

Abstract
Increasing grain yield is a long-term goal in crop breeding to meet the demand for global food security. Heterosis, when a hybrid shows higher performance for a trait than both parents, offers an important strategy for crop breeding. To examine the genetic basis of heterosis for yield in rice, here we generate, sequence and record the phenotypes of 10,074 F2 lines from 17 representative hybrid rice crosses. We classify modern hybrid rice varieties into three groups, representing different hybrid breeding systems. Although we do not find any heterosis-associated loci shared across all lines, within each group, a small number of genomic loci from female parents explain a large proportion of the yield advantage of hybrids over their male parents. For some of these loci, we find support for partial dominance of heterozygous locus for yield-related traits and better-parent heterosis for overall performance when all of the grain-yield traits are considered together. These results inform on the genomic architecture of heterosis and rice hybrid breeding.
Similar content being viewed by others


Main
Over the previous decades, rice-breeding practices have greatly increased grain yield of rice cultivars, largely related to the adoption of semi-dwarf alleles and efficient use of hybrid vigour (heterosis)1,2. In hybrid rice varieties (Oryza sativa), the heterozygous first filial (F1) generation display a yield advantage of 10–20% over their inbred parental lines2,3,4, making heterosis breeding a powerful way to meet global food-security demands. The genomic basis of heterosis has been extensively studied in a number of crops, and explained by several classical models. Previous research of multiple crops has provided support for three non-mutually exclusive hypotheses of dominance, single-locus overdominance and pseudo-overdominance models for genomic loci contributing to heterosis5,6,7,8,9,10,11,12,13.
The genetic basis of heterosis for rice grain yield has remained largely uncharacterized. The second filial (F2) populations generated from the elite hybrids (F1) can provide useful information for heterosis analysis. Assuming a single polymorphic locus contributes to heterosis, it is expected that three genotypes (homozygous male-parent genotype MM, heterozygous genotype MF and homozygous female-parent genotype FF) will be present in F2 with a proportion of 1:2:1 at the local genomic region, allowing for an estimation of heterotic effects. Here we investigated F2 populations from 17 representative hybrid rice crosses, in order to identify loci contributing to yield traits, analyse their heterotic effects and evaluate their contributions to heterotic advantage.
Sequencing, genotyping and genetic mapping
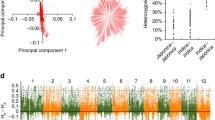
The strategy of this study is briefly described in Extended Data Fig. 1. We have previously constructed a genome map for 1,495 elite hybrid rice varieties13. According to both genomic information and phenotypic performance of the hybrid rice varieties, 17 representative hybrid combinations were selected and the corresponding F1 hybrids were mostly important varieties of super-hybrid rice that were widely cultivated during recent years (Extended Data Fig. 2). We generated 17 sets of large recombinant populations. The resulting 10,074 F2 lines included 9 populations from O. sativa subspecies (ssp.) indica-indica crosses of a three-line system (cytoplasmic male sterility (CMS) for female parental lines, type A, n = 3,947), 6 populations from indica-indica crosses of a two-line system (environmentally sensitive male sterility, type B, n = 3,957), and 2 populations from O. sativa ssp. indica–O. sativa ssp. japonica crosses (type C, n = 2,170). We sequenced the genomes of the 10,074 F2 lines and constructed a dense genotype map with a total of 347,803 recombination events using a high-throughput sequence-based genotyping method14 (Fig. 1a and Extended Data Fig. 3). In the genotype map of each F2 population, the proportion of three genotypes (MM, MF and FF) agreed with expectation of 1:2:1 across the vast majority of the rice genome (Extended Data Fig. 3a).

a, Plots of the first two principal components of 10,074 lines using whole-genome single-nucleotide polymorphisms (SNPs). b, The Manhattan plots of a genome-wide association study (GWAS) for heading date in 10,074 lines. Negative log10 P values from linear mixed model (y axis) are plotted against SNP positions (x axis) on each of 12 rice chromosomes. The genome-wide significant P-value threshold (10−6) is indicated by a horizontal dashed line. The loci with well-characterized genes are indicated near the association peaks. c, Manhattan plot for GWAS of plant height in 10,074 lines. The arrow indicates the newly identified loci for plant height.
The yield traits were analysed using a composite interval-mapping method for each population separately to identify large-effect quantitative trait loci (QTLs) with resolution mostly within 300 kb. The alleles segregating in multiple populations could also be detected through genome-wide associations that combined information across the 17 populations (Fig. 1b, c). The QTLs included 23 candidate genes previously implicated in grain yield, and additional new QTLs for grain yield identified in this work. We were not able to resolve these QTLs to single-gene level, and additional studies will be needed to further map these and identify candidate genes.
For the 15 populations from indica–indica crosses, we identified 474 QTLs for 7 yield traits (Fig. 2a). We then examined the genotyping data for the local genomic region of each of these QTLs to measure the effect of three genotypes (MM, MF and FF) (Methods). The QTLs for grain-yield-related traits showed enrichment with positive dominance effects (the effect of heterozygous genotype was similar to that of homozygous advantageous alleles), especially for plant height and seed-setting rate, providing the basis to maintain or even increase the grain yield, in the case of heterozygous genomes (Fig. 2a).

a, Plots of the dominance effect for 474 QTLs (likelihood of odds (LOD) > 3.5) in the 15 populations underlying seven yield-related traits. b, The genomic locations of seven loci with strong phenotypic effects for grain yield per plant (LOD > 3.5) identified in the population from a cross between 93–11 and Y58S. The alleles with better yield performance that were contributed from the male parent 93–11 and the female parent Y58S are coloured by blue and red, respectively. c, Plots of the dominance effect and the allele effect for the seven QTLs. d, For all QTLs underlying grain yield per plant (LOD > 3.5) in the 15 populations from indica–indica crosses, the proportions of the advantageous alleles contributors (from male parents and female parents) are indicated.
Furthermore, we found that, for most of the grain-yield QTLs, the alleles with higher yield performance were contributed from male parents (parental lines that restored fertility in hybrid rice) (Fig. 2b–d). As the heterozygous state of the QTLs mostly represented positive partial dominance, assuming no interaction between loci, the combined total of the effects of an advantageous allele from only the male parents at most insured that the F1 lines had a yield performance close to their male parents, but that cannot surpass the phenotypic value of that parent. Nevertheless, most hybrids showed a yield advantage of >10% over their male parents, suggesting that the allele from female parents also contributes to heterosis. Hence, we addressed the genetic contributors that led to hybrids outperforming their male parents for each type of hybrid system.
Type-A system: hd3a and tac1
The hybrid rice system of type A, developed in the 1970s, is the most common system, and includes over 75% of known hybrid varieties. For type-A-system hybrid lines, we identified heterosis QTLs to include candidate genes hd3a and tac1 through a combination of genomic mapping and allelic mutations in the coding regions (see Methods). For both candidate genes, the alleles in female parents influenced the yield performance of the hybrid. Hd3a is a rice orthologue of the Arabidopsis thaliana FT gene15. For its orthologue gene in tomato, yield overdominance from SFT heterozygosity has previously been observed5. The allele hd3a that was associated with heterosis had a two-base substitution, which is also associated with delay of flowering in long-day conditions (Fig. 3a). The estimation of allelic effects in type-A populations demonstrated that Hd3a also had large effect on grain yield. We further evaluated the effect of the three genotypes at the Hd3a locus. Compared to the genotype in male parents (Hd3a/Hd3a), the heterozygous state (Hd3a/hd3a) showed an advantage of 7.4% in seed-setting rate (varying from 2.2% to 10.9% in different hybrids) and 9.9% in grain yield per plant (varying from 1.4% to 17.9%) (Fig. 3b). We then examined the effect of the male parents with the hd3a/hd3a genotype on hybrid performance. We noticed that, although the hd3a/hd3a genotype showed an increased grain yield, it also showed delayed rice flowering (extending the growth stage by up to 20%), which would be impractical for agricultural production in most rice-planting areas (Extended Data Fig. 4). Fortunately, the heterozygous state (Hd3a/hd3a) showed an appropriate effect for rice heading: the heterozygous genotype only slightly delayed flowering time compared with the wild-type Hd3a/Hd3a. Hence, when considering both yield and timing, the performance of Hd3a/hd3a was better than either of the two homozygous genotypes.

a, Gene structure of Hd3a and hd3a alleles. b, Plots of the advantage of the heterozygous state (Hd3a/hd3a) over Hd3a/Hd3a for grain yield per plant and yield components in nine type-A populations of 3,947 F2 lines. c, Gene structure of TAC1 and tac1 alleles. d, The performances of tiller angle for three genotypes of the TAC1 gene in 438 F2 lines (mean ± s.e.). The three genotypes are colour-coded in d and e. e, Computational modelling of the planting number per unit area (mu, equivalent to one-fifteenth of a hectare) for TAC1 gene. f, The allele frequencies of hd3a and tac1 in 1,063 pairs of type-A parents and 254 pairs of type-B parents.
The effects of hd3a on yield were weaker in early-season hybrids. This could be because the florigen gene hd3a is under the regulation of multiple floral repressors. As with homozygous early-flowering alleles of multiple genes (including Hd2, Ghd7 and Ghd8) in early-season hybrids, the hd3a allele delayed flowering only 4 days compared with the Hd3a allele, in marked contrast with the ~20 days delay seen in the background of middle-season hybrids. Correspondingly, hd3a did not show any significant improvement in grain yield on this genetic background.
TAC1 has been shown to regulate plant architecture in rice16. The tac1 allele, which has a mutation in the splicing site of the fourth intron, greatly improved plant architecture and allowed more efficient light capture (Fig. 3c). The heterozygous genotype (TAC1/tac1) could produce significantly more seeds (2.6% in grain number per panicle and 1.6% in thousand-seed weight) than the genotype of the male parental genotype (TAC1/TAC1). Because the tac1 allele reduced tiller number (by ~2.5%), grain yield per plant for the TAC1/tac1 was not significantly increased compared to TAC1/TAC1. However, the improved plant architecture seen in the TAC1/tac1 genotype results in the increase of ‘grain yield per unit area’. Compared with the genotype of male parents (TAC1/TAC1), the genotype of hybrids (TAC1/tac1) displayed a change of approximately 2°–3° in tiller angle, allowing an ~15% increase in planting density (Fig. 3d, e). We estimated that the heterozygous states at the two loci (hd3a and tac1) collectively resulted in significantly more seeds (overall 13.3% increase per panicle, P < 10−9) than the genotype of male parents, explaining ~73.5% of heterosis advantage (the percentage of differences between the hybrids and their male parents) in the type-A system.
We further investigated the allelic distribution for these candidate genes in the parental lines of 1,063 hybrid rice varieties of type A. Hd3a and TAC1 were mostly distributed in restorer lines (male parents of hybrids), whereas hd3a and tac1 were in CMS lines (female parents of hybrids) (Fig. 3f). In the 1,063 hybrids of type A, nearly all restorer lines (98.5%) had the ancestral type of Hd3a, whereas most CMS lines (76.7%) had the hd3a allele. Genome-wide measurement of the extent of divergence between restorer and CMS lines in type-A hybrids using the fixation index F-statistics (FST) enabled the identification of highly differentiated loci that included Hd3a and TAC1 (FST = 0.57 and FST = 0.33, respectively, higher than the whole-genome average of FST = 0.11). Other highly differentiated loci included two fertility restorer genes Rf3 (FST = 0.82) and Rf4 (FST = 0.40)17,18, and two yield-related genes Ghd7 (FST = 0.34) and LAX1 (FST = 0.63)19,20. In contrast to hd3a and tac1, the beneficial alleles of the other differentiated loci (for example, Ghd7 and LAX1) were contributed from the restorer lines. We also scanned the genomes of 254 type-B hybrids, in which we found that the heterosis-associated alleles of hd3a and tac1 were present in a small proportion (8% and 10%, respectively).
Notably, the hd3a allele in CMS lines may also have contributed to some reduced performance (Extended Data Fig. 5). Most conventional indica CMS lines carried the low grain-quality alleles of waxy and ALK21,22 and the susceptible allele of the Pi2/Pi9 locus for disease23. hd3a may also affect the introduction of wide-compatibility alleles of S5 for indica–japonica crosses24. Molecular-marker-assisted selection may be useful for improving the CMS lines.
LAX1 and Ghd8 in type-B system
The two-line system (type B), which included thermo-sensitive genic male sterility (TGMS) regulated by the gene tms5 and photoperiod-sensitive genic male sterility (PGMS) regulated by pms3, has greatly facilitated genetic improvements of maternal lines25,26. In the populations from TGMS hybrids, we identified two QTLs that included candidate genes Ghd8 and Tms5 with large effects on grain yield25,27. The alleles of ghd8 and tms5 in female parents corresponded to low production or sterility in the hybrid. Interestingly, the heterozygous state at both loci sometimes showed a marginal overdominance effect for plant height, seed-setting rate and grain yield per plant (Extended Data Fig. 6a, b). Although the two QTLs that included the candidate genes Ghd8 and Tms5 did sometimes show heterotic effects, we were uncertain whether single-gene overdominance or pseudo-overdominance (there could be another nearby gene contributing) was responsible for the heterosis. In one type-A population, we noticed that a QTL underlying several yield components was co-located with the local region around Tms5 (Extended Data Fig. 7a–c), whereas Tms5 itself was not the causal gene (both parents of the cross carried the wild-type allele Tms5, Extended Data Fig. 7d, e). We inferred that the overdominance phenomena observed at the Tms5 locus, as well as the Ghd8 locus, were probably due to pseudo-overdominance conferred by some tightly linked genes with beneficial alleles in female parents.
We identified several beneficial alleles of large effects contributed by female parents in the type-B system, including LAX1 for grain weight20, OsMADS51 for heading date28 and GW3p6 (a newly identified QTL) for grain weight, all with positive partial dominance. These candidate genes were commonly used genetic contributors that led to heterosis performance in type-B hybrids. Moreover, some type-B parental lines have special beneficial alleles, because the two-line hybrid system could provide broad genetic resources for the exploitation of heterosis. The variety ‘Liangyoupeijiu’ (LYP9) generated from a cross between PA64S with a minor background of japonica (for example, containing the japonica-type NAL1 allele)29 and 93–11 was an important PGMS hybrid variety. We found that the alleles contributed by PA64S at a number of loci could collectively explain ~89.2% of the heterosis advantage of LYP9 over 93–11 (Extended Data Fig. 6c and Extended Data Fig. 8). Similarly, in other type-B hybrids, the heterozygous status of only a small number of loci resulted in increased grain yield over the homozygous genotypes of male parents (Extended Data Fig. 6d and Extended Data Fig. 9).
Multiple beneficial alleles of type C
Owing to the identification and application in breeding of wide-compatibility alleles24, the development of intersubspecific hybrids has become feasible in the past few years. Although indica generally has better yield performance in Southern China, Southeast Asia and South Asia, japonica carries many beneficial alleles that are uncommon in indica gene pools (for example, nal1 and tac1)16,29. In each subspecies, there were several large-effect alleles (for example, Sd-1, Hd1, GW6a, OsGI, DEP1)30,31,32,33,34. Positive partial dominance and overdominance effects have served as the major causes of heterosis that make indica–japonica F1 hybrids produce much higher yields than their inbred parental lines (Fig. 4a). Two linked loci on the same chromosome in repulsion phase in two parental lines also tended to result in pseudo-overdominance (for example, GW6a in indica and Hd1 in japonica on chromosome 6; Sd1 in japonica and OsGI/LAX1 in indica on chromosome 1), similar to an example of heterosis for plant height in Sorghum6. In contrast to intrasubspecific hybrids, in which the female parents supplemented only a few key alleles, the female parents in the intersubspecific hybrids often contained many beneficial alleles.

a, Plots of the index of middle-parent heterosis for each QTL underlying grain yield per plant (LOD > 3.5) against the physical positions. The alleles with better performance that were contributed from japonica and indica are coloured by blue and red, respectively. The loci that include previously reported candidate genes are highlighted in bold, and the newly identified QTLs were uniformly named according to the underlying yield component trait, chromosome and physical location. The asterisk indicates the genes with strong overdominance or pseudo-overdominance. b, The linkage mapping of grain number per panicle. LOD values are plotted against the physical positions, and the threshold (3.5) is indicated by a horizontal dashed line. c, The performances of grain yield per plant for three genotypes of the IPA1 gene in 1,037 F2 lines (**P < 0.001, two-tailed t-test).
Plant architecture is a primary factor underlying ‘unit area yield’ in crops. As well as TAC1/tac1, we identified another candidate gene OsSPL14 (also named IPA1)35,36 for regulating plant architecture in an indica–japonica hybrid (Fig. 4b). The rare allele ipa1 strongly enhanced grain number per panicle, whereas the wild-type allele IPA1 performed better in seed-setting rate and panicle number. Heterozygous OsSPL14 (IPA1/ipa1) showed partial dominance for these yield traits (Extended Data Fig. 10). For yield per plant, the heterozygous state of OsSPL14 showed strong overdominance effect, producing higher grain yield than either IPA1/IPA1 or ipa1/ipa1 (P < 0.001, Fig. 4c), which could explain 48.1% of heterosis advantage in this hybrid cross. Comparing the sequence of ipa1 with that of IPA1, we found several variants in the promoter region but no differences in the coding regions, suggesting that the strong overdominance in yield was related to gene expression in the heterozygous state of OsSPL14. These candidate genes for plant architecture (including OsSPL14 and TAC1) may be relevant for understanding balance between early growth and late growth rates and between yield per plant and planting density.
Conclusions
In summary, integrated genomic analyses in large samples have provided insights into the genetic basis of heterosis in rice. Our studies suggest that there are a few loci from female parents that contribute to heterosis within each subgroup, but that these are not universally shared across our representative elite rice lines. The heterozygous state of these loci mostly acted through the way of positive partial dominance, and some loci had to be heterozygous in breeding owing to pseudo-overdominance or ‘overdominance’ in overall performance. The heterotic effects of these loci in rice suggests that additive, partial dominance and overdominance may have been observed owing to allelic dosage effects, consistent with the findings in maize37,38. Further studies are needed to resolve candidate genes contributing to heterosis for yield in rice, as well as their mechanisms.
These insights into the genomic architecture of grain yield in rice will be useful for further work to improve parental lines and create superior hybrids efficiently by optimizing the designs of breeding. Heterosis in maize (and also some other crops) from crosses between two genetically divergent groups may have similar genetic patterns with the intersubspecific hybrids in rice, awaiting further investigations through large-scale genomic approaches. We believe a better understanding of heterosis in crops will help in development of new strategies for meeting global food security.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment.
Sampling and sequencing
From the large collection of hybrid rice (n = 1,495) that had been sequenced and phenotyped, seventeen hybrid combinations were carefully selected to construct F2 populations, generating a total of 10,074 individual lines. The 17 combinations contained all major types of hybrid varieties in rice with a relatively high genetic diversity. The genomic DNA was extracted from the fresh leaf tissue of each F2 plant using the DNeasy Plant Mini Kit (Qiagen). The sequencing library was constructed with an insert size of 200–400 bp using TruePrep Tagment Enzyme (Tn5 transposase, Vazyme), which was then indexed with dual index primers (24 barcodes in one end and 16 barcodes in the other end for the paired-end reads) and amplified using TruePrep Amplify Enzyme39. The indexed DNA samples of 384 F2 lines with different barcodes were then mixed together in an equal molar concentration, followed with the purification by silica membrane column and the size-selection by agarose gel electrophoresis. Each mixture was loaded into one lane of the Illumina HiSeq2500 system. Overall, 10,074 individual lines was sequenced, generating 100-bp paired-end reads of ~1 terabase. Each F2 line had approximately 0.2× genome coverage. The paired-end reads of each F2 line were aligned against the reference genome sequence (IRGSP releases build 4.0 pseudomolecules of rice) individually using the package SMALT (version 0.5.7) with the parameters of ‘-i 700 -j 50 -m 50’. Only the reads mapped uniquely to the reference genome sequence were retained for SNP calling.
Genotype calling
Genotype calling was carried out in any chromosomal region covered by a given number of SNPs. The window size (the number of n consecutive SNPs in a window) was allowed to vary according to the marker density. For most F2 lines from indica–indica crosses, fifteen consecutive SNPs were used as a single window. We applied a sliding window approach to evaluate the raw calls of multiple SNPs and performed the genotype calling using a Bayesian-based algorithm14. The window slid along a chromosome at a step size of one SNP, until all the genotypes were called for the entire chromosome. The recombination map was constructed for each chromosome in 10,074 F2 lines. Detailed procedures in genotyping, recombination breakpoint determination and map construction were implemented in the pipeline SEG-Map with default parameters14.
Phenotyping and linkage analysis
The F2 seeds from the 17 populations were germinated and planted in the experimental fields in Hangzhou, China (at N 30.32, E 120.12) in summer of 2014 from May to October. The phenotyping for this work involved a wide range of grain-yield traits in the field. All the F2 lines were grown in the consecutive farmland with well-distributed soil status in uniform condition with low use of fertilizers. Some hybrids and the restorer lines were also planted to estimate the yield advantage. Heading date was recorded daily as the number of days from sowing to the observation of first inflorescences that emerged above the flag leaf sheath. Plant height was measured from the soil surface to the apex of the tallest panicle. Tiller angle was measured between tiller and the ground level when panicles fully emerged. The yield-related traits, including panicle number, grain number per panicle, seed-setting rate and grain weight per 1,000 grains, were measured in the laboratory following harvest. Grain weight was initially obtained by weighing ~600 fully filled grains, which was then converted to a 1,000-grain weight value. The panicle number and the spikelets produced per panicle were counted manually. In each F2 population, QTL analysis of the yield traits was performed by the composite interval mapping (CIM) method implemented in the software Windows QTL Cartogapher (version 2.5)40 with the cross type of SF2, a window size of 10-cM and a step size of 2 cM. The LOD values were determined on the basis of likelihood ratio tests under a hypothesis allowing both additive and dominance effects. QTLs with LOD value higher than 3.5 were called, of which the physical location was retrieved from the peak location. The newly identified QTLs were uniformly named according to the major trait, chromosome and physical location (for example, GW3p6 represented a QTL for grain weight in the short arm of chromosome 3 within the genomic loci of 6–7 Mb).
GWAS and population genetics analysis
Integrating the ultra-dense genotype map of each F2 population and the haplotype map of their parental lines, the genotype calls were assigned at 1.7 million common SNPs for all the 10,074 F2 line. Principal component analysis of the SNPs was performed using the software EIGENSOFT (version 5.0.1)41, and the first two principal components were used in the analysis of the genetic structure of the 17 populations. Using the combined genotypic information, association analysis was conducted by the mixed model to correct for pedigree relatedness using EMMAX software package42. After filtering the SNPs with high missing data rate (20%) and low minor-allele frequency (2%), a total of 1,482,139 SNPs were used in final associations. The matrix of simple matching coefficients was used to build up the kinship matrix (10,074 × 10,074) for the correction. We defined a whole-genome significance threshold (1 × 10−6) according to permutation tests.
Previously we constructed a genome map for 1,495 elite hybrid rice varieties and their inbred parental lines through whole-genome sequencing13. The whole-genome data of the parental lines of 1,063 hybrid rice varieties of type A and 254 of type B were re-analysed for scanning highly differentiated loci between the group of restorer lines and that of CMS lines. The differentiation statistics (FST) were computed in each 100-kb window across 12 rice chromosomes. For 1,495 elite hybrid rice varieties, we carried out GWAS for heading date, grain number, colouration, grain quality and disease-resistance traits in the previous publication13. The lead SNPs for Hd3a, ALK, Waxy, OsC1, Pi2/Pi9 and TAC1 from previous GWAS13,43 were used to calculate their allele frequencies in the 1,063 pairs of restorer lines and CMS lines in type A.
Allelic variation detection for identified QTLs
In order to identify allelic variation (especially SNPs and indels in coding regions) for the identified QTLs in the large populations, all the Illumina reads were mapped to the reference rice genome with BWA (version 0.7.1)44 using default parameters to generate the BAM files. For the mapped reads, PCR duplicates were removed by the ‘MarkDuplicates’ module in Picard package (version 1.119). To reduce miscalls caused by misalignment in highly polymorphic regions, we realigned the reads at the target loci with the ‘IndelRealigner’ function in the GenomeAnalysisTK (version 3.4.0)45. Taking all of the realigned BAM results together, the sequence variants of high quality were called using ‘UnifiedGenotyper’ in the GenomeAnalysisTK and filtered by the base depths (≥5 at least). Effects of the sequence variants were evaluated according to the gene models of Nipponbare in the RAP-DB (release 2) across the rice genome, with manual checks for the coding structures of the well-characterized genes in rice. The candidate genes from GWAS and linkage analysis were identified based on both the genetic mapping information and the allelic mutations observed from the sequence reads. Using the comprehensive dataset, the allelic variation and their combinations for the key genes (including Hd3a, TAC1, Ehd1, LAX1, Tms5 and OsSPL14) were examined across the 17 populations.
Allele effect estimation
After the QTLs were identified, allele effect was first assessed through a comparison of both homozygous genotypes. The average phenotypic measurements of heterozygous genotypes and homozygous genotypes were further calculated for the estimations of the index of dominance-effect/additive-effect (d/a) and the index of middle parent heterosis for each QTL. Two-tailed t-tests were used to evaluate the significance of the phenotypic contribution of the heterozygous genotypes versus the genotypes of male parents at the key loci, including Hd3a and TAC1. The measurements with a seed-setting rate <20% and grain yield per plant <5 g were excluded from the comparisons to avoid the influence of sterile traits. For the TAC1 gene underlying tiller angle, the planting number per unit area was modelled using the formula n = C/(cosα)2, where n is the plant number per mu (one fifteenth of a hectare), C was an empirical constant (given as 2,500 here for typical inbred indica lines) and α was the tiller angle measured between the tiller and the ground level.
Genetic modelling for heading date
For modelling the genetic architecture of heading date, multiple linear regressions were performed to examine the effects of diverse allelic combinations in four major loci (Hd3a, Ghd7, Hd2 and Ehd1) using the ‘proc reg’ procedure in SAS. The late-flowering alleles of Hd3a and Ehd1 came from female parents and those of Ghd7 and Hd2 were from male parents. Before fitting the model, each marker was recorded. The value 0 was used for the allele from male parents, the value 1 was used for that from female parents and the value for heterozygous genotype depended on the dominance effect of each gene. In the modelling procedure, we only used the genotypic data at the four loci and the phenotypic data for heading date in one type-A population (Guanghui998 × Tianfeng), which resulted in the estimates for recoding, weighting and the intercept. The estimates from this training population were used for the prediction of heading date in the remaining eight populations. The value R was calculated from the correlation between the predicted values from modelling and the observed phenotypic data for heading date in eight populations.
Accession codes
Primary accessions
European Nucleotide Archive
Data deposits
DNA sequencing data are deposited in the European Nucleotide Archive under the accession number PRJEB13735.
References
Wang, Y., Xue, Y. & Li, J. Towards molecular breeding and improvement of rice in China. Trends Plant Sci. 10, 610–614 (2005)
Cheng, S. H., Zhuang, J. Y., Fan, Y. Y., Du, J. H. & Cao, L. Y. Progress in research and development on hybrid rice: a super-domesticate in China. Ann. Bot. 100, 959–966 (2007)
Li, S., Yang, D. & Zhu, Y. Characterization and use of male sterility in hybrid rice breeding. J. Integr. Plant Biol. 49, 791–804 (2007)
Luo, D. et al. A detrimental mitochondrial-nuclear interaction causes cytoplasmic male sterility in rice. Nat. Genet. 45, 573–577 (2013)
Krieger, U., Lippman, Z. B. & Zamir, D. The flowering gene SINGLE FLOWER TRUSS drives heterosis for yield in tomato. Nat. Genet. 42, 459–463 (2010)
Li, X., Li, X., Fridman, E., Tesso, T. T. & Yu, J. Dissecting repulsion linkage in the dwarfing gene Dw3 region for sorghum plant height provides insights into heterosis. Proc. Natl Acad. Sci. USA 112, 11823–11828 (2015)
Hollick, J. B. & Chandler, V. L. Epigenetic allelic states of a maize transcriptional regulatory locus exhibit overdominant gene action. Genetics 150, 891–897 (1998)
Xiao, J., Li, J., Yuan, L. & Tanksley, S. D. Dominance is the major genetic basis of heterosis in rice as revealed by QTL analysis using molecular markers. Genetics 140, 745–754 (1995)
Hua, J. P. et al. Genetic dissection of an elite rice hybrid revealed that heterozygotes are not always advantageous for performance. Genetics 162, 1885–1895 (2002)
Zhou, G. et al. Genetic composition of yield heterosis in an elite rice hybrid. Proc. Natl Acad. Sci. USA 109, 15847–15852 (2012)
Gao, Z.-Y. et al. Dissecting yield-associated loci in super hybrid rice by resequencing recombinant inbred lines and improving parental genome sequences. Proc. Natl Acad. Sci. USA 110, 14492–14497 (2013)
Riedelsheimer, C. et al. Genomic and metabolic prediction of complex heterotic traits in hybrid maize. Nat. Genet. 44, 217–220 (2012)
Huang, X. et al. Genomic analyses of complex traits reveal the genetic basis of heterosis in 1495 hybrid rice varieties. Nat. Commun. 6, 6258 (2015)
Huang, X. et al. High-throughput genotyping by whole-genome resequencing. Genome Res. 19, 1068–1076 (2009)
Kojima, S. et al. Hd3a, a rice ortholog of the Arabidopsis FT gene, promotes transition to flowering downstream of Hd1 under short-day conditions. Plant Cell Physiol. 43, 1096–1105 (2002)
Yu, B. et al. TAC1, a major quantitative trait locus controlling tiller angle in rice. Plant J. 52, 891–898 (2007)
Suresh, P. B. et al. Fine mapping of Rf3 and Rf4 fertility restorer loci of WA-CMS of rice (Oryza sativa L.) and validation of the developed marker system for identification of restorer lines. Euphytica 187, 421–435 (2012)
Tang, H. et al. The rice restorer Rf4 for wild-abortive cytoplasmic male sterility encodes a mitochondrial-localized PPR protein that functions in reduction of WA352 transcripts. Mol. Plant 7, 1497–1500 (2014)
Xue, W. et al. Natural variation in Ghd7 is an important regulator of heading date and yield potential in rice. Nat. Genet. 40, 761–767 (2008)
Komatsu, M., Maekawa, M., Shimamoto, K. & Kyozuka, J. The LAX1 and FRIZZY PANICLE 2 genes determine the inflorescence architecture of rice by controlling rachis-branch and spikelet development. Dev. Biol. 231, 364–373 (2001)
Wang, Z. Y. et al. The amylose content in rice endosperm is related to the post-transcriptional regulation of the waxy gene. Plant J. 7, 613–622 (1995)
Gao, Z. et al. Map-based cloning of the ALK gene, which controls the gelatinization temperature of rice. Sci. China C Life Sci. 46, 661–668 (2003)
Liu, G., Lu, G., Zeng, L. & Wang, G. L. Two broad-spectrum blast resistance genes, Pi9(t) and Pi2(t), are physically linked on rice chromosome 6. Mol. Genet. Genomics 267, 472–480 (2002)
Yang, J. et al. A killer-protector system regulates both hybrid sterility and segregation distortion in rice. Science 337, 1336–1340 (2012)
Zhou, H. et al. RNase Z(S1) processes Ub L40 mRNAs and controls thermosensitive genic male sterility in rice. Nat. Commun. 5, 4884 (2014)
Ding, J. et al. A long noncoding RNA regulates photoperiod-sensitive male sterility, an essential component of hybrid rice. Proc. Natl Acad. Sci. USA 109, 2654–2659 (2012)
Yan, W.-H. et al. A major QTL, Ghd8, plays pleiotropic roles in regulating grain productivity, plant height, and heading date in rice. Mol. Plant 4, 319–330 (2011)
Kim, S. L., Lee, S., Kim, H. J., Nam, H. G. & An, G. OsMADS51 is a short-day flowering promoter that functions upstream of Ehd1, OsMADS14, and Hd3a. Plant Physiol. 145, 1484–1494 (2007)
Fujita, D. et al. NAL1 allele from a rice landrace greatly increases yield in modern indica cultivars. Proc. Natl Acad. Sci. USA 110, 20431–20436 (2013)
Yano, M. et al. Hd1, a major photoperiod sensitivity quantitative trait locus in rice, is closely related to the Arabidopsis flowering time gene CONSTANS. Plant Cell 12, 2473–2484 (2000)
Sasaki, A. et al. Green revolution: a mutant gibberellin-synthesis gene in rice. Nature 416, 701–702 (2002)
Sun, H. et al. Heterotrimeric G proteins regulate nitrogen-use efficiency in rice. Nat. Genet. 46, 652–656 (2014)
Song, X. J. et al. Rare allele of a previously unidentified histone H4 acetyltransferase enhances grain weight, yield, and plant biomass in rice. Proc. Natl Acad. Sci. USA 112, 76–81 (2015)
Izawa, T. et al. Os-GIGANTEA confers robust diurnal rhythms on the global transcriptome of rice in the field. Plant Cell 23, 1741–1755 (2011)
Jiao, Y. et al. Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nat. Genet. 42, 541–544 (2010)
Miura, K. et al. OsSPL14 promotes panicle branching and higher grain productivity in rice. Nat. Genet. 42, 545–549 (2010)
Yao, H., Dogra Gray, A., Auger, D. L. & Birchler, J. A. Genomic dosage effects on heterosis in triploid maize. Proc. Natl Acad. Sci. USA 110, 2665–2669 (2013)
Birchler, J. A., Johnson, A. F. & Veitia, R. A. Kinetics genetics: Incorporating the concept of genomic balance into an understanding of quantitative traits. Plant Sci. 245, 128–134 (2016)
Picelli, S. et al. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res. 24, 2033–2040 (2014)
Wang, S., Basten, C. J. & Zeng, Z. B. Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh. http://statgen.ncsu.edu/qtlcart/WQTLCart.htm (2007)
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006)
Kang, H. M. et al. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42, 348–354 (2010)
Huang, X. et al. Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat. Genet. 44, 32–39 (2011)
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009)
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011)
Acknowledgements
This work was supported by the Chinese Academy of Sciences (XDA08020101) and the National Natural Science Foundation of China (31322028, 91535202, 31421093 and 31301302).
Author information
Authors and Affiliations
Contributions
B.H. conceived the project and its components. X.H. and B.H. designed studies and contributed to the original concept of the project. S.Y. and J.G. contributed the generation of the genetic populations. J.G., Qil.Z., B.C., J.X., N.C. and S.Y. contributed in phenotyping for grain yield traits. W.L., Y.L. J.C., C.Z., D.F., Q.W. and Q.F. performed the genome sequencing. X.H., Qia.Z., Y.Z., L.Z. and T.H. performed genome data analysis and genetic analysis. X.H. and B.H. analysed whole data and wrote the paper.
Corresponding authors
Additional information
Reviewer Information
Nature thanks J. Ross-Ibarra, D. Weigel and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Extended data figures and tables
Extended Data Figure 2 Selection of 17 representative hybrid combinations from 1,495 hybrids.
a, Plots of the first two principal components of 1,495 hybrids. The 17 representative hybrids are coloured in red. b, Plots of the principal component 3 and the principal component 4 of 1,495 hybrids. c, Plots of heading date and plant height of 1,495 hybrids. d, Plots of grain number and seed-setting rate of 1,495 hybrids.
Extended Data Figure 3 High-resolution genotyping of F2 lines by whole-genome sequencing.
a, Aligned recombination maps of 1,105 lines from a cross between Fuhui676 and Guangzhan63-4S. Three genotypes were indicated by blue, red and yellow, respectively (blue, Fuhui676/Fuhui676; red, Guangzhan63-4S/Guangzhan63-4S; yellow, Fuhui676/Guangzhan63-4S). b, The recombination map of a single F2 line from a cross between Fuhui676 × Guangzhan63-4S. The genomic DNA of the line was sequenced in the HiSeq2500 system, and genotyped through the SEG-Map computational algorithm. Detected SNP genotypes in this line were indicated along chromosomes according to their physical locations. A sliding window approach was used for genotype calling, recombination breakpoint determination and map construction. There were several IBS (identity by state) segments shared by both parents (for example, the regions on the chromosomes 10 and 11), which are indicated by the grey boxes.
Extended Data Figure 4 Analysis of flowering time in type-A hybrids.
a, Flowering-time modelling using the genotypes of four genes (Hd3a, Ghd7, Hd2 and Ehd1) known to regulate flowering time. We used the data in one type-A population (Guanghui998 × Tianfeng) for experimentation, and evaluated the accuracy of the modelling in the other eight populations. b, The estimates of the genetic effects of three genotypes of the four genes, in which the flowering time of the homozygous genotypes of male parents was set to be 90 days according to the average performance of restorer lines. The arrows indicated the flowering time for the genotypes Hd3a/hd3a and hd3a/hd3a.
Extended Data Figure 5 The linkage drag around the hd3a locus.
The allelic combinations of six candidate genes on the short arm of chromosome 6 and their frequencies in 1,063 indica CMS lines.
Extended Data Figure 6 QTL for heterosis for yield traits in type-B hybrids.
a, Plots of the dominance effects of three traits at the Tms5 locus in the four type-B hybrids. b, Plots of the dominance effects of three traits at the Ghd8 locus in the four type-B hybrids. c, The yield advantages in the hybrid LYP9 by two QTLs LAX1 and SS3p10 from PA64S (**P < 0.001, two-tailed t-test). The male (M) and heterozygous (H) genotypes are indicated. d, The yield advantages in the hybrid Liangyou676 by two QTLs at the ‘ghd8’ locus and PN3q23 from Guangzhan63-4S (**P < 0.001, two-tailed t-test).
Extended Data Figure 7 Support for pseudo-overdominance at the Tms5 locus.
a, The linkage mapping of fertility in one population of type B. LOD values are plotted against the physical positions, and the threshold (3.5) is indicated by a horizontal dashed line. b, The linkage mapping of panicle number in one population of type A. c, The linkage mapping of yield per plant in the population of type A. d, Gene structure and causal variation site between Tms5 and tms5 alleles in the hybrids of type B. e, No mutations are detected at the Tms5 gene between both parents (Fuhui838 and II32A) of the type-A population.
Extended Data Figure 8 Performances of two QTLs LAX1 and SS3p10 in type-B hybrid LYP9.
a, The linkage mapping of seed-setting rate in the population of type-B hybrid LYP9. LOD values are plotted against the physical positions, and the threshold (3.5) is indicated by a horizontal dashed line. b, The performances of seed-setting rate for three genotypes of the SS3p10 locus in the population of type-B hybrid LYP9. c, The linkage mapping of grain weight in the population of type-B hybrid LYP9. d, The performances of grain weight for three genotypes of the LAX1 locus in the population of type-B hybrid LYP9.
Extended Data Figure 9 Performances of two QTLs, GW3p6 and PN3q23, in type-B hybrid Liangyou676.
a, The linkage mapping of grain weight in the population of type-B hybrid Liangyou676. b, The performances of grain weight for three genotypes of the GW3p6 locus in the population of type-B hybrid Liangyou676. c, The linkage mapping of panicle number in the population of type-B hybrid Liangyou676. d, The performances of panicle number for three genotypes of the PN3q23 locus in the population of type-B hybrid Liangyou676.
Extended Data Figure 10 Partial positive dominance effect of IPA1 for the yield components.
a, The performances of grain number per panicle for three genotypes of the IPA1 gene. b, The performances of panicle number for three genotypes of the IPA1 gene. c, The performances of seed-setting rate for three genotypes of the IPA1 gene.
Rights and permissions
About this article

Cite this article
Huang, X., Yang, S., Gong, J. et al. Genomic architecture of heterosis for yield traits in rice. Nature 537, 629–633 (2016). https://doi.org/10.1038/nature19760
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature19760
- Springer Nature Limited
This article is cited by
-
Improving hybrid rice breeding programs via stochastic simulations: number of parents, number of hybrids, tester update, and genomic prediction of hybrid performance
Theoretical and Applied Genetics (2024)
-
Different contributions of PROG1 and TAC1 to the angular kinematics of the main culm and tillers of wild rice (Oryza rufipogon)
Planta (2024)
-
Ectopic expression of OsWOX9A alters leaf anatomy and plant architecture in rice
Planta (2024)
-
Genetic Diversity and Breeding Signatures for Regional Indica Rice Improvement in Guangdong of Southern China
Rice (2023)
-
Can the Wild Perennial, Rhizomatous Rice Species Oryza longistaminata be a Candidate for De Novo Domestication?
Rice (2023)