

Abstract
Transient receptor potential mucolipin 1 (TRPML1) is a cation channel located within endosomal and lysosomal membranes. Ubiquitously expressed in mammalian cells1,2, its loss-of-function mutations are the direct cause of type IV mucolipidosis, an autosomal recessive lysosomal storage disease3,4,5,6. Here we present the single-particle electron cryo-microscopy structure of the mouse TRPML1 channel embedded in nanodiscs. Combined with mutagenesis analysis, the TRPML1 structure reveals that phosphatidylinositol-3,5-bisphosphate (PtdIns(3,5)P2) binds to the N terminus of the channel—distal from the pore—and the helix–turn–helix extension between segments S2 and S3 probably couples ligand binding to pore opening. The tightly packed selectivity filter contains multiple ion-binding sites, and the conserved acidic residues form the luminal Ca2+-blocking site that confers luminal pH and Ca2+ modulation on channel conductance. A luminal linker domain forms a fenestrated canopy atop the channel, providing several luminal ion passages to the pore and creating a negative electrostatic trap, with a preference for divalent cations, at the luminal entrance. The structure also reveals two equally distributed S4–S5 linker conformations in the closed channel, suggesting an S4–S5 linker-mediated PtdInsP2 gating mechanism among TRPML channels7,8.
Similar content being viewed by others


Main
The mucolipin subfamily of transient receptor potential (TRPML) channels are predominantly localized in the membranes of the late endosomes and lysosomes1,2. Among the three isoforms of TRPML channels (TRPML1–3), TRPML1 is ubiquitously expressed in mammalian cells and has been the subject of extensive study2,9,10,11,12,13,14. TRPML1 is a Ca2+-permeable, non-selective, six-transmembrane tetrameric cation channel, which is thought to be the main lysosomal Ca2+-release channel and is of importance in lysosomal trafficking and signal transduction15,16,17,18,19. Like most TRP channels, TRPML1 is ligand-gated and can be activated by the endolysosomal-specific lipid phosphatidylinositol-3,5-bisphosphate (PtdIns(3,5)P2)7,8, whereas it is inhibited by the plasma membrane-localized phosphoinositide isoform PtdIns(4,5)P214 (Extended Data Fig. 1). This lipid-isoform-dependent gating property defines the compartment-specific channel activity of TRPML1. In humans, loss-of-function mutations in TRPML1 are the direct cause of mucolipidosis type IV, an autosomal recessive lysosomal storage disease characterized by abnormal neurodevelopment, retinal degeneration and iron-deficiency anaemia3,4,5,6. Owing to its direct link to this disease, TRPML1 has been a potential target for small-molecule therapeutic agents, and several synthetic small-molecule agonists have been developed12,20.
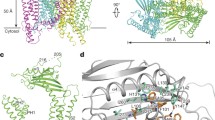
We purified and reconstituted mouse TRPML1 into nanodiscs and determined its structure using single-particle electron cryo-microscopy (cryo-EM) to a resolution of 3.59 Å, using the ‘gold standard’ Fourier shell correlation (FSC) = 0.143 criterion21 (Extended Data Figs 2, 3, 4 and Methods). The cryo-EM density map is of sufficient quality to build models of major parts of the protein (Extended Data Figs 5 and 6). TRPML1 consists of two structural components: the membrane-spanning S1–S6 region embedded in a nanodisc, and the luminal S1–S2 linker that forms a fenestrated canopy atop the channel domain (Fig. 1). Like other classical six-transmembrane tetrameric cation channels, the S1–S6 of each subunit is separated into an S1–S4 voltage-sensing domain and an S5–S6 pore domain, linked by the S4–S5 linker helix. The tetramer assembly is domain-swapped, with the S1–S4 domain interacting with the pore-forming S5–S6 of a neighbouring subunit. As the structure was determined in a lipid environment, a density of poly-acyl chains from closely attached lipid molecules is clearly visible within the grooves between two neighbouring S1–S4 domains (Fig. 1a). The 200-residue, luminal-facing linker domain between S1 and S2 can self-assemble into a tetramer and its crystal structure22 fits readily into our electron microscopy density map, facilitating our model building. Asn230 of the luminal linker domain is glycosylated with visible electron density for the covalently linked N-acetylglucosamine moiety of the sugar (Fig. 1b, c).

a, Side view of a 3D reconstruction of TRPML1 in a nanodisc (grey belt). Channel subunits are coloured individually, with lipid density in grey. b, Side view of a cartoon representation of the TRPML1 structure. N-acetylglucosamine (NAG) molecules are rendered as sticks. c, Cytosolic view of the channel. d, Structure of a single subunit in the same orientation as the cyan-coloured subunit in b.
The S1–S4 domain of TRPML1 is similar to that of other TRP channels, but differs from the canonical voltage-sensing domain of voltage-gated channels (Fig. 2a). It consists of four straight helices tightly packed against each other. Lacking the canonical voltage-sensing arginine residues, the S4 helix runs antiparallel to the S5 helix of a neighbouring subunit and exhibits extensive hydrophobic interactions (Fig. 2a). The S1–S4 domain of TRPML probably remains static, similar to TRPV123,24, and does not undergo any conformational change during channel gating. There are two cytosolic extensions to the S1–S4 domain that are unique to TRPML channels (Fig. 2a, b). First, an Arg/Lys-rich (poly-basic) domain from residues 40 to 62 forms two short clamp-shaped helices, labelled as H1 and H2, right before S1. Second, the linker between S2 and S3 forms a helix–turn–helix (H3–turn–H4) that extends into the cytosol and also makes direct contact with the N-terminal H1 helix (Fig. 2b). The N-terminal poly-basic domain is important for PtdInsP2 binding7,14, and its deletion abolishes PtdIns(3,5)P2 activation of TRPML1 (Extended Data Fig. 1e). The H1 and H2 helices of the poly-basic domain, along with the N terminus of S1, create a positively charged pocket directly beneath the cytosolic membrane surface (Fig. 2b), which is likely to accommodate the inositol trisphosphate head group of membrane-anchored PtdInsP2. Because the PtdInsP2 site is distant from the inner helix-formed channel gate, we consider that PtdInsP2 activation is mediated by the H3–turn–H4 extension, in which the H3 helix interacts with the H1 and the H4 helix interacts with the S4–S5 linker in a specific conformation, as will be discussed later.

a, Right, S1–S4 domain (blue) of the front subunit. The two cytosolic extensions are coloured differently: magenta for H1 and H2 and green for the H3–turn–H4 between S2 and S3. The inset (left) shows inter-subunit contact between the S4 helix (blue) and the neighbouring S5 (pink). b, Expanded view of the poly-basic domain and the interactions between the H1 and H3 helices.
Because the structure was determined in the absence of ligands, the TRPML1 ion conduction pore, consisting of the S5, S6 and two pore helices (P1 and P2), adopts the closed conformation (Fig. 3a, b). The four pore-lining S6 inner helices form a bundle crossing at the cytosolic side with multiple narrow constriction points that prevent the passage of hydrated cations. Surrounding the cytosolic gate, S6 makes tight contact with the N-terminal region of S5 (Fig. 3a, inset), suggesting a coupled movement between S5 and S6 upon channel gating. The Phe, Ile, Tyr, Met and Val residues on S6, just below the selectivity filter region, form a π-helix (Fig. 3a), a structural feature that has also been observed in other TRP channel structures23,25 and may facilitate the bending of the S6 inner helix for channel gating.

a, Ion conduction pore of TRPML1 with front and rear subunits removed for clarity. The central pathway is marked with a dotted mesh. The insets are expanded views of the bundle crossing with atom-to-atom distances in ångstroms and the packing between S5 and S6. The π-helix on S6 is coloured red. b, Pore radius along the central axis. c, Expanded view of the selectivity filter with atom-to-atom distances in ångstroms. S0 density is modelled as Na+ (red sphere). d, Top view of the selectivity filter showing hydrogen-bonding networks between the Asp471 side chain and backbone nitrogen atoms of neighbouring filter residues. e, Top view of the luminal linker domains in the tetramer. Three acidic residues on the luminal pore loop (labelled ‘loop’) form the narrowest part of the central hole. f, Side view of the funnel-shaped central hole and the open central court just above the filter. Front and rear subunits are removed for clarity. The luminal pore loop is coloured red. g, Cross-section through the channel showing the surface electrostatic potential along the ion conduction pathway. The open court at the luminal entrance is accessible by multiple luminal ion passages, indicated by arrows.
The TRPML1 selectivity filter has the sequence 469Asn-Gly-Asp-Asp-Met and adopts a multi-ion configuration (Fig. 3c). The backbone carbonyls of the Asn-Gly-Asp residues all point towards the central pore axis, creating two contiguous ion-binding cages (labelled as S1 and S2)—reminiscent of the two K+ channel sites but with wider dimensions—and probably accommodating partially dehydrated cations. The second Asp (Asp472), the carboxylate side chain of which is pointed upright, is stabilized laterally by a salt bridge with a highly conserved Lys453 between the S5 and P1 helices. Four Asp472 residues in a channel tetramer encircle an ion-binding site (labelled as S0) at the luminal entrance of the channel, where an electron density peak is clearly visible and was modelled as Na+, the only cation in the sample (Fig. 3c). The distance between the S0 ion and the carboxylate oxygen of Asp472 is about 5 Å, and the ion chelation is likely to be mediated by water. TRPML1 is known to be Ca2+ permeable, and Ca2+ permeation lowers channel conductance by blocking monovalent cation currents in a luminal-pH-dependent manner (Extended Data Fig. 7a). Surrounded by acidic residues, we propose that S0 is the Ca2+ blockage site. Indeed, neutralizing Asp472 with Asn results in a markedly diminished luminal Ca2+ block (Extended Data Fig. 7b, c).
The TRPML1 filter is rigidly held in place by two pore helices with tightly packed surroundings. The side-chain carboxylate of Asp471 has an essential role in stabilizing the filter conformation, by generating a hydrogen-bonding network with the backbone amide nitrogen atoms of the neighbouring filter residues (Fig. 3d). The Met473 side chain is extended parallel to the filter and fills the surrounding void (Fig. 3c). The tight packing of the TRPML1 filter is quite different from that of the TRPV1 filter, in which the equivalent P2 helix is replaced by an extended loop, allowing the filter to undergo a marked conformational change upon binding of spider vanillotoxin and gating24. A similar filter conformational change is unlikely to occur in TRPML1.
The four luminal linker domains, each consisting of two long helices and seven strands, form a square-shaped canopy with a central opening above the channel pore (Figs 1b and 3e), a characteristic feature of group 2 TRP channels including TRPML and TRPP25,26 (Extended Data Fig. 8). The α1 and α2 helices, along with the long downward-facing loop (the luminal pore loop) between them, generate a funnel-shaped hole at the centre of the canopy. The tip of the luminal pore loop is clustered with acidic residues and forms the narrowest opening at the bottom of the funnel with a diameter of about 12 Å, wide enough for free ion passage (Fig. 3e, f and Extended Data Fig. 8). The α1 helix is located in the C-terminal region of an exceptionally long S1 helix and, consequently, the luminal canopy is firmly supported by four pillar-like S1 helices over the transmembrane channel with a wide gap, creating an open central court just above the filter with four side windows (Figs 1b and 3f, g) that provide additional luminal ion passages to the ion conduction pore. The open central court is surrounded by an exceptionally large number of acidic residues from the luminal pore loop and the luminal entrance to the filter, creating a highly negative environment (Fig. 3f, g). This feature, along with multiple converging ion passages, renders the central court an electrostatic sink that can favourably attract divalent Ca2+ ions. The enrichment of Ca2+ ions can effectively limit the access of monovalent cations to the filter and, thereby, reduce the permeation of monovalent ions. Indeed, it has been shown that neutralizing the acidic residues on the luminal pore loop weakens the Ca2+ blockage22.
The density of the S4–S5 linker in the overall TRPML1 structure is poorly resolved, indicating dynamic motion of the linker. A focused classification at the transmembrane region yielded two subsets of particles that were reconstructed individually into two structures at 3.64 Å and 3.75 Å resolution, respectively, each adopting a distinct conformation at the S4–S5 linker (Extended Data Fig. 3). The nearly equal distribution of particles for the two structures (9,000 and 11,000 particles, respectively) suggests that the unliganded TRPML1 channel is stable in two closed states (closed I and II), with the S4–S5 linker undergoing conformational equilibrium.
The S4–S5 linker adopts a straight helix in the closed I state, with its N terminus interacting with the S5 helix from the neighbouring subunit (Fig. 4a–c). Four linker helices surround the S6 bundle crossing and stabilize the closed channel pore. It is notable that several gain-of-function mutations similar to the Val432Pro varitint-waddler phenotype15,27,28,29 have been identified on S5 in a proline-scanning mutagenesis study30, including Arg427Pro, Cys430Pro and Cys431Pro, which are all clustered at the interaction interface between S5 and the S4–S5 linker (Fig. 4c). These mutations probably cause a kink in the S5 helix, disrupt the inter-subunit interactions and destabilize the closed channel pore. In the closed II state, the S4–S5 linker undergoes a marked conformational change as compared to the closed I state (Fig. 4a, b). Pivoting at its C terminus, the linker swings away from the central axis and unlocks the cuff around the bundle crossing; the N-terminal half of the linker helix becomes partially unfolded and bulges outward, and the H3–turn–H4 extension of S2/S3 moves slightly towards the centre and engages in direct contact with the bulged linker that is not observed in the closed I state (Fig. 4d). Whereas the S6 inner helices remain in a closed state, the outward movement of the S4–S5 linker starts to pull S5 away from the central axis, and any further movement of S5 would result in a concurrent movement of S6 and pore opening. Therefore, the closed II structure probably represents a transition state in which the channel is primed for opening.

a, Superposition between closed I (purple) and closed II (green) states with H1 and H2 helices omitted for clarity. Arrows indicate the movement of the S4–S5 linker. b, Expanded view of the superposition for one subunit. c, Inter-subunit interactions between N termini of the linker and the neighbouring S5 in the closed I state at the location marked by the purple box in b. d, Interactions between the partially unfolded linker and the H4 helix at the location marked by the green box in b. e, Proposed working model for PtdIns(3,5)P2 activation. The arrows indicate proposed movements upon ligand binding.
As the S4–S5 linker is strategically positioned to couple the voltage- or ligand-induced conformational changes to the opening of the pore in various six-transmembrane channels, it probably has a similar role in PtdInsP2 gating of TRPML1. With its binding site at the N terminus of TRPML1, distal from the S4–S5 linker, PtdIns(3,5)P2 activation is likely to be mediated by the H3–turn–H4 of the S2/S3 extension, in which the H3 helix interacts with H1 and the H4 helix interacts with the bulged S4–S5 linker in the closed II state. We therefore propose a simple gating mechanism, in which the PtdIns(3,5)P2-induced conformational change can be propagated to the outward-bulged S4–S5 linker via the H3–turn–H4 and open the pore (Fig. 4e). This model implies that PtdIns(3,5)P2 activation can occur only when TRPML1 is in the closed II transition state, as the S4–S5 linker helix is decoupled from H3–turn–H4 in the closed I state, and thereby the linker transition between the two closed states is a prerequisite for the PtdInsP2 activation of TRPML.
Methods
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment.
Protein expression, purification and nanodisc reconstitution
Mouse TRPML1 (NCBI RefSeq NM_053177.1) containing a C-terminal thrombin cleavage site followed by a 10× His tag was cloned into a pEZT vector31 and heterologously expressed in HEK293F cells (Life Technologies) using the BacMam system (Thermo Fisher Scientific). The baculovirus was generated in Sf9 cells (Life Technologies) following the standard protocol and used to infect HEK293F cells at a ratio of 1:40 (virus:HEK293F, v/v) and supplemented with 2 mM sodium butyrate to boost protein expression. Cells were cultured in suspension at 37 °C for 48 h and collected by centrifugation at 3,000g. All purification procedures were carried out at 4 °C unless specified otherwise. The cell pellet was re-suspended in buffer A (50 mM Tris pH 8.0, 200 mM NaCl) supplemented with a protease inhibitor cocktail (containing 1 mg ml−1 each of DNase, pepstatin, leupeptin, and aprotinin and 1 mM PMSF) and homogenized by sonication on ice. Mouse TRPML1 was extracted with 1% (w/v) n-dodecyl-β-d-maltopyranoside (DDM; Anatrace) supplemented with 0.02% (w/v) cholesteryl hemisuccinate (CHS; Sigma-Aldrich) by gentle agitation for 2 h. After extraction, the supernatant was collected after a 40-min centrifugation at 48,000g and incubated with Ni-NTA resin (Qiagen) using gentle agitation. After 2 h, the resin was collected on a disposable gravity column (Bio-Rad), washed with buffer B (buffer A + 0.1% DDM + 0.02% CHS) with 20 mM imidazole, and then buffer B. The washed resin was left on-column in buffer B and digested with thrombin (Roche) overnight. After thrombin digestion, the flow-through containing untagged MmTRPML1 was collected, concentrated, and purified by size-exclusion chromatography on a Superose 6 10/300 GL column (GE Heathcare) pre-equilibrated with buffer C (20 mM Tris pH 8.0, 150 mM NaCl, 0.1% DDM, 0.02% CHS). The protein peak was collected, concentrated to 40 μM and reconstituted into lipid nanodiscs following the published protocol24. In brief, MSP1, TRPML1, and lipid (POPC:POPG:POPE = 3:1:1) were mixed at a molar ratio of 4:1:10 respectively, and incubated on ice for 30 min. Detergents were removed by adding Bio-Beads SM2 (Bio-Rad) to a concentration of 100 mg ml−1 followed by gentle agitation. The Bio-Beads were replaced with fresh ones every 4 h, twice in total. After detergent removal, the sample was loaded onto a Superose 6 10/300 GL column pre-equilibrated with buffer D (20 mM Tris pH 8.0 and 150 mM NaCl) and the elution peak corresponding to the reconstituted TRPML1 was collected for electron cryo-microscopy analysis.
Electron microscopy data acquisition
The cryo-EM grids were prepared by applying 3 μl of mouse TRPML1 in nanodiscs (1.3 mg ml−1) to a glow-discharged Quantifoil R1.2/1.3 200-mesh copper holey carbon grid (Quantifoil, Micro Tools GmbH) and blotted for 4.0 s under 100% humidity at 4 °C before being plunged into liquid ethane using a Mark IV Vitrobot (FEI). Micrographs were acquired on a Titan Krios microscope (FEI) operated at 300 kV with a K2 Summit direct electron detector (Gatan), using a slit width of 20 eV on a GIFQuantum energy filter. EPU software (FEI) was used for automated data collection following standard FEI procedure. A calibrated magnification of 46,730× was used for imaging, yielding a pixel size of 1.07 Å on images, with the defocus ranging from −1.2 μm to −3 μm. Each micrograph was dose-fractionated to 30 frames under a dose rate of 4 electrons per pixel per second, with a total exposure time of 15 s, resulting in a total dose of about 50 e− Å−2.
Image processing
Motion correction was performed using the MotionCorr2 program32, and the contrast transfer function parameters of the micrographs were estimated using the GCTF program33. All other steps of image processing were performed using RELION34. Initially, ~1,000 particles were manually picked from a few micrographs. Class averages representing projections of TRPML1 in different orientations were selected from the 2D classification of the manually picked particles, and used as templates for automatic particle picking from the full dataset of 2,974 micrographs. The extracted particles were binned 3 times and subjected to 2 rounds of 2D classification, and a total of 519,463 particles were finally selected for 3D classification and 3D refinement. For the first round of 3D classification, the TRPV1 structure (EMDB code 5778) was used as the reference. One of the 3D classes showed good secondary structural features and is markedly different from TRPV1. It was then selected as the reference for the second round of 3D classification. One of the resulting 3D reconstructions from about 71,000 particles showed improved cryo-EM density and a clear four-fold symmetry with a resolution of ~4 Å after 3D refinement using the un-binned particles (C4 symmetry imposed) and particle polishing. However, part of the transmembrane domain showed poor density, indicative of local structural heterogeneity. Therefore, we performed a focused 3D classification with density subtraction35 in order to improve the density of the transmembrane domain. In this approach, the density corresponding to the soluble domain of the channel as well as the belt-like density from the nanodisc was subtracted from the original particles. The subsequent 3D classification on the modified particles was carried out by applying a mask around the transmembrane domain and having all of the orientations fixed at the value determined in the 3D refinement. This round of classification yielded 6 classes of particles, among which one major class (~50,000 particles) showed poor density in the transmembrane region and was discarded. After a new 3D refinement using the particles from the 5 classes (~20,000 total particles), the resolution of the map was improved from 4 Å to 3.59 Å. In addition, these 5 classes of particles can be partitioned into 2 groups with 9,000 and 11,000 particles, respectively. 3D refinement of each group yielded structures at 3.64 Å (closed I state) and 3.75 Å (closed II state) resolution, respectively, each showing distinct conformations at the S4–S5 linker. All resolutions were estimated by applying a soft mask around the protein density and the gold-standard Fourier shell correlation (FSC) = 0.143 criterion. ResMap36 was used to calculate the local resolution map.
Model building, refinement and validation
For the soluble domain, the structure of human TRPML1 I–II linker (PDB code: 5TJC) was docked into the cryo-EM map of mouse TRPML1 followed by manual adjustment and mutation of residues that differed between mouse and human TRPML1. For the transmembrane domain, de novo atomic model building was conducted in Coot37. Amino acid assignment was achieved based mainly on the clearly defined densities for bulky residues (Phe, Trp, Tyr, and Arg). Models were refined against summed maps using phenix.real_space_refine38 implemented in PHENIX39. The model was validated using previously described methods40 to avoid overfitting. The final structure models include residues 40–198, 215–285, and 296–527. About 40 residues at the amino terminus and 50 residues at the carboxy terminus are disordered and not modelled. The geometry statistics of the models were generated using MolProbity41. Pore radii were calculated using the HOLE program42. All the figures were prepared in PyMol43 or Chimera44.
Electrophysiology
By replacing the two di-leucine motifs (L15L and L577L) responsible for lysosomal targeting with alanine residues13,45, mouse TRPML1 (TRPML1–4A) can be overexpressed and trafficked to the plasma membrane in HEK293 cells, allowing for direct measurement of channel activity by patching the plasma membrane. In this setting, the extracellular side is equivalent to the luminal side of TRPML1 in endosomes or lysosomes. The constitutively active V432P mutant, equivalent to the gain-of-function mutation of the mouse TRPML3 in the varitint-waddler (Va) phenotype15,27,28,29, can be trafficked to the plasma membrane without replacing the di-leucine motifs, and therefore V432P and V432P/D472N mutants were both constructed on the background of the wild-type channel. The N-terminal truncation mutant was constructed on the background of TRPML1–4A. DNA fragments encoding mouse TRPML1 and its mutants were cloned into a pEGFP-C2 vector using XhoI and SacII restriction sites, as described2. N-terminal eGFP-tagged mouse TRPML1 or its mutant were expressed in HEK293 cells grown in a six-well tissue culture dish by transiently transfecting 2 μg of DNA plasmid using Lipofectamine 2000 (Thermo Fisher Scientific). Then, 24 to 48 h after transfection, cells were dissociated by trypsin treatment and kept in complete serum-containing medium; the cells were re-plated onto 35 mm tissue culture dishes and kept in a tissue culture incubator until recording. Patch clamp in the whole-cell or inside-out configuration was used to measure TRPML1 activity on the HEK plasma membrane. The standard bath solution for whole-cell current recording contained (in mM): 145 sodium methanesulfonate, 5 NaCl, 1 MgCl2, 1 CaCl2 or nominal Ca2+ free, 10 HEPES buffered with Tris, pH 7.4, or 10 MES buffered with Tris, pH 4.6; and the pipette solution contained (in mM): 140 caesium methanesulfonate, 5 NaCl, 5 MgCl2, 10 EGTA, 10 HEPES buffered with Tris, pH 7.4. For the measurement of luminal Ca2+-concentration-dependent blockage, the bath solution contained (in mM): 145 sodium methanesulfonate, 5 NaCl, 1 MgCl2, 10 HEPES buffered with Tris, pH 7.4, with 0.5 EGTA for 0 Ca2+, or 0.1, 1, 10 Ca2+ without EGTA. The bath solution for inside-out configuration contained (in mM): 140 potassium methanesulfonate, 5 NaCl, 2 MgCl2, 0.4 CaCl2, 1 EGTA, 10 HEPES buffered with Tris, pH 7.4; and the pipette solution contained (in mM): 145 sodium methanesulfonate, 5 NaCl, 1 MgCl2, 0.5 EGTA, 10 MES buffered with Tris, pH 4.6. The patch pipettes were pulled from Borosilicate glass (Harvard Apparatus) and heat polished to a resistance of 2–5 MΩ (2–3 MΩ for inside-out patch, and 3–5 MΩ for whole-cell current recoding). Data were acquired using an AxoPatch 200B amplifier (Molecular Devices) and a low-pass analogue filter set to 1 kHz. The current signal was sampled at a rate of 20 kHz using a Digidata 1322A digitizer (Molecular Devices) and further analysed with pClamp 9 software (Molecular Devices). After the patch pipette attached to the cell membrane, the giga seal (5–10 GΩ) was formed by gentle suction. The whole-cell configuration was formed by short zap or suction to rupture the patch. The holding potential was set to 0 mV. The whole-cell and inside-out macroscopic current recordings were obtained using voltage pulses ramped from −140 mV to +50 mV over a duration of 800 ms. The sample traces for single-channel recording or the I–V curves of macroscopic currents shown in each figure were obtained from recordings on the same patch. The lipid ligands used in this study are phosphatidylinositol-3,5-bisphosphate diC8 (PtdIns(3,5)P2 diC8, Echelon), phosphatidylinositol-4,5-bisphosphate diC8 (PtdIns(4,5)P2 diC8, Echelon) and mucolipin synthetic agonist 1 (ML-SA1, Sigma). All data points are mean ± s.e.m. (n ≥ 5).
Data availability
The cryo-EM density maps of the mouse TRPML1 in nanodiscs have been deposited in the Electron Microscopy Data Bank (EMDB) under accession numbers EMD-8883 (overall), EMD-8881 (closed I) and EMD-8882 (closed II). Atomic coordinates have been deposited in the Protein Data Bank (PDB) under accession numbers 5WPV (overall structure), 5WPQ (closed I state) and 5WPT (closed II state).
References
Cheng, X., Shen, D., Samie, M. & Xu, H. Mucolipins: intracellular TRPML1-3 channels. FEBS Lett. 584, 2013–2021 (2010)
Dong, X. P. et al. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 455, 992–996 (2008)
Bargal, R. et al. Identification of the gene causing mucolipidosis type IV. Nat. Genet. 26, 118–123 (2000)
Bassi, M. T. et al. Cloning of the gene encoding a novel integral membrane protein, mucolipidin—and identification of the two major founder mutations causing mucolipidosis type IV. Am. J. Hum. Genet. 67, 1110–1120 (2000)
Nilius, B., Owsianik, G., Voets, T. & Peters, J. A. Transient receptor potential cation channels in disease. Physiol. Rev. 87, 165–217 (2007)
Sun, M. et al. Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Hum. Mol. Genet. 9, 2471–2478 (2000)
Dong, X. P. et al. PI(3,5)P2 controls membrane trafficking by direct activation of mucolipin Ca2+ release channels in the endolysosome. Nat. Commun. 1, 38 (2010)
Feng, X. et al. Drosophila TRPML forms PI(3,5)P2-activated cation channels in both endolysosomes and plasma membrane. J. Biol. Chem. 289, 4262–4272 (2014)
Chandra, M. et al. A role for the Ca2+ channel TRPML1 in gastric acid secretion, based on analysis of knockout mice. Gastroenterology 140, 857–867.e1 (2011)
Cheng, X. et al. The intracellular Ca2+ channel MCOLN1 is required for sarcolemma repair to prevent muscular dystrophy. Nat. Med. 20, 1187–1192 (2014)
Puertollano, R. & Kiselyov, K. TRPMLs: in sickness and in health. Am. J. Physiol. Renal Physiol. 296, F1245–F1254 (2009)
Shen, D. et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat. Commun. 3, 731 (2012)
Vergarajauregui, S. & Puertollano, R. Two di-leucine motifs regulate trafficking of mucolipin-1 to lysosomes. Traffic 7, 337–353 (2006)
Zhang, X., Li, X. & Xu, H. Phosphoinositide isoforms determine compartment-specific ion channel activity. Proc. Natl Acad. Sci. USA 109, 11384–11389 (2012)
Xu, H., Delling, M., Li, L., Dong, X. & Clapham, D. E. Activating mutation in a mucolipin transient receptor potential channel leads to melanocyte loss in varitint-waddler mice. Proc. Natl Acad. Sci. USA 104, 18321–18326 (2007)
Venkatachalam, K., Wong, C. O. & Zhu, M. X. The role of TRPMLs in endolysosomal trafficking and function. Cell Calcium 58, 48–56 (2015)
Lee, J. H. et al. Presenilin 1 maintains lysosomal Ca2+ homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Reports 12, 1430–1444 (2015)
Kilpatrick, B. S., Yates, E., Grimm, C., Schapira, A. H. & Patel, S. Endo-lysosomal TRP mucolipin-1 channels trigger global ER Ca2+ release and Ca2+ influx. J. Cell Sci. 129, 3859–3867 (2016)
Cao, Q. et al. BK channels alleviate lysosomal storage diseases by providing positive feedback regulation of lysosomal Ca2+ release. Dev. Cell 33, 427–441 (2015)
Chen, C. C. et al. A small molecule restores function to TRPML1 mutant isoforms responsible for mucolipidosis type IV. Nat. Commun. 5, 4681 (2014)
Rosenthal, P. B. & Henderson, R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 333, 721–745 (2003)
Li, M. et al. Structural basis of dual Ca2+/pH regulation of the endolysosomal TRPML1 channel. Nat. Struct. Mol. Biol. 24, 205–213 (2017)
Liao, M., Cao, E., Julius, D. & Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504, 107–112 (2013)
Gao, Y., Cao, E., Julius, D. & Cheng, Y. TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 534, 347–351 (2016)
Shen, P. S. et al. The structure of the polycystic kidney disease channel PKD2 in lipid nanodiscs. Cell 167, 763–773.e11 (2016)
Nilius, B. & Owsianik, G. The transient receptor potential family of ion channels. Genome Biol. 12, 218 (2011)
Grimm, C. et al. A helix-breaking mutation in TRPML3 leads to constitutive activity underlying deafness in the varitint-waddler mouse. Proc. Natl Acad. Sci. USA 104, 19583–19588 (2007)
Kim, H. J. et al. Gain-of-function mutation in TRPML3 causes the mouse varitint-waddler phenotype. J. Biol. Chem. 282, 36138–36142 (2007)
Nagata, K. et al. The varitint-waddler (Va) deafness mutation in TRPML3 generates constitutive, inward rectifying currents and causes cell degeneration. Proc. Natl Acad. Sci. USA 105, 353–358 (2008)
Dong, X. P. et al. Activating mutations of the TRPML1 channel revealed by proline-scanning mutagenesis. J. Biol. Chem. 284, 32040–32052 (2009)
Morales-Perez, C. L., Noviello, C. M. & Hibbs, R. E. Manipulation of subunit stoichiometry in heteromeric membrane proteins. Structure 24, 797–805 (2016)
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017)
Zhang, K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016)
Scheres, S. H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012)
Bai, X. C., Rajendra, E., Yang, G., Shi, Y. & Scheres, S. H. Sampling the conformational space of the catalytic subunit of human γ-secretase. eLife 4, e11182 (2015)
Kucukelbir, A., Sigworth, F. J. & Tagare, H. D. Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014)
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010)
Afonine, P. V., Headd, J. J., Terwilliger, T. C. & Adams, P. D. New tool: phenix.real_space_refine. Computational Crystallography Newsletter 4, 43–44 (2013)
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010)
Amunts, A. et al. Structure of the yeast mitochondrial large ribosomal subunit. Science 343, 1485–1489 (2014)
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 (2010)
Smart, O. S., Neduvelil, J. G., Wang, X., Wallace, B. A. & Sansom, M. S. HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 14, 354–360, 376 (1996)
The PyMOL Molecular Graphics System v.1.8 (Schrödinger, LLC, 2015)
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004)
Grimm, C. et al. Small molecule activators of TRPML3. Chem. Biol. 17, 135–148 (2010)
Acknowledgements
We thank N. Nguyen for manuscript preparation. Single-particle cryo-EM data were collected at the University of Texas Southwestern (UTSW) Medical Center Cryo-Electron Microscopy Facility. We thank D. Nicastro and Z. Chen for support in facility access and data acquisition. Negatively stained sample screening was performed at the UTSW Electron Microscopy core. This work was supported in part by the Howard Hughes Medical Institute (Y.J.) and by grants from the National Institutes of Health (GM079179 to Y.J.; NS062792 and AR060837 to H.X.) and the Welch Foundation (grant I-1578 to Y.J.). X.B. is supported by the Cancer Prevention and Research Initiative of Texas and Virginia Murchison Linthicum Scholar in Medical Research fund.
Author information
Authors and Affiliations
Contributions
Q.C. and J.S. prepared the samples; Q.C., J.S., J.G. and X.B. performed data acquisition, image processing and structure determination; W.Z. performed electrophysiology; H.X. provided DNA materials and constructive advice; all authors participated in research design, data analysis, and manuscript preparation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information Nature thanks S. Hansen, C. Ulens and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Figure 1 Ligand activation of TRPML1 overexpressed in HEK293 cells.
a, Macroscopic currents of plasma membrane-localized TRPML1–4A in an inside-out patch in the presence and absence of ligands in bath solution (cytosolic). TRPML1 can be activated by PtdIns(3,5)P2 and mucolipin synthetic agonist 1 (ML-SA1), yielding inwardly rectified cation currents. PtdIns(3,5)P2 and ML-SA1 activations are synergistic, indicating non-overlapping activation sites between the two ligands. b, Macroscopic currents of TRPML1–4A in the whole-cell configuration. The pipette (cytosolic) solution contained 50 μM PtdIns(3,5)P2 (black trace). Addition of 10 μM ML-SA1 into the bath solution (extracellular/luminal side) yielded a much larger current, suggesting that ML-SA1 can also activate the channel from the luminal side. c, Sample traces of single-channel currents recorded at −120 mV in an inside-out excised patch showing PtdIns(3,5)P2 and ML-SA1 activation. The patch contained multiple channels (N ≥ 5). N, total number of channels; Po, single-channel open probability; NPo, total open probability (the number of channels multiplied by single-channel open probability); C, closed state; O, open state, with the number indicating open events from multiple channels. d, Sample traces of single-channel currents recorded at −120 mV in an inside-out excised patch showing PtdIns(4,5)P2 (in bath solution) inhibition of TRPML1. e, Single-channel recordings of N-terminal truncation mutation of TRPML1–4A in an inside-out patch. Deletion of the poly-basic domain abolishes PtdIns(3,5)P2 activation, confirming its participation in PtdInsP2 binding; the ML-SA1 activation remains intact in this mutant, confirming distinct activation sites between PtdInsP2 and the small-molecule agonist.
Extended Data Figure 2 Structure determination of mouse TRPML1 in nanodiscs.
a, Representative micrograph of TRPML1 in a nanodisc. b, Two-dimensional class averages. c, Euler angle distribution of particles used in the final three-dimensional reconstruction, with the heights of the cylinders corresponding to the number of particles. d, Gold-standard FSC curves of the final 3D reconstructions. e, Final density maps coloured by local resolution.
Extended Data Figure 4 Data collection, structure refinement and model validation.
a, Data collection and model refinement statistics. b, FSC curves for cross-validation between the maps and the models. Curves for model versus summed map in blue (full), for model versus half map in green (work), and for model versus half map not used for refinement in black (free).
Extended Data Figure 5 Sample electron microscopy density maps (blue mesh) for various parts of the channel.
The maps are low-pass filtered to 3.59 Å and sharpened with a temperature factor of −120 Å2.
Extended Data Figure 6 Sequence alignment of TRPML channels.
Secondary structure assignments are based on the mouse TRPML1 structure. Purple crosses mark the basic residues important for PtdInsP2 binding and the blue dot marks the location of V432P mutation in TRPML1.
Extended Data Figure 7 Luminal Ca2+ and pH modulation of TRPML1.
a, Sample traces (I–V curves) of whole-cell currents from the constitutively active TRPML1(V432P) mutant recorded with various luminal (bath solution) Ca2+ concentrations [Ca2+] and pH values. Inset shows normalized channel currents at −120 mV. Data are mean ± s.e.m. of five measurements. 0 mM [Ca2+] here refers to nominally Ca2+-free medium in the recording. b, Sample traces of whole-cell currents of the TRPML1(V432P/D472N) mutant recorded at various luminal (bath solution) [Ca2+] and pH values. Inset shows normalized channel currents at −120 mV. Data are mean ± s.e.m. of five measurements. c, Luminal [Ca2+]-dependent blockage of inward currents in TRPML1(V432P) and TRPML1(V432P/D472N) measured at −120 mV and pH 7.4. In summary, 1 mM Ca2+—close to the lysosomal Ca2+ concentration—can markedly reduce the channel current; lowering the pH can partially alleviate Ca2+ blockage, probably by protonating the Ca2+-binding acidic residues; in the absence of Ca2+, however, lower pH by itself has an inhibitory effect on channel conductance. Neutralizing Asp472 with Asn diminishes the luminal Ca2+ blocking.
Extended Data Figure 8 Structural comparison between the S1–S2 linker domains of TRPML1 and PKD2, a member of the TRPP family.
a, Side view of TRPML1 luminal linker domain atop the channel with open side windows. The front subunit is highlighted in blue. b, Side view of PKD2 polycystin domain. The polycystin domain has an extra hairpin loop (red) that clogs the side window, making the central hole the only extracellular passage to the filter. There is an extra helix–turn motif (magenta) between S3 and S4 in PKD2 that extends upright and provides extra contact between the polycystin domain and the transmembrane domain. c, The luminal pore loop (magenta) between α1 and α2 points downwards in TRPML1, generating a funnel-shaped central hole with a constriction of 12 Å. d, The luminal pore loop (red) in PKD2 points upwards and generates a central hole with an inverted funnel shape. The front and rear subunits are removed in panels c and d for clarity.
Supplementary information
Rights and permissions
About this article

Cite this article
Chen, Q., She, J., Zeng, W. et al. Structure of mammalian endolysosomal TRPML1 channel in nanodiscs. Nature 550, 415–418 (2017). https://doi.org/10.1038/nature24035
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature24035
- Springer Nature Limited
This article is cited by
-
Unexpected inhibition of the lipid kinase PIKfyve reveals an epistatic role for p38 MAPKs in endolysosomal fission and volume control
Cell Death & Disease (2024)
-
Nanoscale one-dimensional close packing of interfacial alkali ions driven by water-mediated attraction
Nature Nanotechnology (2024)
-
Regulating ion affinity and dehydration of metal-organic framework sub-nanochannels for high-precision ion separation
Nature Communications (2024)
-
Mutation of TRPML1 Channel and Pathogenesis of Neurodegeneration in Haimeria
Molecular Neurobiology (2024)
-
TRP (transient receptor potential) ion channel family: structures, biological functions and therapeutic interventions for diseases
Signal Transduction and Targeted Therapy (2023)