
Abstract
Objective
To investigate the changes in cerebral glucose metabolism in patients with posttraumatic cognitive impairment after memantine therapy.
Methods
We performed serial F-18 fluorodeoxyglucose positron emission tomography studies before and after memantine therapy (20 mg per day) on 17 patients with posttraumatic cognitive impairment using statistical parametric mapping analysis. In addition, covariance analysis was performed to identify regions, where changes in regional cerebral glucose metabolism correlated significantly with increased Mini-Mental Status Examination scores.
Results
Statistical parametric mapping analysis demonstrated that, compared with baseline, significantly increased cerebral glucose metabolism occurred in both inferior, middle and superior frontal gyri, both angular gyri, both precuneus, the right middle cingulum, the left inferior parietal lobule, the left fusiform gyrus, the left precentral gyrus, the left paracentral lobule, and the left lingual gyrus after memantine therapy (P uncorrected < 0.005). In contrast, cerebral glucose metabolism was significantly decreased in both cerebellum, the left thalamus, the left olfactory, the right middle temporal gyrus, the right amygdala, and the right insula (P uncorrected < 0.005). In the correlation analysis, improvements in Mini-Mental Status Examination scores after memantine therapy were significantly associated with increased cerebral glucose metabolism in the inferior and middle frontal gyri and the inferior parietal lobule of the left hemisphere (P corrected < 0.0001).
Conclusions
Our findings indicate that the prefrontal and the parietal association cortices may be the relevant structures for the pharmacological response to memantine therapy in patients with posttraumatic cognitive impairment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cognitive impairments are among the most common consequences of traumatic brain injury (TBI) at all levels of severity. Although following TBI, persons may experience impairments in any cognitive domain, posttraumatic cognitive impairments most often include disturbances of arousal and attention, executive dysfunctions, memory impairments, language deficits, and social communication disturbances [1–3] and such cognitive impairments may arise from direct injury to the neural network consisting of the cerebral cortex, subcortical structures, or brainstem elements, or from secondary neuronal damage, including cytotoxic damage and neurotransmitter excitotoxicity [4].
Although the neurobiological sequences of TBI remain incompletely understood, pharmacologic strategies to ameliorate cognitive impairment following TBI have been based on the theory that disrupted neurotransmitter systems are responsible for the cognitive deficits and that modulation of these systems with a receptor agonist or antagonist may subsequently improve cognitive function [5]. In addition, even though there is no accepted pharmacologic intervention for posttraumatic cognitive impairment, treatment regimen has consisted of a variety of stimulants [6], cholinesterase inhibitors [7], dopaminergic agents [8], and antidepressants [9].
Memantine is a noncompetitive antagonist at the N-methyl-d-aspartate (NMDA) receptor, and has been used to treat patients with Alzheimer’s disease (AD) [10]. It is clinically well tolerated and has no major side effects or interactions with other, commonly used pharmacological substances [11]. Recently, the potential use of memantine for the treatment of TBI has been raised [12, 13], but clinical reports of its effects on TBI are still lacking. In the current study, we performed a serial F-18 fluorodeoxyglucose positron emission tomography (F-18 FDG-PET) study analysis before and after memantine therapy in patients with posttraumatic cognitive impairment. We used statistical parametric mapping (SPM) to evaluate whether cognitive improvements were reflected in changes in cerebral glucose metabolism after memantine therapy, thus offering insight into the neural basis of cognitive dysfunction in patients with TBI.
Materials and methods
Subjects
We consecutively enrolled 18 patients who were diagnosed with a posttraumatic cognitive impairment. TBI was determined by initial brain computed tomography or magnetic resonance imaging at the time of initial injury and medial history of patients. The severity of cognitive impairment was assessed using the Mini-Mental Status Examination (MMSE). Exclusion criteria included evidence of premorbid neurological disease or psychiatric disorder, the presence of impaired consciousness with created an inability to accurately evaluate cognitive function, and the concurrent use of other cognition-enhancing agents.
All patients with posttraumatic cognitive impairment underwent a MMSE evaluation at the beginning of the study and at the 8-week follow-up. F-18 FDG-PET imaging studies were performed upon initial evaluation as a baseline and at 8 weeks after the start of medication. During the treatment, the dosage of memantine was increased from 10 mg (5 mg twice per day) to 20 mg (10 mg twice per day) daily over a 4-week periods, the dosage of memantine was then maintained at 20 mg for the duration of the study. Of the 18 patients initially diagnosed with posttraumatic cognitive disorders, one patient was excluded from this analysis due to follow-up loss. All subjects or their family members gave informed consent, and all procedures were performed with the approval of the Institutional Review Board for Clinical Studies of our institution.
F-18 FDG-PET images acquisition
For all subjects, the first F-18 FDG-PET scan was performed prior to initiation of memantine therapy, and the second F-18 FDG-PET scan was performed after the full 8-week course of memantine. Subjects were scanned using a GE Advance PET scanner (GE, Milwaukee, WI, USA) with an intrinsic resolution of 4.8 mm full width at half maximum and simultaneous imaging of 50 contiguous transverse planes with a thickness of 3.3 mm for a longitudinal field of view of 14.5 cm. After fasting for at least 6 h, subjects received 555 MBq of F-18 FDG intravenously. All subjects were instructed to rest comfortably for 30 min with their eyes closed and ears unplugged in a quiet room, then acquisition of emission image was performed during 15 min. An 8-min transmission scan was performed using triple Ge-68 rod sources to correct for attenuation. Gathered data were reconstructed in a 128 × 128× 35 matrix with a pixel size of 1.95 × 1.95 × 4.25 mm by means of a filtered back-projection algorithm employing a trans-axial 8.5 mm Hanning filter and an 8.5 mm axial Ramp filter.
Data analysis
Spatial preprocessing and statistical analysis of PET images were performed using SPM software (SPM2, Institute of Neurology, University College London, UK) on MATLAB software version 7.1 (Mathworks Inc., Natick, MA, USA). The F-18 FDG-PET templates for all of the scans were created by averaging all F-18 FDG-PET images and spatially normalizing with the Montreal Neurological Institute (MNI, McGill University, CA) standard PET template using a nonlinear transformation of SPM2. Spatially, normalized images were then smoothed by convolution using an isotropic Gaussian kernel with a 12 mm full width half maximum to increase the signal-to-noise ratio and to accommodate the subtle variations in anatomical structure. The effects of global metabolism were removed by normalizing the count of each voxel to the mean count of the brain (proportional scaling in SPM). To identify brain regions where cerebral glucose metabolism increased following memantine treatment, the pre- and posttreatment F-18 FDG-PET images were compared voxel-to-voxel (by two-tailed paired t test), and an uncorrected P value of <0.005 and an extended threshold (K e) > 50 was considered statistically significant. In addition, covariance analysis was performed to identify regions where increased changes in cerebral glucose metabolism were significantly correlated with increases in MMSE scores using a single-subject covariate model. Regions reaching a corrected P value of <0.0001 were considered significant in the covariance analysis with >50 continuous voxels in cluster size. For visualization of the t score statistics, the significant voxels were projected onto the 3D-rendered brain or a standard high-resolution MRI template provided by SPM2, thus allowing for anatomical identification. Anatomical labeling of significant voxels was performed using the automated anatomic labeling SPM toolbox [14], which was based on anatomy provided by the MNI.
Statistical analysis
A Wilcoxon signed-rank paired test was used to compare the changes in MMSE after memantine treatment; P values of <0.01 were considered statistically significant. Statistical analyses were performed using SAS 9.1.3.
Results
Patient demographics are summarized in Table 1. Data are given as mean ± SD. The patients consisted of 13 males and 4 females with a mean age of 37.8 ± 18.8 years (range 20–73 years). Ten cases were predominately caused by diffuse axonal injury and seven cases primarily resulted from cerebral contusion; the mean time from injury was 6.8 ± 7.7 months (range 1–28 months). All patients were right handed and the mean duration of education was 12.6 ± 3.5 years (range 6–16 years). The mean MMSE score at baseline was 19.0 ± 8.9 (range 2–27). Following 8-week of memantine therapy, the mean MMSE score improved significantly in 22.5 ± 7.6 (range 5–30; mean change, 3.5 points; P = 0.0001). When analyzing the change in subitems of the MMSE, the subitems of attention and calculation, as well as language, were significantly improved following memantine therapy (mean changes in score of 0.8 and 1.3, respectively; P = 0.002 and 0.003, respectively), although no significant improvements in the subitems of orientation (0.8 points), registration (0.2 points), recall (0.4 points) were founded.
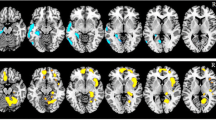
The SPM analysis demonstrated significantly increased cerebral glucose metabolism following an 8-week memantine treatment was in the middle frontal gyrus, the superior frontal gyrus, the inferior frontal gyrus, the precuneus, the middle cingulum, and the angular gyrus in the right hemisphere and in the inferior parietal lobule, the fusiform gyrus, the precentral gyrus, the inferior frontal gyrus, the superior frontal gyrus, the angular gyrus, the middle frontal gyrus, the paracentral lobule, and the lingual gyrus in the left hemisphere when compared with the baseline evaluation (P uncorrected < 0.005; Fig. 1; Table 2). In contrast, cerebral glucose metabolism was significantly decreased in the bilateral cerebellum, the left thalamus, the left olfactory bulb, the right middle temporal gyrus, the right amygdala, and the right insula areas (P uncorrected < 0.005; Fig. 2; Table 2). In the covariance analysis between the changes in cerebral glucose metabolism and MMSE score, improved MMSE score after memantine treatment was significantly correlated with increases in cerebral glucose metabolism in the left inferior parietal lobule, the left inferior frontal gyrus, and the left middle frontal gyrus (P corrected < 0.0001; Fig. 3; Table 3).

Statistical parametric maps demonstrating the spatial distribution of significant increases in cerebral glucose metabolism following memantine therapy in patients with posttraumatic cognitive impairment. Displayed voxels are significant at P uncorrected < 0.005

Statistical parametric maps demonstrating the spatial distribution of significant decreases in cerebral glucose metabolism following memantine therapy in patients with posttraumatic cognitive impairment. Displayed voxels are significant at P uncorrected < 0.005

Statistical parametric maps demonstrating the correlation of increased regional cerebral glucose metabolism and increase in Mini-Mental Status Examination scores. Displayed voxels are significant at P corrected < 0.0001. R right, L left
Discussion
We investigated the changes in cerebral glucose metabolism of patients with posttraumatic cognitive impairment after memantine therapy. Our findings were (1) the cognitive function as measured by MMSE scores significantly improved after memantine treatment, (2) when compared with baseline evaluation, memantine therapy significantly increased cerebral glucose metabolism in the prefrontal and the parietal association cortices, (3) in the covariance analysis, cerebral glucose metabolism in the left inferior and middle frontal cortices and the left inferior parietal lobule was significantly correlated with MMSE scores after memantine therapy.
Memantine was recently approved by the European Union and by the FDA for the treatment of moderate to severe AD and may also have efficacy for the treatment of vascular dementia [15]. The proposed mechanism of memantine is a specific, moderate affinity, uncompetitive NMDA receptor antagonism, and its pharmacological rationale was based on the assumption that inhibition of pathological glutamatergic activity. In TBI, neuronal damage from pathologically excessive glutamate stimulation in the cortical area appears to significantly contribute to posttraumatic cognitive impairment [16–18]. However, clinical studies demonstrating the effects of NMDA receptor antagonist for the treatment of posttraumatic cognitive impairment are still lacking.
Recently, positive effects of memantine treatment for neurodegenerative diseases [19], stroke [20], neuropathic pain [21], and schizophrenia [22] have been reported. However, memantine is under investigation as a potential treatment for other diseases as well, including traumatic and hypoxic brain injuries [23, 24]. In AD, memantine monotherapy or combined with cholinesterase inhibitors improved attention, memory, and language functions [19, 25]. Based on the current findings, increased cerebral glucose metabolism in the frontal lobe, including the dorsolateral prefrontal cortex, and in the parietal lobe, including the precuneus, the angular gyrus, and the inferior parietal lobule, may be associated with the previously reported cognitive enhancing effects of memantine. A possible explanation for these findings is that the restorative capacity of memantine may particularly affect attention, executive function, and working memory by increasing cerebral glucose metabolism in the frontal and the parietal cortices.
Cerebral hypometabolism associated with cognitive impairment after TBI has been demonstrated diffuse or focal lesions in the frontal, parietal, temporal, and occipital cortices [26, 27]. There are a few preliminary studies [22, 28] demonstrating a enhancing effect of memantine on the frontal cerebral metabolism in schizophrenia [22] and alcohol-induced dementia [28], but not much is known about the underlying mechanism of memantine’s effect on cognition and cerebral metabolism in TBI. The current study was the first to investigate the changes in cerebral glucose metabolism following memantine therapy in posttraumatic cognitive impairment. The current findings demonstrated diffuse increased cerebral glucose metabolism in the prefrontal and the parietal association area after memantine treatment; this change could be related to cognitive improvements in the setting of TBI.
NMDA receptors in the central nervous system have been proposed to be widely spread along the neuronal surface, particularly at the cerebral cortex, dentate gyrus, cerebellum, substantia nigra, and spinal cord [29], although the exact distribution depends on the local neuronal environment. The current findings suggest that memantine treatment, which acts as a blockade for NMDA receptor activity, significantly decreased cortical glucose metabolism in the cerebellum, hippocampus, and some cerebral cortices where NMDA receptors are concentrated. Studies addressing the underlying mechanism of decreased cerebral glucose metabolism after memantine therapy and how decreased cortical metabolism following memantine treatment affects cognitive function in TBI are indicated.
Several limitations of the current study design should be considered. First limitation relates to the lack of evaluation into the metabolic recovery of cortex in the control group, which represents the natural course of cortical metabolic change in TBI. Therefore, normative data for the control group should also be acquired in further studies. The second limitation was related to the heterogeneous etiology and duration of TBIs. The pathophysiologic mechanisms of TBI in the current population consisted of diffuse axonal injuries or cerebral contusions. The current findings need to be validated by future studies enrolling patients with a more homogenous set of injury etiologies, such as diffuse axonal injury or cerebral contusion. In addition, detailed cognitive evaluations were not addressed. Although the MMSE is a simple and universally applied scale that can be easily and rapidly performed by a clinician specializing in neurorehabilitation, the MMSE is not sensitive for detecting cognitive impairments of the frontoparietal subdomains. Therefore, a detail correlation analysis between the changes in cognitive subdomains and cerebral glucose metabolism could not be carried and will need to be addressed in the future studies.
Conclusion
Our findings indicate that the prefrontal and the parietal association cortices, which are parts of the neural network for cognitive processes, may be the relevant structures responsible for the pharmacological response to memantine treatment in patients with posttraumatic cognitive impairment. In addition, our results remain only preliminary until the current findings will be confirmed by further studies with control data.
References
Waxweiler RJ, Thurman D, Sniezek J, Sosin D, O’Neil J. Monitoring the impact of traumatic brain injury: a review and update. J Neurotrauma. 1995;12:509–16.
Arciniegas D, Adler L, Topkoff J, Cawthra E, Filley CM, Reite M. Attention and memory dysfunction after traumatic brain injury: cholinergic mechanisms, sensory gating, and a hypothesis for further investigation. Brain Inj. 1999;13:1–13.
Arciniegas DB, Held K, Wagner P. Cognitive impairment following traumatic brain injury. Curr Treat Options Neurol. 2002;4:43–57.
Arciniegas DB, Silver JM. Pharmacotherapy of posttraumatic cognitive impairments. Behav Neurol. 2006;17:25–42.
Phillips JP, Devier DJ, Feeney DM. Rehabilitation pharmacology: bridging laboratory work to clinical application. J Head Trauma Rehabil. 2003;18:342–56.
Challman TD, Lipsky JJ. Methylphenidate: its pharmacology and uses. Mayo Clinic Proc. 2000;75:711–21.
Masanic CA, Bayley MT, VanReekum R, Simard M. Open-label study of donepezil in traumatic brain injury. Arch Phys Med Rehabil. 2001;82:896–901.
Ben Smal D, Samuel C, Rouy-Thenaisy K, Rgnault J, Azouvi P. Bromocriptine in traumatic brain injury. Brain Inj. 2006;20:111–5.
Silver JM, McAllister TW, Arciniegas DB. Depression and cognitive complaints following mild traumatic brain injury. Am J Psychiatry. 2009;166:653–61.
Areosa SA, Sherriff F, McShane R. Memantine for dementia. Cochrane Database Syst Rev 2005:CD003154.
Parsons CG, Danysz W, Quack G. Memantine is a clinically well tolerated N-methyl-d-aspartate (NMDA) receptor antagonist—a review of preclinical data. Neuropharmacology. 1999;38:735–67.
Rao VL, Dogan A, Todd KG, Bowen KK, Dempsey RJ. Neuroprotection by memantine, a non-competitive NMDA receptor antagonist after traumatic brain injury in rats. Brain Res. 2001;911:96–100.
Muir KW. Glutamate-based therapeutic approaches: clinical trials with NMDA antagonists. Curr Opin Pharmacol. 2006;6:53–60.
Tzourio-Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Etard O, Delcroix N, et al. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. Neuroimage. 2002;15:273–89.
Orgogozo J, Rigaud A, Stffler A, Mbius H, Forette F. Efficacy and safety of memantine in patients with mild to moderate vascular dementia: a randomized, placebo-controlled trial (MMM 300). Stroke. 2002;33:1834–9.
Mukhin A, Fan L, Faden AI. Activation of metabotropic glutamate receptor subtype mGluR1 contributes to post-traumatic neuronal injury. J Neurosci. 1996;16:6012–20.
Zipfel GJ, Babcock DJ, Lee JM, Choi DW. Neuronal apoptosis after CNS injury: the roles of glutamate and calcium. J Neurotrauma. 2000;17:857–69.
Regner A, Alves LB, Chemale I, Costa MS, Friedman G, Achaval M, et al. Neurochemical characterization of traumatic brain injury in humans. J Neurotrauma. 2001;18:783–92.
Pomara N, Ott BR, Peskind E, Resnick EM. Memantine treatment of cognitive symptoms in mild to moderate Alzheimer disease: secondary analyses from a placebo-controlled randomized trial. Alzheimer Dis Assoc Disord. 2007;21:60–4.
Berthier ML, Green C, Lara JP, Higueras C, Barbancho MA, Dvila G, et al. Memantine and constraint-induced aphasia therapy in chronic poststroke aphasia. Ann Neurol. 2009;65:577–85.
Rogers M, Rasheed A, Moradimehr A, Baumrucker SJ. Memantine (Namenda) for neuropathic pain. Am J Hosp Palliat Care. 2009;26:57–9.
Cerullo MA, Adler CM, Strakowski SM, Eliassen JC, Nasrallah HA, Nasrallah AT. Memantine normalizes brain activity in the inferior frontal gyrus: a controlled pilot fMRI study. Schizophr Res. 2007;97:294–6.
Sencer A, Arica O, Kirin T, Grgl A, Aktan D. Effects of memantine and MK-801 on ischemia in an experimental model of acute subdural hematoma. Neurol Res. 2008;30:497–503.
Liu C, Lin N, Wu B, Qiu Y. Neuroprotective effect of memantine combined with topiramate in hypoxic–ischemic brain injury. Brain Res. 2009;1282:173–82.
Riepe MW, Adler G, Ibach B, Weinkauf B, Tracik F, Gunay I. Domain-specific improvement of cognition on memantine in patients with Alzheimer’s disease treated with rivastigmine. Dement Geriatr Cogn Disord. 2007;23:301–6.
Nakayama N, Okumura A, Shinoda J, Nakashima T, Iwama T. Relationship between regional cerebral metabolism and consciousness disturbance in traumatic diffuse brain injury without large focal lesions: an FDG-PET study with statistical parametric mapping analysis. J Neurol Neurosurg Psychiatry. 2006;77:856–62.
Shiga T, Ikoma K, Katoh C, Isoyama H, Matsuyama T, Kuge Y, et al. Loss of neuronal integrity: a cause of hypometabolism in patients with traumatic brain injury without MRI abnormality in the chronic stage. Eur J Nucl Med Mol Imaging. 2006;33:817–22.
Preuss UW, Bahlmann M, Bartenstein P, Schtz CG, Soyka M. Memantine treatment in alcohol dementia: rapid PET changes and clinical course. Eur Neurol. 2001;45:57–8.
Groc L, Bard L, Choquet D. Surface trafficking of N-methyl-d-aspartate receptors: physiological and pathological perspectives. Neuroscience. 2009;158:4–18.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kim, Y.W., Shin, JC. & An, Ys. Changes in cerebral glucose metabolism in patients with posttraumatic cognitive impairment after memantine therapy: a preliminary study. Ann Nucl Med 24, 363–369 (2010). https://doi.org/10.1007/s12149-010-0360-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12149-010-0360-3