). Amphetamine effects on acoustic startle and prepulse inhibition in 90 healthy adults: Physiological and genetic predictors. Neuropsychopharmacology 39: S177.
Swerdlow NR, Eastvold A, Gerbranda T, Uyan KM, Hartman P, Doan Q et al (2000). Effects of caffeine on sensorimotor gating of the startle reflex in normal control subjects: impact of caffeine intake and withdrawal. Psychopharmacology 151: 368–378.
Swerdlow NR, Eastvold A, Karban B, Ploum Y, Stephany N, Geyer MA et al (2002a). Dopamine agonist effects on startle and sensorimotor gating in normal male subjects: Time course studies. Psychopharmacology 161: 189–201.
Swerdlow NR, Light GA, Cadenhead KC, Sprock J, Hsieh MH, Braff DL (2006a). Startle gating deficits in a large cohort of patients with schizophrenia: Relationship to medications, symptoms, neurocognition and level of function. Arch Gen Psychiatry 63: 1325–1335.
Swerdlow NR, Light GA, Sprock J, Calkins ME, Greene MF, Greenwood TA et al (2013). Deficient prepulse inhibition in schizophrenia detected by the multi-site COGS. Schizophr Res 152: 503–512.
Swerdlow NR, Stephany N, Shoemaker JM, Ross L, Wasserman LC, Talledo J (2002b). Effects of amantadine and bromocriptine on startle and sensorimotor gating: parametric studies and cross-species comparisons. Psychopharmacology (Berl) 164: 82–92.
Swerdlow NR, Talledo JA, Sutherland AN, Nagy D, Shoemaker JM (2006b). Antipsychotic effects on prepulse inhibition in normal ‘low gating’ humans and rats. Neuropsychopharmacology 31: 2011–2021.
Swerdlow NR1, Weber M, Qu Y, Light GA, Braff DL (2008). Realistic expectations of prepulse inhibition in translational models for schizophrenia research. Psychopharmacology 199: 331–388.
Swerdlow NR, van Bergeijk DP, Bergsma F, Weber E, Talledo J (2009). The effects of memantine on prepulse inhibition. Neuropsychopharmacology 34: 1854–1864.
Takahashi H, Rissling AJ, Pascual-Marqui R, Kirihara K, Pela M, Sprock J et al (2012). Neural substrates of normal and impaired preattentive sensory discrimination in large cohorts of nonpsychiatric subjects and schizophrenia patients as indexed by MMN and P3a change detection responses. Neuroimage 66C: 594–603.
Umbricht D, Koller R, Vollenweider FX, Schmid L (2002). Mismatch negativity predicts psychotic experiences induced by NMDA receptor antagonist in healthy volunteers. Biol Psychiatry 51: 400–406.
Umbricht D, Schmid L, Koller R, Vollenweider FX, Hell D, Javitt DC (2000). Ketamine-induced deficits in auditory and visual context-dependent processing in healthy volunteers: implications for models of cognitive deficits in schizophrenia. Arch Gen Psychiatry 57: 1139–1147.
Vollenweider FX, Barro M, Csomor PA, Feldon J (2006). Clozapine enhances prepulse inhibition in healthy humans with low but not with high prepulse inhibition levels. Biol Psychiatry 60: 597–603.
Wilcock G, Möbius HJ, Stöffler A, MMM 500 group (2002). A double-blind, placebo-controlled multicentre study of memantine in mild to moderate vascular dementia (MMM500). Int Clin Psychopharmacol 17: 297–305.
Willenborg B, Schmoller A, Caspary J, Melchert UH, Scholand-Engler HG, Jauch-Chara K (2011). Memantine prevents hypoglycemia-induced decrements of the cerebral energy status in healthy subjects. J Clin Endocrinol Metab 96: E384–E388.
Zdanys K, Tampi RR (2008). A systematic review of off-label uses of memantine for psychiatric disorders. Prog Neuropsychopharmacol Biol Psychiatry 32: 1362–1374.
Zuckerman M, Bone RN, Neary R, Mangelsdorf D, Brustman B (1974). What is the sensation seeker? Personality trait and experience correlates of the Sensation-Seeking Scales. J Consulting Clin Psychol 39: 308–321.
Acknowledgements
We thank Ms Sarah Lamb and Ms Laura Gaddis for their technical assistance in data collection, and Ms Michelle Breier for assistance in data collation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Rights and permissions
About this article

Cite this article
Swerdlow, N., Bhakta, S., Chou, HH. et al. Memantine Effects On Sensorimotor Gating and Mismatch Negativity in Patients with Chronic Psychosis. Neuropsychopharmacol 41, 419–430 (2016). https://doi.org/10.1038/npp.2015.162
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2015.162
- Springer Nature Switzerland AG
We’re sorry, something doesn't seem to be working properly.
Please try refreshing the page. If that doesn't work, please contact support so we can address the problem.