

Abstract
The modulation of ion channel activity by lipids is increasingly recognized as a fundamental component of cellular signalling. The transient receptor potential mucolipin (TRPML) channel family belongs to the TRP superfamily1,2 and is composed of three members: TRPML1–TRPML3. TRPMLs are the major Ca2+-permeable channels on late endosomes and lysosomes (LEL). They regulate the release of Ca2+ from organelles, which is important for various physiological processes, including organelle trafficking and fusion3. Loss-of-function mutations in the MCOLN1 gene, which encodes TRPML1, cause the neurodegenerative lysosomal storage disorder mucolipidosis type IV, and a gain-of-function mutation (Ala419Pro) in TRPML3 gives rise to the varitint–waddler (Va) mouse phenotype4,5,6. Notably, TRPML channels are activated by the low-abundance and LEL-enriched signalling lipid phosphatidylinositol-3,5-bisphosphate (PtdIns(3,5)P2), whereas other phosphoinositides such as PtdIns(4,5)P2, which is enriched in plasma membranes, inhibit TRPMLs7,8. Conserved basic residues at the N terminus of the channel are important for activation by PtdIns(3,5)P2 and inhibition by PtdIns(4,5)P28. However, owing to a lack of structural information, the mechanism by which TRPML channels recognize PtdIns(3,5)P2 and increase their Ca2+ conductance remains unclear. Here we present the cryo-electron microscopy (cryo-EM) structure of a full-length TRPML3 channel from the common marmoset (Callithrix jacchus) at an overall resolution of 2.9 Å. Our structure reveals not only the molecular basis of ion conduction but also the unique architecture of TRPMLs, wherein the voltage sensor-like domain is linked to the pore via a cytosolic domain that we term the mucolipin domain. Combined with functional studies, these data suggest that the mucolipin domain is responsible for PtdIns(3,5)P2 binding and subsequent channel activation, and that it acts as a ‘gating pulley’ for lipid-dependent TRPML gating.
Similar content being viewed by others

Main
Our studies used TRPML3 from the common marmoset Callithrix jacchus (TRPML3WT, 97% sequence identity to human TRPML3; Extended Data Fig. 1) harbouring a putative glycosylation knockout mutation (Asn138Gln, TRPML3NQ). Whole-cell electrophysiological recording shows that TRPML3WT is fully functional and that the Asn138Gln mutation does not appreciably affect channel function6,9,10,11 (Extended Data Fig. 2). Single particle cryo-EM analyses produced a 3D reconstruction of TRPML3NQ at an overall resolution of approximately 2.9 Å (See Methods and Extended Data Fig. 3), which allowed for de novo model building (Fig. 1, Extended Data Figs 3, 4 and Extended Data Table 1). Several non-protein densities were resolved in the cryo-EM map, which we assigned as lipid molecules (Fig. 1a, Extended Data Fig. 5). Lipid molecules were observed at analogous sites in the structures of two other TRP channels, polycystin-2 (PKD2)12,13 and TRPV114.

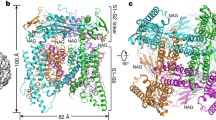
a, b, Cryo-EM reconstruction (a) and model (b) of TRPML3 viewed from the extracytosolic side (left) and from the membrane plane (right). c, Topology diagram delineating the protein domains and their secondary structures. d–f, Detailed view of the ECD (histidines implicated in pH sensing indicated by green spheres) (d), a TRPML3 protomer (e) and the mucolipin domain (f).
TRPML3 can be divided into three domains: the extracytosolic domain (ECD), the transmembrane channel domain (TMD), and the cytosolic domain (Fig. 1). The ECD comprises two β-sheets and two extracytosolic helices, which tetramerize into a ring that caps the extracytosolic side of the channel (Fig. 1d), and is structurally similar to the ECDs of TRPML115 and PKD212,13,16 (Extended Data Fig. 6). Unlike PKD2, in which the ECD interacts extensively with the TMD, there are minimal interactions between the ECD and TMD in TRPML312,13,16 (Extended Data Fig. 6). Three histidines within the ECD have been shown by mutagenesis to play an important role in pH sensing by TRPML39. The locations of these critical residues, at the ECD–ECD and ECD–TMD interfaces, suggest that pH sensing may involve histidine-mediated structural rearrangements (Fig. 1d).
The architecture of the TRPML3 TMD resembles that of bacterial voltage-gated Na+ channel, comprising a domain-swapped homotetramer with a voltage sensor-like domain (VSLD) made up of transmembrane segments S1–S4 and a pore formed by S5, S6, and two pore helices (P1 and P2) (Fig. 1e). The TMD has three prominent features. First, TRPML3 does not contain a TRP domain, a hallmark of most TRP channels17,18,19,20. Second, S4 in TRPML3 is entirely α-helical, in marked contrast with other TRP channels that contain a 310-helix13,17,18,19,21 (Extended Data Fig. 6). The 310-helix appears to confer mobility for vanilloid-dependent conformational changes on TRPV118. Third, multiple transmembrane segments protrude out of the membrane, with S1 extending about 18 Å into the lysosomal lumen and S2 extending about 24 Å into the cytosol (IS2) (Fig. 1a, b). The S1 extension establishes a larger interaction interface with the pore than seen in TRPV1, TRPV2 or PKD213,18,19 (Extended Data Fig. 6).
The cytosolic domain is a defining feature of TRPMLs (Fig. 1f). IS2 interacts with short intracellular helices connected to S1 and S3 (IS1a, IS1b and IS3). Notably, a short helix (IS6), which is connected to S6, interacts with IS1, IS2 and IS3 to form an intracellular helical bundle that we refer to as the mucolipin domain (MLD; Fig. 1).
The tetrameric ECD ring forms a vestibule above the transmembrane domain with central and lateral openings, both of which are too large to function as barriers for ion flow (Extended Data Fig. 6). The selectivity filter is located between the two pore helices, formed by the backbone carbonyls of Asn456 and Gly457 and the sidechains of Asp458 and Asp459 (Fig. 2 and Extended Data Fig. 7). Notably, we observed three cryo-EM density peaks (2.9–9.9σ) within the selectivity filter that we term sites 1–3. Owing to the ion composition in the sample (150 mM Na+), we tentatively assigned these peaks as sodium ions (Fig. 2). The minimum radius of the selectivity filter is 2.2 Å, which is wide enough to accommodate partly hydrated Ca2+ or Na+. The hydrated site 1 ion is coordinated by the carboxylate groups of Asp458 and Asp459, site 2 mainly by the backbone carbonyl groups of Gly457, and site 3 mainly by the backbone carbonyl groups of Asn456. There are three additional putative ion peaks in the cavity below the selectivity filter, which we term sites 4–6.

a, Radius along the permeation pathway. b, Six sodium ions (purple) were built into cryo-EM density peaks (red mesh). c, Proposed main sodium-occupied states, with most populated states highlighted in blue. SF, selectivity filter. d–f, Selectivity filter comparison with CaVAb (PDB ID: 5KLB23) (d) and NaVM (PDB ID: 5BZB24) (f), with the TRPML3 ion densities shown as red mesh (e), with atomic coordinates of respective ions shown as red spheres.
The C-terminal portions of the four S6 helices come into close proximity and form a constriction we term the S6 gate (Fig. 2). The minimum radius of the S6 gate at the side chains of Ile498 is about 1 Å, allowing the passage of dehydrated ions only. This conformation may represent a closed state, although a resting state that allows a low basal current at negative potential cannot be ruled out (Extended Data Fig. 2). A π helix—a rare helix with a wider turn (4.1 residues per turn) than an α-helix (3.6 residues per turn)—is observed in the middle of S6, introducing a kink that redirects the C-terminal end of the helix towards the pore to form the S6 gate. Several TRP channels contain a π helix in S613,17,22, and this structural feature has been proposed to function as a hinge that endows S6 with a gating capacity19.
TRPML3 is highly selective for Ca2+ over K+, and appears to be more selective for Ca2+ over Na+, but the molecular basis of its ion selectivity is not well understood9. Unlike Ca2+ or Na+, K+ would fit in the selectivity filter only in its dehydrated form and cannot optimally interact with the selectivity filter; this is likely to contribute to the high selectivity of TRPML3 for Ca2+ over K+. Notably, the configuration of the selectivity filter, which is composed of the two backbone carbonyls and two aspartate residues per subunit, is reminiscent of the engineered Ca2+-selective voltage-gated ion channel CaVAb23. High-resolution structures of CaVAb23 and the bacterial sodium channel NaVM24 reveal selectivity filters containing three ion-binding sites, similar to that of TRPML3 (Fig. 2). In both CaVAb and NaVM, the electrostatic energetics of site 1 was suggested to be important for conferring ion selectivity23,24. Consistent with this idea, mutating Asp459 to alanine rendered TRPML3 less selective and permeant for calcium25. In the TRPML3 reconstruction, the Na+ density peak at site 1 is appreciably weaker than those at sites 2 and 3. Because sites 2 and 3 are unlikely to be occupied at the same time, owing to their proximity (2.8 Å), approximately one Na+ ion can occupy the selectivity filter (at either site 2 or site 3), while two Ca2+ ions could occupy the selectivity filter simultaneously (at sites 1 and 2 or at sites 1 and 3), further contributing to the Ca2+ selectivity of TRPML3. We also suggest that sites 4 and 5 are occupied in an alternating fashion along with sites 2 and 3, respectively. The main populated states for Na+ ions in the selectivity filter are summarized in Fig. 2c.
In contrast to TRPV1 and TRPV2, the selectivity filter of TRPML3 is part of an extensive structural network that resembles the selectivity filters of CaVAb and NaVM, which do not act as gates. Therefore, we propose that the S6 gate is solely responsible for channel gating in TRPML3.
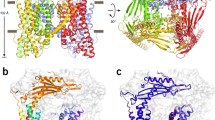
Using isothermal titration calorimetry (ITC), we found that dioctanoyl (diC8)-PtdIns(3,5)P2 binds to TRPML3NQ with a dissociation constant (Kd) of 2.5 μM (Fig. 3d). There is a large pocket between the VSLD and the MLD, formed by IS1a, IS1b and IS2, with an entrance lined by basic amino acids (Fig. 3). Distinct sets of these residues in IS1a and IS1b have been implicated in binding of PtdIns(3,5)P2 and PtdIns(4,5)P2 by TRPML17,8. PtdIns(3,5)P2 occupies this pocket in our docking studies with the headgroup positioned at the pocket entrance (Extended Data Fig. 5e, f). Point mutations in the putative PtdIns(3,5)P2 binding site (Arg58Ala, Lys62Ala, Tyr342Ala and Arg305Ala) result in a substantial reduction in diC8-PtdIns(3,5)P2 binding and a triple mutation (Lys52Ala/Arg58Ala/Lys62Ala) results in no measureable binding by ITC (Fig. 3d, Extended Data Fig. 2, Extended Data Table 2).

a, b, The putative PtdInsP2 binding site is located within a pocket of the MLD (a) where many basic residues (green) localize (b). c, IS6 fits tightly into a hydrophobic pocket formed by IS1a, IS3 and IS2; key residues shown in green. d, Mutations of the binding site substantially reduce diC8-PtdIns(3,5)P2 binding. Representative ITC data of three technical replicates; see Extended Data Fig. 8 for details. e, f, Whole lysosomal current–voltage relationships (e) and current amplitudes at −100 mV (f), in the presence and absence of PtdIns(3,5)P2. Data are presented as mean ± s.e.m., with each individual data point overlaying the bar. Number of lysosomes recorded (technical replicates) are in parentheses. WT, wild type. Statistical tests were performed to compare the sizes of PtdIns(3,5)P2-activated currents in the mutants with those in TRPML3NQ. **P < 0.01, two-tailed Student’s t-test; NS, not significant. P values: 0.0007 for Lys52Ala/Arg58Ala/Lys62Ala, 0.007 for Arg58Ala, 0.002 for Arg305Ala, 0.002 for Phe524Ala. Confidence intervals (95%): 232.8 (low)/642.7(high) for Lys52Ala/Arg58Ala/Lys62Ala, 139.4/650.5 for Arg58Ala, 202.3/660.4 for Arg305Ala, 168.1/585.7 for Phe524Ala. g, A model by which the MLD (red) propagates signals from the ECD or PtdInsP2 (green) binding to the S6 gate. Protons (yellow) and PtdIns(4,5)P2 induce a conformational change in S2 that relays through the gating pulley (MLD) to bending of S6 at the p-helix to close the pore, while PtdIns(3,5)P2 binding induces a distinct change in S2 that leads to pore opening.
Electrophysiological recordings from lysosomes showed that the Arg58Ala point mutation substantially reduced the activation of TRPML3 current by PtdIns(3,5)P2 and the triple mutant (Lys52Ala/Arg58Ala/Arg62Ala) showed no PtdIns(3,5)P2-mediated activation (Fig. 3e, f and Extended Data Fig. 2). Notably, a point mutation of Arg305 (Arg305Ala), a residue located on S2 that has not been previously implicated in PtdIns(3,5)P2 binding7,8, abolished PtdIns(3,5)P2-mediated activation (Fig. 3e, f). To test whether the effect of the Arg305Ala mutation is due to altered expression or targeting of TRPML3, we introduced the gain-of-function mutation Ala419Pro10 to the Arg305Ala mutant. The Ala419Pro mutation probably locks S6 in an open conformation owing to the position of Ala419 near the p-helix on S6 (Extended Data Fig. 8). The Arg305Ala/Ala419Pro mutant generated substantial inwardly rectifying currents in the presence or absence of PtdIns(3,5)P2 (Fig. 3e, f), suggesting that the Arg305Ala is properly expressed and the Ala419Pro mutation bypasses PtdIns(3,5)P2-dependent activation. Together, our data show that the pocket of the MLD serves as a PtdIns(3,5)P2 binding site, and that interaction of PtdIns(3,5)P2 with Arg305 in S2 may be key to activation of TRPML3.
IS1a, IS2 and IS3, which form the core of the MLD, create a docking site for three conserved hydrophobic residues from IS6 (Leu521, Phe524 and Ile525) (Fig. 3c). The architecture of the MLD and its linkage to S6 via IS6 appear to enable direct communication of PtdInsP2 binding to the S6 gate. Mutation of Phe524 to alanine substantially reduced PtdIns(3,5)P2-mediated TRPML3 currents in lysosomal electrophysiological recordings (Fig. 3e, f). The introduction of Ala419Pro into the Phe524Ala mutant restored the channel current, confirming proper channel assembly and trafficking (Fig. 3e, f).
Together, our findings reveal the structural basis for ion conduction by TRPML3, and suggest a model in which TRPML gating may be controlled by discriminative binding of different PtdInsP2 species. We speculate that binding of PtdIns(3,5)P2 induces a conformational change in S2, which pulls on S6 via the MLD to open the S6 gate. By contrast, binding of PtdIns(4,5)P2 probably causes a different S2 movement that leads to gate closure (Fig. 3g). We propose that S2 is the primary mobile unit associated with phosphoinositide binding and that the MLD acts as a gating pulley, based on our findings that (i) Arg305 in S2 appears to be important for binding of and activation by PtdIns(3,5)P2; (ii) S2 is connected to the MLD as a continuous helix (IS2); (iii) the facts that S1 interacts extensively with the pore and S4 is entirely α-helical make these two helices unlikely candidates as a mobile unit; and (iv) interaction of Phe524 in IS6 with the MLD appears to be important for PtdIns(3,5)P2-dependent activation of the channel. Our proposed gating pulley model is based on several assumptions and further study of PtdInsP2-bound TRPML structures will be required to test this model. Finally, given the high sequence conservation, the PtdInsP2-dependent gating mechanism is likely to be conserved between TRPML1 and TRPML3 (Extended Data Fig. 1), with our structure providing the framework to elucidate the molecular basis of mutations that cause mucolipidosis type IV (ML-IV)26,27,28,29 (Extended Data Fig. 8).
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized, and investigators were not blinded to allocation during experiments and outcome assessment.
Protein expression and purification
To identify a stable TRPML3 orthologue for structure determination, 31 orthologues were subjected to an expression screen. Genes were codon-optimized for expression in Spodoptera frugiperda (Sf9) cells. Mutant constructs were prepared using the Quikchange mutagenesis kit (Agilent). For expression, constructs were cloned into a pFastBac vector with a C-terminal FLAG- and His-tag, cleavable by PreScission Protease (PPX). Constructs were transformed into DH10Bac Escherichia coli cells for bacmid preparation, which were isolated from 2 ml overnight cultures by isopropanol precipitation. Virus was prepared according to the manufacturer’s manual (Invitrogen, Bac-to-Bac). Protein was expressed in Sf9 cells at a cell density of 1.5 M ml−1 and grown at 27 °C for 60–65 h. Cells were harvested by centrifugation at 1,710 g and resuspended in lysis buffer (50 mM Tris pH 8, 150 mM NaCl, 2 mg ml−1 iodoacetamide, 14.3 mM β-mercaptoethanol, 1 μg ml−1 leupeptin, 1.5 μg ml−1 pepstatin, 0.66 μg ml−1 aprotinin and 1mM phenylmethylsulfonyl fluoride). Cells were lysed by homogenization or sonication (3 × 30 pulses) and protein was extracted in 40 mM dodecyl maltopyranoside (DDM) and 4 mM cholesteryl hemisuccinate (CHS) Tris salt (Anatrace) for 1 h at 4 °C under gentle agitation. The insoluble fraction was removed by centrifugation. Anti-FLAG M2 resin (Sigma-Aldrich) was applied to the lysate and protein binding performed for 1 h at 4 °C under gentle agitation. The resin was packed onto a column and washed with 5 column volumes of DDM buffer (20 mM Tris pH 8, 150 mM NaCl, 1 mM DDM, 0.1 mM CHS and 2 mM DTT) and 10 column volumes of DM wash buffer (20 mM Tris pH 8, 150 mM NaCl, 4 mM decyl maltopyranoside (DM), 0.1 mM CHS and 2 mM DTT). Protein was eluted with 5 column volumes of DDM buffer supplemented with 100 μg ml−1 FLAG peptide. The protein was concentrated to 1 mg ml−1 and the tag was cut overnight with PPX at 4 °C. Protein was exchanged into DM buffer by concentration and 10 times dilution in DM buffer. Subsequently, the protein was applied to a Superose-6 size exclusion column in DM buffer. Peak fractions were concentrated and PMAL-C8 was added at 1:3 mass ratio of TRPML3:PMAL-C8 and incubated overnight. Biobeads were added (15 mg ml−1) and incubated for 4 h at 4 °C. A final size exclusion chromatography step was performed in HEPES pH 7.4, 75 mM NaCl and 75 mM KCl to remove excess amphipols.
EM sample preparation
For negatively-stained TRPML3, 4 μl amphipol-solubilized protein (0.04 mg ml−1) was applied to a freshly glow-discharged 400 Cu-Rh Maxtaform grid (Electron Microscropy Services) that had been coated with a thin layer of carbon. After incubation on grid for ~90 s, excess solution was wicked away using Whatman #1 filter paper and 5 μl ~1% (w/v) uranyl formate solution was immediately applied directly to the grid. After ~10 s, excess stain was wicked away and the grid surface was washed three times with 25 μl 1% (w/v) UF solution to yield thorough embedding of the sample. Following the final wash the grid was blotted to dryness and allowed to air-dry in a fume hood.
For cryo-EM, 3 μl purified amphipol-solubilized TRPML3 (0.5 mg ml−1) was applied to a freshly plasma-cleaned (75% argon/25% oxygen atmosphere, 15 W for 6 s in a Gatan Solarus) UltrAuFoil R1.2/1.3 300-mesh grid (Electron Microscopy Services) and manually blotted using filter paper (Whatman No.1) for ~4 s (≥95% relative humidity, 4 °C) immediately before plunge freezing in liquid ethane cooled by liquid nitrogen.
EM image acquisition and data processing
All EM data were acquired using the Leginon automated data acquisition program30 and all image pre-processing was performed concurrently with data collection using the Appion image-processing pipeline31. Images of negatively stained TRPML3 were acquired on a Tecnai Spirit (FEI) transmission electron microscope (TEM) operating at 120 keV. Images were collected at a nominal magnification of 52,000× on a Tietz TVIPS F416 camera, corresponding to a pixel size of 2.05 Å per pixel at specimen level. All micrographs were collected with an electron dose of ~25 electrons per Å2 with a defocus range of −0.8 to −1.6 μm. Two hundred and sixty-five images were used for automated particle picking using Difference of Gaussians (DoG) picker32 to yield 17,080 particles. CTF estimation was performed using CTFFind433 and whole-image phase-flipping was performed using EMAN34 before particle extraction. Seventeen thousand and eighty 2× binned particles were extracted and subjected to reference-free 2D classification using multivariate statistical analysis (MSA) and multi-reference alignment (MRA)35 in the Appion pipeline. Classes representing intact particles were isolated and a resulting stack of 8,928 particles were subjected to 3D classification (k = 3, tau fudge = 2) using RELION v1.436 with a 60 Å low-pass filtered map of EMD-5778 as an initial model. The best class representing 3,457 particles was then refined (C4 symmetry imposed) using the final outputted map from 3D classification as an initial model.
Images of frozen-hydrated TRPML3 were collected on a Titan Krios (FEI) operating at 300 keV equipped with a K2 Summit direct electron detector operating in super-resolution mode. Two thousand, two hundred and fifty-nine movies were collected at a nominal magnification of 22,500× with a physical pixel size of 1.31 Å per pixel (0.655 Å per pixel super-resolution pixel size) using a defocus range of −0.8 to −2.8 μm. Each movie (48 frames) was acquired using a dose rate of ~9 electrons per pixel per second over 12 s yielding a cumulative dose of ~63 electrons per Å2. The MotionCor frame alignment program37 was used to motion-correct the Fourier-binned 2 × 2 (1.31 Å per pixel) super-resolution frames using a 3-frame running average window. Aligned images were used for CTF determination using CTFFIND433 and only micrographs yielding CC estimates better than 0.5 at 4 Å resolution were kept. DoG picker32 was used to automatically pick particles from the first 50 micrographs yielding a stack of 20,571 particles binned by 4 (5.24 Å per pixel, 64-pixel box size) which were then subjected to reference-free 2D classification using MSA/MRA35 in the Appion pipeline. The best eight classes were then used as template for particle-picking against the entire data set using FindEM38. A final particle 4× binned stack of 839,127 particles was extracted (5.24 Å per pixel, 64-pixel box size) using RELION v1.436 and subjected to reference-free 2D classification. Classes that exhibited high-resolution features were then selected (498,612 particles) for 3D refinement into a single class using the negative stain reconstruction as an initial model. Following refinement, particles were re-centred and re-extracted without binning (1.31 Å per pixel, 256-pixel box size) and subjected to 3D auto-refinement with C4 symmetry imposed. The refinement was continued using a soft-mask (3 pixel extension, 5 pixel soft cosine edge) generated from a volume contoured to display the full density39. These particles were then subjected to 3D classification (k = 4, tau fudge = 8) without angular or translational searches using a soft mask contoured to show only the protein density. The highest resolution class (104,084 particles) was then further refined using the same soft protein-only mask to yield a 3.53 Å resolution reconstruction as determined by gold-standard 0.143 Fourier shell correlation (FSC)40,41. Subsequent movie-refinement (with a running average of seven frames) was followed by particle polishing (using a standard deviation of 300 pixels on the inter-particle distance and a three-frame B factor running average) with a soft mask generated against the full molecule. These ‘shiny’ particles were then refined using a soft protein-only mask to yield a final reconstruction determined to 3.02 Å resolution (gold-standard FSC). Super-resolution movies were then aligned using MotionCor2 on 5 × 5 tiled frames with a B factor of 100 applied and dose-weighting being performed42. The refined particles coordinates from the final highest-resolution class were used to extract 2× binned particles (1.31 Å per pixel, 256-pixel box size) and subjected to 3D auto-refinement using RELION 2.043. Particles were re-centred and re-extracted from unbinned, dose-weighted summed images (0.655 Å per pixel, 512-pixel box size) and subjected to 3D auto-refinement using a soft protein-only mask. The final resolution was estimated to 2.94 Å using phase-randomization to account for the convolution effects of a solvent mask on the FSC between the two independently refined half maps44. The ‘blocres’ function in the Bsoft package45 was used to perform local resolution estimation, based on the two half-volumes outputted from the RELION 3D auto-refinement. All display images were generated using UCSF Chimera46.
Model building and refinement
The model of TRPML3 was built de novo into the electron density map in Coot, using the TRPV2 structure (PDB ID: 5AN8) as a guide for the channel region47. Idealized helices and β-strands were placed in the EM density and rigid body refined. Bulky sidechains were built and used to determine the register of helices and β-strands. Subsequently, the loops were built to connect the secondary structural elements. Side chains were built into the EM density adhering to optimal rotamer conformations as indicated by EM density. The structure was real-space refined in Coot47 constrained to ideal geometry and secondary structure where appropriate. Several non-protein densities were observed into which CHS molecules and partial phospholipid molecules were built, based on the shape of the density and buffer composition. Finally, the model was iteratively real-space refined using the Phenix command line by global minimization and rigid body refinement with a high weight on ideal geometry and restrained to secondary structure48. The B factors were set to 30 Å2 for the protein and 50 Å2 for lipid molecules, after which the B factors were refined using the Phenix command line. The model was further manually optimized in Coot for adherence to geometric restraints using Molprobity (http://molprobity.biochem.duke.edu/) to identify problematic regions. After we built our model, the crystal structure of the TRPML1 ECD fragment (PDB ID: 5TJA) was reported which we used to confirm the accuracy of our model. The final model excludes 32 residues at the N terminus, two short loops in the ECD (residues 148–153 and residues 197–200), and 27 residues at the C terminus. FSCs of the half maps against the refined model agreed well with each other, showing that our model is not over-refined (Extended Data Fig. 3). Radius along the permeation pathway was calculated using HOLE49.
Cell culture and transfections
HEK293T cells (62312975 – ATCC) were grown in DMEM supplemented with 10% FBS (Gibco or Atlanta Biologicals), 1% penicillin/streptomycin (Gibco) and were sustained in 5% CO2 atmosphere at 37 °C. The cell lines used in this study have not been further authenticated. The cells for whole-cell electrophysiological recordings were tested for mycoplasma contamination. Cells between passage 5 and 30 grown in 40-mm wells (or 20-mm wells for lysosomal recordings) were transiently transfected at 30–50% confluency. For whole-cell recording, 0.8 μg pEG BacMam plasmid50 encoding TRPML3 and 0.2 μg pcDNA plasmid encoding GFP were transfected using FuGene6 (Promega). Transfection mixtures of DNA were incubated for 1.5 h at room temperature and added to each corresponding well. Transfected cells were reseeded onto 12-mm round glass coverslips (Fisher) 24 h post transfection and used 12–24 h later for electrophysiological experiments. For lysosomal recordings, GFP-tagged TRPML3 at its C terminus (TRPML3–GFP) was subcloned into pEG BacMam plasmid50. Each 20-mm well was transfected with 0.5 μg plasmid encoding TRPML3–GFP using PolyJet (SignaGen Laboratories) following the manufacturer’s protocol.
Lysosomal electrophysiology
Whole-lysosome patch clamp recordings were done as previously described51. In brief, transfected cells were re-plated onto 12-mm coverslips ~36 h after transfection. Lysosomes were enlarged with vacuolin-1 (1 μM, overnight) and were released from cells using glass pipettes. Polished glass pipettes used for patch-clamp recordings had resistances of 5–8 MΩ. Pipette solution (lumen) contained (in mM) 145 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, 10 MES and 10 glucose (pH adjusted to 4.6 with NaOH). Bath solution contained (in mM) 140 K-gluconate, 4 NaCl, 2 MgCl2, 0.39 CaCl2, 1 EGTA and 10 HEPES (pH adjusted to 7.2 with KOH). Recordings were done 48–72 h after transfection. Signals were amplified and filtered at 1 kHz using a MultiClamp 700B amplifier, and digitized at 5 kHz with a Digidata 1400A digitizer, both controlled with Clampex 10.4 (from Molecular Devices). Lysosomes were held at 0 mV. Current–voltage relationships were obtained using a ramp protocol from −100 mV to +50 mV in 1 s. Data were analysed using Clampfit 10.4 (Molecular Devices), Excel (Microsoft) and Origin (Origin Laboratory).
Whole-cell recordings
Whole-cell voltage-clamp experiments were performed with glass electrodes pulled from borosilicate glass capillaries (Sutter Instruments) with a final resistance of 2–5 MΩ. The intracellular pipette solution contained (in mM) 150 CsCl, 2 MgCl2, 5 EGTA, 10 HEPES, 4 Mg-ATP, adjusted to pH 7.2 (CsOH), and osmolarity of 280 ± 5 mOsm l−1. Glass coverslips were loaded into an open bath chamber (RC-26G, Warner Instruments) and all extracellular solutions were applied with a gravity-driven perfusion system that could completely exchange the bath solutions within 10–20 s.
Whole-cell current responses were elicited every 5 s with a continuous repeating voltage ramp protocol (holding potential 0 mV, 400-ms voltage ramp from 100 to −100 mV). Current responses were low-pass filtered at 2 kHz (Axopatch 200B), digitally sampled at 10 kHz (Digidata 1440A), converted to digital files in Clampex10.7 (Molecular Devices) and stored on an external hard drive for offline analyses (Clampfit10.7).
Cells were first bathed in an extracellular solution containing (in mM) 140 NaCl, 5 KCl, 2 MgCl2, 1 CaCl 10 mM HEPES at pH 7.4 (NaOH) and osmolarity of 290 ± 5 mOsm l−1 followed by a 0 Na solution containing (in mM) 150 NMDG, 10 HEPES at pH 7.4 (HCl) and osmolarity of 290 ± 5 mOsm l−1. Bath solutions containing either 20 μM or 80 μM 5-mesityl-3-oxa-4-azatricyclo[5.2.1.0∼2,6∼]dec-4-ene (SN-2; Specs; prepared daily from DMSO stocks (10 mM) stored at −80 °C; final DMSO 0.8%) were applied until the current at −100 mV no longer increased in amplitude. Leak conductance was assessed at the end of each recording by the residual current at −100 mV after application of the 0 Na solution.
Isothermal titration calorimetry
All mutants were prepared in the TRPMLNQ background using Quikchange mutagenesis kit (Agilent). Protein was expressed and purified as described previously, except that CHS was omitted from all buffers and the sample was kept in DDM buffer at all times. After binding, the FLAG resin was loaded onto a gravity column and washed with 10 column volumes of DDM buffer. The sample was eluted in DDM buffer supplemented with 100 μg ml−1 FLAG peptide. The tag was cut overnight and protein was further purified by Superose-6 size exclusion chromatography in DDM buffer. Protein was concentrated to ~80 μM for ITC experiments performed on a MicroCal ITC200 system. 800 μM diC8-PtdIns(3,5)P2 was titrated 1.6 μl at a time into an 80-μM protein solution. The curves were fit to a single-site binding isotherm using the Malvern Instruments software.
Docking of PtdIns(3,5)P2 to TRPML3
Docking of PtdIns(3,5)P2 to TRPML3WT was performed using AutoDock Vina52. The initial coordinates of 1-arachidolyl,2-stearyl-sn-glycero-3-phospho-(1-d-myo-inositol 3,5-bisphosphate) molecule was generated from the SMILES string using PHENIX.eLBOW, and stereochemistry was manually corrected in PHENIX.REEL. In particular, the myo- configuration of the inositol ring and double bonds in the lipid tail were examined and corrected if necessary. Before docking, the sidechains of residues K41, K52, R56, R58, K59, K62, R305, and Y342 of TRPML3WT were set as flexible. Docking was carried out over a search space of 70 × 56 × 110 Å that covers the entire putative PtdInsP2 binding cavity formed by IS1, IS2, IS3 and IS6. Seven out of the nine output poses exhibited similar conformations, where the 3′- and 5′-phosphates of PtdIns(3,5)P2 interact with the positively charged residues in the pocket, especially R52, R58, and K62. One representative docking result was shown in Extended Data Fig. 6. Docking results were visualized and analysed with PyMol.
Data availability
The sequence of TRPML3CJ can be found in the National Center for Biotechnology Information (NCBI) under accession code JAB18777.1. The coordinates are deposited in the Protein Data Bank (PDB) with the accession code 5W3S and the electron density maps have been deposited in the Electron Microscopy Data Bank (EMDB) under accession code EMD-8764.
Accession codes
Primary accessions
Electron Microscopy Data Bank
GenBank/EMBL/DDBJ
Protein Data Bank
References
Ramsey, I. S., Delling, M. & Clapham, D. E. An introduction to TRP channels. Annu. Rev. Physiol. 68, 619–647 (2006)
Julius, D. TRP channels and pain. Annu. Rev. Cell Dev. Biol. 29, 355–384 (2013)
Xu, H. & Ren, D. Lysosomal physiology. Annu. Rev. Physiol. 77, 57–80 (2015)
Bargal, R. et al. Identification of the gene causing mucolipidosis type IV. Nat. Genet. 26, 118–123 (2000)
Di Palma, F. et al. Mutations in Mcoln3 associated with deafness and pigmentation defects in varitint-waddler (Va) mice. Proc. Natl Acad. Sci. USA 99, 14994–14999 (2002)
Xu, H., Delling, M., Li, L., Dong, X. & Clapham, D. E. Activating mutation in a mucolipin transient receptor potential channel leads to melanocyte loss in varitint-waddler mice. Proc. Natl Acad. Sci. USA 104, 18321–18326 (2007)
Dong, X. P. et al. PI(3,5)P2 controls membrane trafficking by direct activation of mucolipin Ca2+ release channels in the endolysosome. Nat. Commun. 1, 38 (2010)
Zhang, X., Li, X. & Xu, H. Phosphoinositide isoforms determine compartment-specific ion channel activity. Proc. Natl Acad. Sci. USA 109, 11384–11389 (2012)
Kim, H. J. et al. A novel mode of TRPML3 regulation by extracytosolic pH absent in the varitint-waddler phenotype. EMBO J. 27, 1197–1205 (2008)
Kim, H. J. et al. Gain-of-function mutation in TRPML3 causes the mouse Varitint-Waddler phenotype. J. Biol. Chem. 282, 36138–36142 (2007)
Grimm, C. et al. Small molecule activators of TRPML3. Chem. Biol. 17, 135–148 (2010)
Wilkes, M. et al. Molecular insights into lipid-assisted Ca2+ regulation of the TRP channel Polycystin-2. Nat. Struct. Mol. Biol. 24, 123–130 (2017)
Shen, P. S. et al. The structure of the polycystic kidney disease channel PKD2 in lipid nanodiscs. Cell 167, 763–773 (2016)
Gao, Y., Cao, E., Julius, D. & Cheng, Y. TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 534, 347–351 (2016)
Li, M. et al. Structural basis of dual Ca2+/pH regulation of the endolysosomal TRPML1 channel. Nat. Struct. Mol. Biol. 24, 205–213 (2017)
Grieben, M. et al. Structure of the polycystic kidney disease TRP channel Polycystin-2 (PC2). Nat. Struct. Mol. Biol. 24, 114–122 (2017)
Paulsen, C. E., Armache, J. P., Gao, Y., Cheng, Y. & Julius, D. Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature 520, 511–517 (2015)
Cao, E., Liao, M., Cheng, Y. & Julius, D. TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 504, 113–118 (2013)
Zubcevic, L. et al. Cryo-electron microscopy structure of the TRPV2 ion channel. Nat. Struct. Mol. Biol. 23, 180–186 (2016)
Gregorio-Teruel, L. et al. The integrity of the TRP domain is pivotal for correct TRPV1 channel gating. Biophys. J. 109, 529–541 (2015)
Saotome, K., Singh, A. K., Yelshanskaya, M. V. & Sobolevsky, A. I. Crystal structure of the epithelial calcium channel TRPV6. Nature 534, 506–511 (2016)
Liao, M., Cao, E., Julius, D. & Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504, 107–112 (2013)
Tang, L. et al. Structural basis for Ca2+ selectivity of a voltage-gated calcium channel. Nature 505, 56–61 (2014)
Naylor, C. E. et al. Molecular basis of ion permeability in a voltage-gated sodium channel. EMBO J. 35, 820–830 (2016)
Kim, H. J., Yamaguchi, S., Li, Q., So, I. & Muallem, S. Properties of the TRPML3 channel pore and its stable expansion by the Varitint-Waddler-causing mutation. J. Biol. Chem. 285, 16513–16520 (2010)
Venkatachalam, K., Wong, C. O. & Zhu, M. X. The role of TRPMLs in endolysosomal trafficking and function. Cell Calcium 58, 48–56 (2015)
AlBakheet, A. et al. A novel mutation in a large family causes a unique phenotype of Mucolipidosis IV. Gene 526, 464–466 (2013)
Tüysüz, B., Goldin, E., Metin, B., Korkmaz, B. & Yalçinkaya, C. Mucolipidosis type IV in a Turkish boy associated with a novel MCOLN1 mutation. Brain Dev. 31, 702–705 (2009)
Bach, G. Mucolipidosis type IV. Mol. Genet. Metab. 73, 197–203 (2001)
Suloway, C. et al. Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 151, 41–60 (2005)
Lander, G. C. et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J. Struct. Biol. 166, 95–102 (2009)
Voss, N. R., Yoshioka, C. K., Radermacher, M., Potter, C. S. & Carragher, B. DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. J. Struct. Biol. 166, 205–213 (2009)
Rohou, A. & Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015)
Ludtke, S. J., Baldwin, P. R. & Chiu, W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 128, 82–97 (1999)
Ogura, T., Iwasaki, K. & Sato, C. Topology representing network enables highly accurate classification of protein images taken by cryo electron-microscope without masking. J. Struct. Biol. 143, 185–200 (2003)
Scheres, S. H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012)
Li, X. et al. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 10, 584–590 (2013)
Roseman, A. M. FindEM—a fast, efficient program for automatic selection of particles from electron micrographs. J. Struct. Biol. 145, 91–99 (2004)
Scheres, S. H. Beam-induced motion correction for sub-megadalton cryo-EM particles. eLife 3, e03665 (2014)
Scheres, S. H. & Chen, S. Prevention of overfitting in cryo-EM structure determination. Nat. Methods 9, 853–854 (2012)
Rosenthal, P. B. & Henderson, R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 333, 721–745 (2003)
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017)
Kimanius, D., Forsberg, B. O., Scheres, S. H. & Lindahl, E. Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. eLife 5, e18722 (2016)
Chen, S. et al. High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy 135, 24–35 (2013)
Heymann, J. B. & Belnap, D. M. Bsoft: image processing and molecular modeling for electron microscopy. J. Struct. Biol. 157, 3–18 (2007)
Goddard, T. D., Huang, C. C. & Ferrin, T. E. Visualizing density maps with UCSF Chimera. J. Struct. Biol. 157, 281–287 (2007)
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004)
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010)
Smart, O. S., Neduvelil, J. G., Wang, X., Wallace, B. A. & Sansom, M. S. HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 14, 354–360, 376 (1996)
Goehring, A. et al. Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat. Protocols 9, 2574–2585 (2014)
Cang, C. et al. mTOR regulates lysosomal ATP-sensitive two-pore Na+ channels to adapt to metabolic state. Cell 152, 778–790 (2013)
Trott, O. & Olson, A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 (2010)
Acknowledgements
EM data were collected at The Scripps Research Institute (TSRI) electron microscopy facility. We thank C.-G. Cheong and S. Thomas for initial TRPML3 biochemistry, J.-C. Ducom for computational support, B. Anderson for microscope support, S. Chowdhury for aiding in preliminary EM analyses, A. Kuk for help with the docking study, and R. Brennan and M. Schumacher for providing access to ITC equipment. This work was supported by the National Institutes of Health (NIH) (R35NS097241 to S.-Y.L., DP2EB020402 to G.C.L., R01NS055293 and R01NS074257 to D.R.). G.C.L is supported as a Searle Scholar and a Pew Scholar. M.A.H. was supported by a Helen Hay Whitney Foundation postdoctoral fellowship. Computational analyses of EM data were performed using shared instrumentation funded by NIH S10OD021634.
Author information
Authors and Affiliations
Contributions
M.H. conducted biochemical optimization of TRPML3 for structure determination and model building under the guidance of S.-Y.L. M.A.H. conducted all electron microscopy experiments and the single-particle 3D reconstruction under the guidance of G.C.L. J.W. performed all the lysosome electrophysiology recordings under the guidance of D.R. Y.S. and W.F.B. carried out PtdIns(3,5)P2 binding and whole-cell patch recordings, respectively, under the guidance of S.-Y.L. S.-Y.L., G.C.L., D.R., M.H. and M.A.H. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information Nature thanks C. Ulens and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Figure 1 Sequence alignment of TRPMLs.
Secondary structure elements are indicated by rectangles (helices) and arrows (β-strands). Coloured as in Fig. 1. Residues with sequence conservation are highlighted in grey, the selectivity filter in green, the p-helix in yellow, the pH-sensing histidines in red, and putative PtdInsP2 binding residues in blue. Potential glycosylation sites are indicated by an orange branch.
Extended Data Figure 2 Functional characterization of the TRPML3 channel and representative ITC raw data and binding isotherms for diC8-PtdIns(3,5)P2 interacting with TRPML3 mutants.
a, b, Representative whole-cell current traces recorded with repeated voltage ramps (from −100 to +100 mV; 400 ms; 5-s intervals between ramps) from HEK293T cells transfected with TRPML3WT (a) or TRPML3NQ (b) at basal currents in 140 mM Na (orange), 0 mM Na (black), and during application of either 20 μM (green) or 80 μM (blue) of the TRPML3 agonist SN-2. c, Averaged inward current sizes at −100 mV normalized to cell capacitance (pA/pF) (TRPML3WT: n = 6 biologically independent experiments; TRPML3NQ: n = 6; open circles represent individual experimental data points). No significant differences (NS) in the current density at 20 μM (P = 0.89) and 80 μM (P = 0.75) SN-2 was determined between TRPML3WT and TRPML3NQ (two-tailed Student’s t-test, P > 0.05). Confidence intervals (95%): −2.43 (low)/1.88(high) for 0 Na+, −6.18/4.31 for 140 Na+, −62.69/56.84 for 20 μM SN-2 and −150.61/121.17 for 80 μM SN-2. Bar graph and error bars denote means ± s.e.m. d, e, Whole lysosomal current–voltage traces from TRPML3WT (d) and TRPML3NQ R58A (e), in the presence and absence of PtdIns(3,5)P2. Average inward currents are shown in Fig. 3f. f, For each TRPML3 mutant, raw ITC data and fitted binding isotherms are shown. Putative binding site mutants (Arg58Ala, Lys62Ala, Tyr342Ala, Arg305Ala and Lys52Ala/Arg58Ala/Lys62Ala) show a substantially reduced binding affinity while two negative control mutations (Lys59Ala and Lys326Ala) do not affect binding appreciably. NQ refers to N138Q mutant background. All titrations were performed in triplicate (technical replicates). Representative data are shown. Mean thermodynamic parameters for triplicate titrations are shown in Extended Data Table 2. Mean Kd values for each triplicate are as follows: NQ 2.5 μM, NQ R58A 11.8 μM, NQ K59A 4.4 μM, NQ K62A 11.3 μM, NQ R305A 11.8 μM, NQ K326A 4.0 μM, NQ Y342A 9.4 μM, NQ K52A/R58A/K62A not determined. Owing to the low heat associated with binding in many TRPML3 mutants, only the Kd values for NQ, NQ K59A, and NQ K326A were reliably measured.
Extended Data Figure 3 Cryo-EM data collection, processing, and validation.
a, b, Representative micrograph of TRPML3 in vitreous ice (a). We collected 2,259 movies of TRPML3. Only images exhibiting Thon rings beyond 4 Å were used for image processing, as assessed by a 1D plot (b). c, Per-frame radiation damage weighting was applied by estimating the average per-frame B factor for all movies in the TRPML3 data set (above). The frequency-dependent weights used to generate the final stack of summed particle images are shown below. d, Representative 2D class averages. e, Initial classification of particles into three 3D classes (II), one of which (class 1) was used for subsequent 3D refinement. After performing the particle polishing step in RELION (III), per-particle CTF was estimated, particles were re-extracted with a box size of 512 × 512, and then refined in RELION to produce the final reconstruction (IV). f, Fourier shell correlation plot calculated from independently refined half-maps. g, FSC curves calculated between the atomic model and the final map (black line), and between the model and each half-map (orange and blue lines). h, Local resolution estimates of the final reconstructions calculated using BSOFT45. i, Worm representation with the ASU coloured according to the per-residue Cα root mean square deviation (r.m.s.d.) value (Å), the rest of the molecule is coloured wheat. j, Histogram of the per-residue Cα r.m.s.d. values calculated from the top 10 refined atomic models with the mean per-residue Cα r.m.s.d. value shown as a black vertical bar.
Extended Data Figure 4 Quality of cryo-EM density of key elements in the structure.
The structural elements are shown in cartoon representation with side chains as sticks, coloured as in Fig. 1. The cryo-EM density is shown as blue mesh.
Extended Data Figure 5 Putative lipid densities in the TRPML3 map.
a, Four lipid densities per protomer were resolved in the cryo-EM density and corresponding lipid molecules were built. An additional lipid density was observed near S4 of the VSLD and S5 of the pore domain, but the lipid molecule was not built owing to ambiguity of the density. b, Two cholesteryl hemisuccinate (CHS) molecules were built in the crevice formed by S5 and S6, and the third CHS molecule was fit alongside the N-terminal end of S1. In between the CHS molecules an elongated density was observed, into which a long lipid tail was built, probably from either a phospholipid or a fatty acid molecule. c, d, Lipid densities in analogous locations were reported for TRPV1 (c, PDB ID: 5IRZ14) and PKD2 (d, PDB ID: 5MKE12). e, f, PtdIns(3,5)P2 was docked onto the TRPML3 structure, showing that the phosphoinositol headgroup docks to the basic pocket between the VSLD and MLD interacting primarily with the side-chains of Lys52, Arg58, Lys62, and Tyr342, while the acyl chains penetrate through a tunnel in the cytosolic domain to a cavity formed by S3, S4 and S6. Residues interacting with the docked PtdIns(3,5)P2 are shown in green (f). g, PtdIns(3,5)P2 (blue sticks) docked to a location in the TRPML3 structure (blue cartoon representation) distinct from the phosphoinositide (green sticks) found in the TRPV1 structure (PDB ID: 5IRZ14, green cartoon representation) in the pocket formed by S3–S5.
Extended Data Figure 6 Structural comparison of TRPML3 with other channels.
a, The ECD from TRPML3 (red ribbon) superimposes well on the crystal structure of the ECD from TRPML1 (brown ribbon, PDB ID: 5TJA15), with a Cα r.m.s.d. of 1.8 Å. Views are shown from the membrane plane (left) and the extracytosolic side of the membrane (right). b, The polycystin domain from PKD2 (blue ribbon, PDB ID: 5T4D13) adopts a similar fold to the ECD from TRPML3 (red ribbon). The structural elements of PKD2 are labelled. c, The TRPML3 ECD lies ~9 Å away from the channel domain, which leads to limited interactions between the ECD and VSLD. d, The PKD2 TOP/polycystin domain lies directly on top of the channel domain (PDB ID: 5T4D13) and forms extensive interactions with the pore. e, More extensive interactions are formed between S1 and the remainder of the channel in TRPML3 than in other TRP channels (buried surface areas ~3,000 Å2 and 1,500–2,000 Å2, respectively). The interaction interface is shown in green. f, In many TRP channels a short 310-helix is present in S4. By contrast, S4 in TRPML3 is completely α-helical, suggesting a more static nature.
Extended Data Figure 7 Detailed view of the selectivity filter.
a, Four cryo-EM density peaks observed near Asp458 and Asp459 are attributed to water molecules, based on coordination distances (~3.2 Å). TRPML3 is shown in cartoon representation with the selectivity filter residues shown as sticks. Cryo-EM density is shown in orange mesh for water molecules (red spheres) and red mesh for sodium ions (purple spheres). b, Interatomic distances (Å) between ions and coordinating side-chain and backbone atoms. c–e, Selectivity filter comparison of TRPML3 (c) with TRPV1 (PDB ID: 3J5P22) (d) and TRPV6 (PDB ID: 5IWP21) (e), with respective ions shown as spheres (purple for Na+ and green for Ca2+).
Extended Data Figure 8 The locations of the varitint-waddler and ML-IV-causing mutations in the TRPML3 structure.
a, Overview of the TRPML3 structure with the location of the varitint-waddler mutation Ala419Pro indicated by a green sphere and the p-helix coloured magenta. TRPML3 is shown as cartoon representation, coloured as in Fig. 1. b, Detailed view of the location of Ala419. Ala419 is positioned in the middle of S5 near the p-helix on S6, such that mutation to a proline is likely to disrupt normal S6 bending, locking it in an ‘open gate’ conformation. c, d, Overview of the TRPML3 structure with the location of ML-IV-causing mutations indicated by red spheres, shown from the membrane plane (c) and the extracytosolic side of the membrane (d). Unless otherwise indicated, residues are labelled using TRPML1 numbering. The locations of varitint-waddler mutations are represented by cyan spheres. While the mutations on the ECD are associated with mild phenotypes and affect channel assembly, there are many missense mutations that cause more severe phenotypes and are localized to the channel region. These mutations can be categorized into three groups according to location: mutations around pore helix 1 (group 1), mutations around S5 near A419 (group 2), and mutations within the VSLD around the PtdInsP2-binding site (group 3). On the basis of our structural and functional studies, we can infer that group 1 mutations potentially disrupt ion conduction and selectivity, group 2 mutations disturb gating, and group 3 mutations affect either PtdInsP2 binding (R403C in TRPML1) or S2 motion associated with gating (T308P and D362Y in TRPML1). Future structural and functional studies of these mutations will shed light on the molecular basis of pathogenic mutations that lead to ML-IV.
Supplementary information
Rights and permissions
About this article

Cite this article
Hirschi, M., Herzik Jr, M., Wie, J. et al. Cryo-electron microscopy structure of the lysosomal calcium-permeable channel TRPML3. Nature 550, 411–414 (2017). https://doi.org/10.1038/nature24055
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature24055
- Springer Nature Limited
This article is cited by
-
Lysosomal cation channel TRPML1 suppression sensitizes acute myeloid leukemia cells to chemotherapeutics by inhibiting autophagy
Molecular and Cellular Biochemistry (2024)
-
TRP (transient receptor potential) ion channel family: structures, biological functions and therapeutic interventions for diseases
Signal Transduction and Targeted Therapy (2023)
-
Cytoskeletal regulation of a transcription factor by DNA mimicry via coiled-coil interactions
Nature Cell Biology (2022)
-
Cryo-EM structure of type 1 IP3R channel in a lipid bilayer
Communications Biology (2021)
-
Visualizing structural transitions of ligand-dependent gating of the TRPM2 channel
Nature Communications (2019)