
Abstract
The Wild Abortive (WA) system is the major cytoplasmic male sterility (CMS) source for hybrid rice production in indica rice and its fertility restoration is reported to be controlled by two major loci viz. Rf3 on chromosome 1 and Rf4 on chromosome 10. With the availability of the rice genome sequence, an attempt was made to fine map, develop candidate gene based markers for Rf3 and Rf4 and validate the developed marker system in a set of known restorer lines. Using polymorphic markers developed from microsatellite markers and candidate gene based markers from Rf3 and Rf4 loci, local linkage maps were constructed in two mapping populations of ~1,500 F2 progeny from KRH2 (IR58025A/KMR3R) and DRRH2 (IR68897A/DR714-1-2R) hybrids. QTLs and their interactions for fertility restoration in Rf3 and Rf4 loci were identified. The identified QTL in both mapping populations together explained 66–72 % of the phenotypic variance of the trait suggesting their utility in developing a marker system for identification of fertility restorers for WA-CMS. Sequence comparison of the two candidate genes from the Rf3 and Rf4 regions in male sterile (A) and restorer (R) lines showed 2–3 bp indels and a few substitutions in the Rf3 region and indels of 327 and 106 bp in the Rf4 region respectively. The marker system identified in the present study was validated in 212 restorers and 34 maintainers along with earlier reported markers for fertility restoration of WA-CMS. Together DRCG-RF4-14 and DRCG-RF4-8 for the Rf4 locus and DRRM-RF3-5/DRRM-RF3-10 for the Rf3 locus showed a maximum efficiency of 92 % for identification of restorers.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Hybrid rice technology is one of the promising, sustainable and proven technologies for enhancing the rice productivity with a yield advantage of 15–20 % over inbred varieties. Rice hybrids occupy more than 50 % of rice area in China and are being adopted in other rice growing countries (Virmani et al. 2003). For the development of rice hybrids, mostly the three-line system consisting of cytoplasmic male sterile (CMS) line (A), a maintainer line (B) and a restorer line (R) is being adopted. CMS can be restored by fertility restorer (Rf) genes associated with nuclear genes mostly encoding pentatricopeptide repeat (PPR) proteins (Hanson and Bentolila 2004). In rice, though 20 independent CMS cytoplasms have been reported, only three CMS systems viz. Wild Abortive (WA), Boro Tai (BT) and Honglian (HL) are mostly deployed for commercial hybrid seed production (Li and Yuan 2000; Fujii and Toriyama 2009). Of these, the WA system is widely used CMS source for indica rice accounting for about 90 % of the rice hybrids produced in China and 100 % of the hybrids developed outside China (Sattari et al. 2007). Inheritance of fertility restoration in the WA-CMS system has been extensively investigated and reported genetic analyses have shown that the number, position and effects of the Rf genes are variable depending upon the materials and methods used. Genetic studies on fertility restoration in WA-CMS lines showed that restoration is monogenic (Shen et al. 1996); digenic (Bharaj et al. 1991); digenic with different types of interactions (Govinda Raj and Virmani 1988; Sohu and Phul 1995; Sharma et al. 2001; Waghmode and Mehta 2011); trigenic (Kumar and Chakrabarti 1983; Sarkar et al. 2002) and trigenic interactions (Huang 1987; Hossain et al. 2010).
With the advent of molecular mapping, several research groups have identified chromosomal locations of the Rf genes for various CMS systems in rice. Rf1 gene restoring BT-CMS has been cloned and demonstrated to be the locus consisting of pentatricopeptide genes (Wang et al. 2006). In CMS system derived from Lead rice (LD-CMS), Rf2 has been identified in japonica on chromosome 2 and fine mapped (Shinjyo and Sato 1994; Fujii and Toriyama 2009). Huang et al. (2000) fine mapped the Rf5 (t) on chromosome 10 for HL-CMS and another locus Rf6 (t) was also identified on chromosome 10 for the same cytoplasm (Liu et al. 2004). Another Rf gene for CMS-Chinese Wild (cw) rice was fine mapped on chromosome 4 (Fujii and Toriyama 2009).
Zhang et al. (1997) mapped the Rf3 locus for WA-CMS on chromosome 1 and Yao et al. (1997) confirmed its location and also mapped the Rf4 locus on chromosome 10. Four QTLs on chromosomes 1, 7, 10 and 11 were identified for fertility restoration of WA-CMS (Zhuang et al. 2000). The role of the Rf4 locus in fertility restoration and its location on chromosome 10 has been identified by many research groups (Tan et al. 1998; Jing et al. 2001; Zhang et al. 2002; Mishra et al. 2003; Singh et al. 2005; Ahmadikhah and Karlov 2006; Ahmadikhah et al. 2007; Sheeba et al. 2009; Ngangkham et al. 2010). A major QTL for fertility restoration of WA-CMS associated with the Rf3 region was identified in IR36 and IR60966 (Ahmadikhah et al. 2007; Ahmadikhah and Alavi 2009). Both Rf3 and Rf4 loci have been identified in different donors using rice microsatellite (RM) or simple sequence repeat (SSR) markers (He et al. 2002; Bazrkar et al. 2008; Sattari et al. 2008; Nematzadeh and Kiani 2010). Ngangkham et al. (2010) fine mapped the Rf4 locus in Basmati restorer line PRR78 within an interval of 0.8 cM and developed candidate gene (CG) based marker from PPR3 gene based on sequence information derived from japonica (Nipponbare). Thus, fertility restoration of WA-CMS appears to be controlled by different Rf loci in different restorer lines unlike other CMS systems. However, two loci (Rf3 and Rf4) on chromosomes 1 and 10 appear to be consistent in restoring the fertility across most of the studies. Attempts were made to use these two loci for marker assisted selection (MAS) to identify restorer lines possessing Rf genes for WA-CMS to expedite phenotype-based screening. Nas et al. (2003) developed a single-gene MAS system based on sequence tagged site (STS) marker, RG140/PvuII (STS-RG140) for Rf3 on chromosome 1. Using two STS markers system (RG140/PvuII and S10019/BstUI) for Rf3 and Rf4 loci, 13 restorer lines were surveyed (Sattari et al. 2007). Microsatellite (RM6100/RM25654) and CG based marker systems (TMPPR3) were also evaluated for their selection efficiency (Sheeba et al. 2009; Ngangkham et al. 2010). From the foregoing studies, it appears that Rf4 is a major locus for fertility restoration of WA-CMS in most of the cases and Rf3 in some studies. Therefore, the present study was undertaken to fine map Rf3 and Rf4 loci, to study the contribution and their interaction for fertility restoration of WA-CMS and to validate selection efficiency of the developed markers in a set of restorer lines and compare with reported markers.
Materials and methods
Plant material
Two mapping populations of ~1,500 F2 progeny derived from hybrids viz. KRH2 (IR58025A/KMR3R) and DRRH2 (IR68897A/DR714-1-2R) were studied for mapping of Rf3 and Rf4 loci. These segregating populations and their parents were grown during Kharif 2009 (wet season: July to October) at the research farm of Directorate of Rice Research (DRR farm), Hyderabad, India following recommended package of practices. A set of 212 restorer lines and 34 maintainer lines collected from International Rice Research Institute (IRRI), Philippines and other institutions from India were also grown under the same conditions. For progeny test, F3 plants from fertile F2 (>90 %) were grown during Kharif 2010. The KRH2 F2 population was earlier mapped for the Rf4 locus by Sheeba et al. (2009).
Phenotyping and genotyping
Three spikelets per plant were collected and the pollen fertility was determined using 1 % I-KI stain (Virmani et al. 1997). Mean of observations from three microscopic fields was noted and calculated as percentage of the pollen fertility based on Standard Evaluation System (IRRI 1996) and F2 individual plants were grouped into completely sterile (0 %), partially sterile (1–30 %), partially fertile (>30–60 %) and fertile (>60 %) (Govinda Raj and Virmani 1988). For genetic studies, the partial fertile and partial sterile groups were combined as semi-fertile group and analyzed for Chi-square test. For progeny test, 10 F3 seeds were randomly selected from F2 plants (>90 % pollen fertility) and analyzed by markers associated with fertility restoration. Based on the segregation pattern of the closely associated markers, the F2 plants were considered as fertile homozygotes. Total genomic DNA from fresh and young leaves of parents, segregating populations, known restorer and maintainer lines was isolated using modified protocol of Zheng et al. (1995).
Primer design
Based on the position of the STS-RG140 marker reported (5,095,628 bp) on the Rf3 locus (Zhang et al. 1997); 2 Mb genomic region (4–6 Mb) was targeted for identification of microsatellites and design of primers on chromosome 1 derived from Nipponbare sequence, Accession No. NC_008394 (http://www.ncbi.nlm.nih.gov/genbank/). For the Rf4 locus, the marker RM6100 was reported at 1,837,2167 bp (Singh et al. 2005), 2 Mb region of chromosome 10 of Nipponbare (NC_008403) was targeted (17–19 Mb) for designing microsatellite primers. In addition to the sequence of japonica subspecies, corresponding indica sequence from genomic library of IR24 (AB110443) was also obtained (Komori et al. 2004). The repeats were identified using software ‘Tandem Repeat Finder’ (Benson 1999) and primers were designed using Primer 3.0 (http:frodo.wi.mit.edu/primer3/) (ESM Table 1).
Candidate gene information was obtained from japonica sequence (NC_008394, Pseudomolecule 6.1, http://rice.plantbiology.msu.edu/) from 4 to 6 Mb of chromosome 1. Annotated indica sequence from IR24 BAC clone (AB110443) of chromosome 10 (Komori et al. 2004) was downloaded from http://www.ncbi.nlm.nih.gov/genbank/. Based on the sequences of candidate genes along with their upstream regions (1 kb), two to six primer pairs were designed per gene ensuring the coverage of complete gene along with upstream region (~1 kb) using Primer 3.0 software (http://frodo.wi.mit.edu). All the primers were synthesized at Integrated DNA Technologies Inc. (Iowa City, IA) (ESM Table 2).
PCR amplification
For amplification of the microsatellite markers, PCR was carried out in Thermal Cycler (Bio-Rad C1000) using 40 ng template DNA, 0.125 mM dNTPs each, 2.5 p moles of forward and reverse primers, 1 U Taq polymerase (Bangalore Genei, India) and 1× Taq buffer (Bangalore Genei, India) in a total volume of 10 μl reaction. The cycling conditions included initial denaturation at 94 °C for 4 min, followed by 30 s at 94 °C, 30 s at 55 °C, and 1 min at 72 °C for 35 cycles, followed by a final extension at 72 °C for 7 min. Amplified products were resolved in 8 % polyacrylamide gel using mini vertical polyacrylamide gels (CBS Scientific Co. Inc., USA).
For candidate gene based markers, a 20 μl PCR reaction mixture was prepared containing 50 ng template DNA, 0.25 mM dNTPs each, 0.5 pmol of forward and reverse primers, 2 U Taq DNA polymerase (Bangalore Genei, India) 1× Taq buffer (Bangalore Genei, India) and 5 M betaine (Sigma, USA) (Henke et al. 1997), The cycling conditions comprised initial denaturation at 94 °C for 4 min, followed by 1 min at 94 °C, 1 min at 55 °C, and 2 min at 72 °C for 35 cycles, followed by a final extension at 72 °C for 7 min. PCR products were resolved in 2.5–3.5 % Metaphor® agarose gel (Lonza, USA). Gels were stained in 0.5 mg/ml ethidium bromide and documented using Alpha Imager 1220 (Alpha Innotech, USA).
Parental polymorphism
Four parents of the two mapping populations were surveyed with 29 Rice Microsatellite (RM) primers, 56 designed SSR primers and 23 CG based primers from chromosome 1 for the Rf3 region. For the Rf4 region, 50 SSR and 21 CG primers identified from IR24 BAC clone (AB110443) were used for parental polymorphism. In addition, 23 RM primers along with the reported marker RM6100 (Singh et al. 2005) and 58 SSR primers designed from Nipponbare genome (17–19 Mb region) were also assayed in the parents.
Selective genotyping
The markers polymorphic between the parents were subjected to selective genotyping (Nandi et al. 1997) in two phenotypically distinct classes for pollen fertility comprising 22 sterile (pollen fertility: 0 %) and 22 fertile F2 individuals (pollen fertility: 100 %). Recombination frequencies were calculated for the two classes (Allard 1956) and the marker allele frequencies were compared between the two tails of progeny for the two classes. Only the markers with >0.25 difference between the absolute allele frequency values of the two tails were considered for further analysis.
QTL analysis
The markers that exhibited low recombination frequency (>0.25) in selective genotyping procedure were analyzed in 1,580 and 1,576 F2 individuals of KRH2 and DRRH2, respectively, to construct genetic maps using the MapDisto v. 1.7 software (Lorieux 2007). Linkage groups were obtained with >3.0 LOD and of 0.3 r max for Rf3 and Rf4 loci in both the populations and map distances were calculated using Kosambi mapping function (Kosambi 1944). Since the evaluation criterion for measuring fertility restoration in pollen of WA-CMS system is continuous (IRRI 1996); the data from the present study was analyzed as continuous data for QTL analysis using QTL Cartographer 2.5 (Wang et al. 2010). The interaction between the identified QTLs and their effects in both the mapping populations was analyzed using QTLNetwork 2.1 software (Yang et al. 2008).
Validation of markers
To assess the selection efficiency of the markers in restorer lines, the identified markers from the present study were screened in 212 restorers and 34 maintainer lines. All the lines were phenotypically evaluated at DRR farm and assessed with markers from Rf3 and Rf4 loci identified in this study along with all the reported markers.
Sequencing
The polymorphic products derived from four CG based primers from the parents of KRH2 and DRRH2, along with male sterile line APMS6A and two restorer lines 1,005 and C20R were also subjected to sequence analysis. PCR products were eluted from 1 % agarose gel, purified using Wizard® SV Gel and PCR Clean-Up System (Promega); cloned in pGEM-T easy vector (Promega) and sequenced using an ABI Prism 3700 automated DNA sequencer (Perkin Elmer, MA) by Bioserve Biotechnologies, India. The derived consensus sequences were compared between the A and R lines and also with the sequences of japonica subspecies (Nipponbare) using CLUSTALW multiple sequence alignment tool employing BIOEDIT software (http://www.ebi.ac.uk/Tools/msa.clustaw2/; http://www.mbio.ncsu.edu/BioEdit/bioedit.html). The sequence difference as indels (insertions/deletions) and Single Nucleotide Polymorphisms (SNPs) between the A and R lines were considered for polymorphism.
Results
Phenotyping
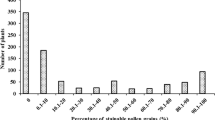
Based on the extent of the stain taken up by the pollen, out of 1,576 individuals of KRH2 population (mean: 65.6 %), 578 plants showed complete sterility (0 %), 57 showed partial sterility (1–30 %), 161 showed partial fertility (>30–60 %) and 780 showed high fertility (>60 %). Among 1,580 F2 individuals of DRRH2 population (mean: 67.0 %), 166 showed complete sterility (0 %), 153 showed partial sterility (1–30 %), 173 showed partial fertility (>30–60 %) and 1,088 showed high fertility (>60 %) as shown in Table 1.
Genetics
Analysis with Chi-square test for three groups (Fully fertile >60 %; Semi-fertile 59–1 % and Complete sterile) suggested the involvement of three genes in controlling fertility restoration. In KRH2, a segregation ratio of 32:9:23 (χ2 = 0.379; P value = 0.8273) and in DRRH2, a segregation ratio of 44:13:7 (χ2 = 0.351; P value = 0.8389) was obtained (Table 1).
In silico analysis and design of markers based on microsatellites and candidate genes
Bioinformatics of the Rf3 region revealed 185 genes; of which only four genes viz. mitochondrial-processing peptidase subunit alpha (LOC_Os01g09560), pollen-specific protein (LOC_Os01g09670), and two pentatricopeptide (PPR) genes, (LOC_Os01g10090; LOC_Os01g10800) were considered as candidate genes in the present study. Bioinformatics of the Rf4 region in IR24 (indica) showed four PPR genes, which were considered as candidate genes in the present study. For the Rf3 locus, for 2 Mb sequence (4–6 Mb) of chromosome 1 from japonica (Nipponbare), with the designed microsatellite markers in this study, the density of the microsatellite markers has increased from one RM marker per 69 kb (IRGSP 2005) to one marker per 23.5 kb. For the Rf4 locus, the density of microsatellite markers increased from one marker per 87 kb to one marker per 24.7 kb with the designed markers from japonica (Nipponbare) sequence. For the same locus, using indica sequence (IR24), 50 additional microsatellite markers designed which further increased the marker density to one marker per 15.2 kb. Twenty-three markers designed for the candidate genes has increased the density of markers to one marker per 18.5 kb for the Rf3 locus and 21 markers designed for the candidate genes has increased the overall density to one marker per 13.1 kb for the Rf4 locus (ESM Tables 1 and 2).
Parental polymorphism and selective genotyping
For KRH2 parents, 15 out of 29 RM markers (51.7 %) and 19 from 56 designed SSRs (33.9 %) and for DRRH2 parents, 10 out of 29 RM markers (34.5 %) and 17 out of 56 designed SSR markers (30.4 %) were polymorphic for the Rf3 locus. Two candidate genes viz. mitochondrial-processing peptidase subunit alpha (LOC_Os01g09560) and the pollen-specific protein (LOC_Os01g09670) showed polymorphism for the four parents (8.7 %). For the Rf4 locus, 7 out of 23 RM markers (30.4 %) and 13 out of 108 designed SSR markers (12 %) were polymorphic between the KRH2 parents. For DRRH2, 6 out of 23 RM markers (26.1 %) and 15 designed SSR markers out of 108 markers (13.8 %) were polymorphic (Table 2). Two CG based markers viz. DRCG-RF4-8 (PPR683) and DRCG-RF4-14 (PPR762) based on IR24 (indica) were polymorphic between the parents of both the populations (9.5 %) (ESM Fig. 1).
Out of 23 markers designed for four candidate genes and their corresponding upstream regions (~1 kb) in Rf3 region, polymorphism with a narrow resolution was observed in two genes viz. LOC_Os01g09560 (mitochondrial-processing peptidase subunit alpha, mitochondrial precursor, putative, expressed) and LOC_Os01g09670 (pollen-specific protein SF21, putative expressed).
From selective genotyping based on the recombination frequency and resolution, a total of seven markers viz. RM10305, RM10318, DRRM-RF3-5, DRRM-RF3-6, DRRM-RF3-10, DRRM-RF3-15 and DRRM-RF3-24 for the Rf3 locus and seven markers viz. RM6100, DRRM-RF4-72, DRRM-RF4-74, DRRM-RF4-10, DRRM-RF4-20, DRCG-RF4-8 and DRCG-RF4-14 for the Rf4 locus were selected in KRH2 population. For DRRH2 population, six markers viz. RM10287, DRRM-RF3-5, DRRM-RF3-6, DRRM-RF3-10, DRRM-RF3-24 and DRRM-RF3-27 for the Rf3 locus and six markers viz. RM6100, DRRM-RF4-72, DRRM-RF4-74, DRRM-RF4-10, DRCG-RF4-8 and DRCG-RF4-14 for the Rf4 locus were selected for mapping analysis. The resolution of the two polymorphic candidate gene markers from the Rf3 region in segregating populations was too poor to assign heterozygous status and hence, was not considered for further analysis.
Local linkage maps
Two individual linkage groups for Rf3 and Rf4 loci for each population were constructed (Tables 3, 4; Fig. 1). In KRH2 population for the Rf3 locus, a 10.4 cM genetic map was generated and the designed SSR marker DRRM-RF3-10 exhibited highly significant association with the Rf trait (P = 0.0, χ2 = 853.6), RM10318 marker exhibited moderate association (P = 0.02, χ2 = 7.64) and DRRM-RF3-6 exhibited low significance with the trait (P = 0.02, χ2 = 7.83). For the Rf4 locus, a 14.1 cM genetic map was generated and all the markers used in this study exhibited correlation with the Rf trait. CG based markers i.e. DRCG-RF4-8 (χ2 = 239.9) and DRCG-RF4-14 (χ2 = 351.2), showed significant association with high χ2 values and with low P values (P = 0.0). The reported marker for Rf4, RM6100 also exhibited association with the trait (P = 0.00067, χ2 = 14.61).

Local linkage maps of F2 population for Rf loci; a KRH2 population chromosome 1; b KRH2 population chromosome 10; c DRRH2 population chromosome 1; d DRRH2 population chromosome 10
For DRRH2 population, a 11.7 cM genetic map was generated for the Rf3 locus and two designed SSR markers DRRM-RF3-5 and DRRM-RF3-10 exhibited highly significant association with the Rf trait (P = 0.0, χ2 = 32.1, 101.0). For the Rf4 locus, a 11.4 cM genetic map was generated and the designed CG based markers DRCG-RF4-8 (χ2 = 463.6) and DRCG-RF4-14 (χ2 = 747.6) exhibited good association with the trait (P = 0.0).
QTL analysis
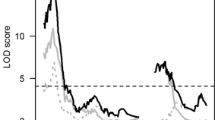
Two QTLs on chromosome 10 and two QTLs on chromosome 1 for fertility restoration were detected in the two mapping populations using Composite Interval Mapping (CIM) and Multiple Interval Mapping (MIM) of QTL Cartographer (Wang et al. 2010) (Table 5). Though QTLs identified with MIM method were considered, both methods identified the QTLs with similar confidence levels in the present study. In KRH2, two QTLs were identified on chromosome 1 viz. a QTL (qWARF-1-1) between the designed markers DRRM-RF3-10 and DRRM-RF3-5 (LOD 16.1) with additive and dominance effects (a = −11.35, d = −1.34) and another QTL (qWARF-1-2) close to DRRM-RF3-15 marker (LOD 4.7, a = −7.68, d = −4.91). In the same population, two QTLs were detected for the Rf4 locus on chromosome 10. One QTL (qWARF-10-1) between the CG based markers DRCG-RF4-8 and DRCG-RF4-14 (LOD 5.6, a = −5.18, d = 8.18) and the other QTL (qWARF-10-2) between the reported marker RM6100 (Singh et al. 2005) and designed marker DRRM-RF4-20 with LOD 3.3 (a = 2.26, d = −5.35) were identified.
In DRRH2 population, one QTL (qWARF-1-1) was detected between the designed marker DRRM-RF3-10 and DRRM-RF3-6 with LOD score 4.1 (a = −2.37, d = 7.95) for the Rf3 locus. Two QTLs were identified for the Rf4 locus in the same population, a QTL (qWARF-10-1) in between the CG based marker DRCG-RF4-14 and reported marker RM6100 (LOD 5.3, a = −1.22, d = −10.98) and another QTL (qWARF-10-2) between the designed markers DRRM-RF4-74 and DRRM-RF4-72 with LOD threshold value of 3.4 and additive and dominance effects (a = 1.84, d = 1.70).
QTL interactions
Based on the QTL Network analysis, one QTL (qWARF-1-1) in the Rf3 locus and two QTLs in the Rf4 locus (qWARF-10-1 and qWARF-10-2) were identified in KRH2 population (Fig. 2a). A QTL for the Rf3 locus (qWARF-1-1) associated with DRRM-RF3-10 marker with 31 % of the phenotypic variance was identified. For the Rf4 locus (qWARF-10-1), a QTL comprising CG marker DRCG-RF4-14 flanking another CG marker DRCG-RF4-8 with 41 % of the phenotypic variance was identified and a QTL (qWARF-10-2) comprising reported marker RM6100 with DRRM-RF4-10 was also identified. Two epistatic interactions among the QTLs viz. qWARF-1-1 and qWARF-10-1 and also between qWARF-10-1 and qWARF-10-2 were detected (Table 5).

Significant epistatic QTLs on chromosome 10 and 1 for fertility restoration trait by QTLNetwork 2.1; a KRH2 population: Rf3 locus-qWARF-1-1 association with DRRM-RF3-10; Rf4 locus-qWARF-10 flanked by DRCG-RF4-14 and DRCG-RF4-8 and qWARF-10-2 association with RM6100; b DRRH2 population: Rf3 locus-qWARF-1-1 QTL flanked by DRRM-RF3-10 and DRRM-RF3-6, Rf4 locus-qWARF-10-1 QTL is on DRCG-RF4-14 and flanked by DRCG-RF4-8 marker and qWARF-10-2 was flanked by DRRM-RF4-10 and RM6100
A total of three QTLs, one in the Rf3 locus and two in the Rf4 locus were predicted by QTL Network analysis in DRRH2 population (Fig. 2b). For the Rf3 locus, one QTL (qWARF-1-1) flanked by DRRM-RF3-10 and DRRM-RF3-6 markers with 23 % of the phenotypic variance. Among the two QTLs in the Rf4 locus, a QTL (qWARF-10-1) was represented by the CG based marker DRCG-RF4-14 and flanked by CG based marker DRCG-RF4-8 with 43 % phenotypic variance and another QTL (qWARF-10-2) was identified between DRRM-RF4-10 and RM6100. Epistatic interactions were observed between qWARF-1-1 and qWARF-10-1, and between qWARF-10-1 and qWARF-10-2.
Sequencing of the candidate genes
In the Rf3 locus, sequencing of 1,501 bp polymorphic PCR product derived from the primer set of DRCG-Rf3-2F—DRCG-Rf3-2R positioned from 2 to 1,502 bp of LOC_Os01g09560 gene (5,361 bp) encoding putative Mitochondrial-processing peptidase subunit alpha, mitochondrial precursor of A and R lines showed two (TT) base pair deletion at 907 and 908 bp position in the first intron in R lines (Fig. 3a) in addition to 16 SNPs between A and R lines. The sequence of 472 bp PCR product from LOC_Os01g09670 putative Pollen-specific protein SF21 (3,002 bp) derived from the marker DRCG-Rf3-13 positioned from 1 to 472 bp showed two deletions in the gene viz. two base pairs deletion (GA) at 29 and 30 bp positions and three base pairs deletion (GAG) in A lines at 77 to 79 bp position in exon 1 (Fig. 3b). For the Rf4 locus, sequencing of polymorphic (1,095 bp and 770 bp) PCR products of the marker DRCG-RF4-8, positioned from 221 bp upstream to 549 bp genic region of PPR683 showed 6 bp deletion in APMS6A, IR58025A and IR68897A and a 327 bp deletion in KMR3, 1,005 and DR714-1-2R lines spanning exon 1 and intron 1. The restorer line C20R also gave similar product size with CMS lines i.e. 1,095 bp along with 6 bp deletion as observed in A lines (Fig. 3c). The CLUSTALW multiple sequence alignment comparison with Nipponbare genes (NC_008403) showed 6 bp deletion in all the three A lines CMS and C20R line and a 327 bp deletion in three R lines. Sequencing of polymorphic products (887 bp and 782 bp) of the marker DRCG-RF4-14 positioned from 1,207 to 1,989 bp of PPR762 showed 106 bp deletion spanning exon 1 in three R lines (Fig. 3d). The product of restorer line C20R was 887 bp, similar to that of A lines. Indel (insertions/deletions) polymorphisms between the A and R lines were considered for the analysis.


a CLUSTALW alignment comparison of sequences derived from PCR products amplifying an expected 1,501 bp spanning intron 1 of Mitochondrial-processing peptidase subunit alpha gene (LOC_Os01g09560) from primer set: DRCG-Rf3-2F—DRCG-Rf3-2R in the Rf3 region. b CLUSTALW alignment comparison of sequences derived from PCR products amplifying an expected 472 bp spanning exon 1 of pollen-specific protein SF21 gene (LOC_Os01g09670) from primer set: DRCG-Rf3-13 in the Rf3 region. c CLUSTALW alignment comparison of sequences derived from PCR products amplifying 1,095 bp (A line) and 770 bp (R line) spanning exon 1 and intron 1 of PPR683 gene from primer set: DRCG-RF4-8 in the Rf4 region. d CLUSTALW alignment comparison of sequences derived from PCR products amplifying 887 bp (A line) and 782 bp (R line) spanning exon 1 of PPR762 gene from primer set: DRCG-RF4-14 in the Rf4 region
Validation of linked markers
Out of six markers i.e. RG140/PvuII, RM10305, RM10318, DRRM-RF3-5, DRRM-RF3-6 and DRRM-RF3-10 used in validation of the Rf3 locus, DRRM-RF3-10 marker showed maximum selection accuracy of 57.3 % in 212 restorer lines and 88 % in 34 maintainer lines (Table 6) (ESM Table 3). Out of five markers surveyed for validation of the Rf4 locus, candidate gene marker DRCG-RF4-14 exhibited maximum selection accuracy of 86.8 % in 212 restorer lines and 91.2 % in maintainer lines. The reported CG based marker TMPPR3 (Ngangkham et al. 2010) targeting the same gene also exhibited identical selection accuracy. The combination of the Rf3 and Rf4 loci markers i.e. DRRM-RF3-5/DRRM-RF3-10 and DRCG-RF4-14 exhibited 92 % selection accuracy in restorer lines.
Discussion
Despite the economic importance of WA-CMS in hybrid rice technology, neither the mechanism of WA-CMS nor its fertility restoration has been elucidated clearly. One to three genes with interactions were reported for the fertility restoration of WA-CMS (Govinda Raj and Virmani 1988; Bharaj et al. 1991; Hossain et al. 2010). In the present study, both the mapping populations showed continuous variation in pollen fertility ranging from 0 to 100 %. Genetic analysis of the two mapping populations fitted well to the trigenic ratio indicating the involvement of three genes with interaction in the fertility restoration of WA-CMS. Trigenic ratios in fertility restoration were earlier reported in some restorer lines (Kumar and Chakrabarti 1983; Sohu and Phul 1995; Hossain et al. 2010).
From the compilation of the genetic and mapping studies for fertility restoration for WA-CMS, it is observed that the variation is continuous as inferred from the phenotype of the pollen fertility suggesting the involvement of QTLs or more than three genes in the trait; however the classification of the data into discreet groups is influencing the genetic ratios ranging from monogenic to digenic with interactions to trigenic with interactions.
Several microsatellite markers have been developed in the present study increasing the resolution of one marker per 18.5 kb in the Rf3 region and 13.1 kb in the Rf4 region which is very high as compared to earlier mapping studies. Out of 185 and 189 annotated genes in the Rf3 and Rf4 regions, only PPR genes and genes directly involved in pollen biosynthesis were considered as putative candidate genes for primer design in the present study. However, with identification of genes involved in fertility restoration of Chinese wild (CW) type of CMS (Fujii and Toriyama 2009) and Lead Rice-type of CMS (Itabashi et al. 2011) necessitates inclusion of a wide range of candidate genes other than PPR genes in future studies. The use of sequence of candidate genes in the Rf4 region from IR24 indica BAC clone (AB110443), which is reported to be a strong restorer for WA-CMS with two dominant genes (Gao 1981) has resulted in two polymorphic CG based primers (PPR683 and PPR762). Based on the same BAC clone, Ahmadikhah and Karlov (2006) also identified a set of polymorphic primers co-segregating with fertility restoration of WA-CMS. The functional role of these polymorphic candidate genes in fertility restoration of WA-CMS needs further validation.
Using selective genotyping approach, with 36 and 29 polymorphic primers in KRH2 and DRRH2, only 14 and 12 markers showed a difference >0.25 between the absolute value of the two tails comprising fertile (>90 %) and sterile (0 %) groups. Though the parental polymorphism was clear in most of the cases, the resolution of PCR products determining heterozygosity in the mapping population was very less. Most of the mapping studies for fertility restoration trait followed unidirectional selective genotyping as recessive class analysis (Jing et al. 2001; Bazrkar et al. 2008). Genotyping of individuals from sterile class or both classes (sterile and fertile) with polymorphic markers appears to be a promising strategy for the identification of loci for fertility restoration.
In the present study, two linkage maps were constructed for the two mapping populations accommodating about ~78,000 data points for about ~3,000 individuals and 26 markers using MapDisto (Lorieux 2007). The integration of genetic map and physical map in terms of sequence showed 392.5 kb (from 4,966,407 to 5,358,932 bp) for the Rf3 region on chromosome 1. For KRH2, the region corresponded to 37.7 kb/cM and for DRRH2; it was 33.5 kb/cM. For the Rf4 locus, the combination of the genetic map and sequence information showed 246.7 kb (from 19,157,925 to 19,426,628 bp) and the region corresponded to 19.1 kb/cM in KRH2 and 23.6 kb/cM in DRRH2. Based on sequence information derived mapping, Ngangkham et al. (2010) suggested location of the restorer gene limited to a physical interval of 163.6 kb in the Rf4 region. Thus the fine mapping information and markers generated in the present study can be used for identification of candidate genes in a marker delimited region for fertility restoration as well for MAS.
In both populations, a QTL (qWARF-1-1) with high LOD threshold was detected in the Rf3 locus using QTL Cartographer. In KRH2 population a second QTL (qWARF-1-2) was also identified. The location of the QTL (5,232,060 bp) identified on chromosome 1 in the present study has not been coinciding with any of the reported QTLs on chromosome 1 other than Rf3 and hence, needs further validation (Shen et al. 1998; Ahmadikhah et al. 2007; Ahmadikhah and Alavi 2009; Nematzadeh and Kiani 2010). For the Rf4 region, two QTLs were mapped in both mapping populations. One QTL (qWARF-10-1) spanned two candidate gene markers and the other QTL (qWARF-10-2) encompassed the most reported marker RM6100. The two QTLs are at the reported Rf4 region across the studies (Zhang et al. 1997; Tan et al. 1998; Jing et al. 2001; Bazrkar et al. 2008; Sattari et al. 2008; Ngangkham et al. 2010). The contribution and interaction of the identified QTLs in the present study were analyzed using QTLNetwork software (Yang et al. 2008). A QTL (qWARF-1-1) explaining 31 % of phenotypic variance in KRH2 and 23 % of phenotypic variance in DRRH2 was identified for the Rf3 locus. In the Rf4 region, a QTL (qWARF-10-1) was identified with two candidate gene markers explaining phenotypic variance of 41 % in KRH2 and 43 % in DRRH2. In both mapping populations, the interactions were identified between one QTL of Rf3 (qWARF-1-1) and two QTLs of Rf4 (qWARF-10-1; qWARF-10-2). In rice, QTLNetwork 2.1 was earlier used for analysis of QTLs and their interactions for cold tolerance (Juan et al. 2010). Out of several earlier studies, the effects of Rf3 and Rf4 together were explained only by Sattari et al. (2008) and Sheeba et al. (2009). From the analysis of two QTL identification softwares in two mapping populations, two loci together explained 65–75 % phenotypic variance of the trait suggesting their utility in developing a marker system. However, development of markers for a putative third locus or more QTLs with smaller effects viz. qWARF-1-2 and qWARF-10-2 as identified in the present study is needed for marker system achieving 100 % efficiency.
Sequence analysis of PCR products of DRCG-RF4-8 targeting PPR683 showed a 6 bp deletion in A lines and a 327 bp deletion in R lines in comparison with sequence of IR24 BAC clone (AB110443). Association of the deletion in this gene with fertility restoration of WA-CMS is being reported for the first time. Sequence analysis of PCR products targeting PPR762 and comparison of sequences identified a deletion of 106 bp in R lines. Similar deletion in the same gene was also reported by Ngangkham et al. (2010). Sequence analysis of Rf1a and Rf1b fertility restorer genes of BT cytoplasm showed that substitutions are responsible for the functional variation (Wang et al. 2006); hence the functional significance of these deletions at the PPR genes of the Rf4 locus needs to be deciphered. However for MAS and pyramiding, the structural polymorphism identified at the candidate genes can be used as marker system.
A total of 212 restorers and 34 maintainer lines were assayed for selection efficiency using eight designed markers and three reported markers of fertility restoration in WA-CMS. Marker combination of DRCG-RF4-14/DRCG-RF4-8/TMPPR3 for the Rf4 locus and DRRM-RF3-5/DRRM-RF3-10 for Rf3 showed maximum efficiency of 92 % in identification of restorers. The earlier attempts for MAS have used STS markers (Nas et al. 2003; Sattari et al. 2007) and either Rf3 or Rf4 markers (Nas et al. 2003; Sheeba et al. 2009; Ngangkham et al. 2010), only one study used two STS markers from Rf3 and Rf4 loci with selection accuracy of 100 % in 13 R lines (Sattari et al. 2007). Selection accuracy of 94.9 % in a set of 21 restorer lines with RM6100 from Rf4 was reported by Sheeba et al. (2009). A series of markers based on microsatellites and candidate genes identified for the Rf3 and Rf4 regions in the present study could be useful to identify the restorer lines with 92 % efficiency.
The present study, Rf3 and Rf4 loci were fine mapped, their interaction in fertility restoration of WA-CMS was shown to range from 66 to 72 % of the total phenotypic variance suggesting the involvement of three or more loci and the developed marker system was validated in 212 restorer lines with 92 % efficiency for identification of restorer lines for WA-CMS. However, the candidate genes in Rf3 and Rf4 loci still need to be identified along with the other loci controlling fertility restoration of WA-CMS. Attempts are underway in our laboratory to further fine map these two loci along with identification of other Rf genes controlling fertility restoration of WA-CMS.
References
Ahmadikhah A, Alavi M (2009) A cold-inducible modifier QTL affecting fertility restoration of WA CMS in rice. Int J Genet Mol Biol 1:89–93
Ahmadikhah A, Karlov GI (2006) Molecular mapping of fertility restoration gene Rf4 for WA-cytoplasmic male sterility in rice. Plant Breed 125:363–367
Ahmadikhah A, Karlov GI, Nematzadeh GH, Ghasemi Bezdi K (2007) Inheritance of the fertility restoration and genotyping of rice lines at the restoring fertility (Rf) loci using molecular markers. Int J Plant Prod 1(1):13–21
Allard RW (1956) Formulas and tables to facilitate the calculation of recombination values in heredity. Hilgardia 24:235–278
Bazrkar L, Ali AJ, Babaeian NA, Ebadi AA, Allahgholipour M, Kazemitavar K, Nematzadeh G (2008) Tagging of four fertility restorer loci for wild abortive cytoplasmic male sterility system in rice (Oryza sativa L.) using microsatellite markers. Euphytica 164:669–677
Benson G (1999) Tandem repeat finder: a program to analyze DNA sequences. Nuclic Acids Res 27:573–580
Bharaj TS, Bains SS, Sidhu GS, Gagneja MR (1991) Genetics of fertility restoration of ‘Wild Abortive’ cytoplasmic male sterility in rice (Oryza sativa L.). Euphytica 56:199–203
Fujii S, Toriyama K (2009) Suppressed expression of RETROGRADE-REGULATED MALE STERILITY restores pollen fertility in cytoplasmic male sterile rice plants. Proc Natl Acad Sci USA 106(23):9513–9518
Gao MW (1981) A preliminary analysis of genotype of hybrid hsien rice with wild rice cytoplasm. Acta Genet Sin 8(1):66–74
Govinda Raj K, Virmani SS (1988) Genetics of fertility restoration of WA type cytoplasmic male sterility in rice. Crop Sci 28:787–792
Hanson MR, Bentolila S (2004) Interactions of mitochondrial and nuclear genes that affect male gametophytic development. Plant Cell 16:S154–S169
He GH, Wang WM, Liu GQ, Hou L, Xiao YH, Tang M, Yang ZL, Pei Y (2002) Mapping of two fertility restoring genes for WA cytoplasmic male sterility in Minghui63 using SSR markers. Acta Genet Sin 29(9):798–802
Henke W, Herdel K, Jung K, Schnorr D, Loening SA (1997) Betaine improves the PCR amplification of GC rich DNA sequences. Nucleic Acids Res 25(19):3957–3958
Hossain MD, Singh AK, Zaman F (2010) Genetics of fertility restoration of ‘WA’ based cytoplasmic male sterility system in rice (Oryza sativa L.) using indica/japonica derivative restorers. Sci Asia 36:94–99
Huang ZS (1987) Genetics analysis of indica rice CMS male sterile resloring gene. Zhonghua Nongye Yanjiu 36(2):37–150
Huang QY, He QY, Jing RC, Zhu RS, Zhu YG (2000) Mapping of the nuclear fertility restorer gene for HL cytoplasmic male sterility in rice using microsatellite markers. Chin Sci Bull 45:430–432
IRRI (International Rice Research Institute) (1996) Standard evaluation system for rice, 4th edn. IRRI, Los Banos (Philippines)
Itabashi E, Iwata N, Fujii S, Kazama T, Toriyama K (2011) The fertility restorer gene, Rf2, for Lead Rice-type cytoplasmic male sterility of rice encodes a mitochondrial glycine rich protein. Plant J 65(3):359–367
Jing R, Li X, Yi P, Zhu Y (2001) Mapping of fertility restoring genes of rice WA cytoplasmic male sterility using SSLP markers. Bot Bull Acad Sin 42:167–171
Juan JZ, Yu xiang Z, Dali Z, Liang yong MA, Xi ming LI, Bing xin LIU, Chang deng Y (2010) Identification of QTLs for rice cold tolerance identified at plumule and 3-leaf-seedling stages using QTLNetwork software. Rice Sci 17(4):282–287
Komori T, Ohta S, Murai N, Takakura Y, Kuraya Y, Suzuki S, Hiei Y, Imaseki H, Nitta N (2004) Map-based cloning of a fertility restorer gene, Rf-1, in rice (Oryza sativa L.). Plant J 37:315–325
Kosambi DD (1944) The estimation of map distance from recombination value. Ann Eugen 12:172–175
Kumar S, Chakrabarti SN (1983) Genetic and cytogenetic analysis of spikelet sterility in indica japonica crosses in Oryza sativa L. Indian J Genet Plant Breed 60:441–450
Li JM, Yuan LP (2000) Hybrid rice: genetics, breeding, and seed production. Plant Breed Rev 17:15–158
Liu XQ, Xu X, Tan YP, Li SQ, Hu J, Huang JY, Yang DC, Li YS, Zhu YG (2004) Inheritance and molecular mapping of two fertility restoring loci for Honglian gametophytic cytoplasmic male sterility in rice (Oryza sativa L.). Mol Gen Genomics 271:586–594
Lorieux M (2007) MapDisto: a free user friendly program for computing genetic maps. Computer demonstration given at the Plant and Animal Genome XV conference, Jan 13–17 2007, San Diego, CA. URL: http://mapdisto.free.fr/
Mishra GP, Singh RK, Mohapatra T, Singh AK, Prabhu KV, Zaman FU, Sharma RK (2003) Molecular mapping of a gene for fertility restoration of wild abortive (WA) cytoplasmic male sterility using a basmati rice restorer line. J Plant Biochem Biotechnol 12:37–42
Nandi S, Subudhi PK, Senadhira D, Manigbas NL, Sen Mandi S, Huang N (1997) Mapping QTLs for submergence tolerance in rice by AFLP analysis and selective genotyping. Mol Gen Genet 255:1–8
Nas TMS, Casal CL, Li Z, Virmani SS (2003) Application of molecular markers for identification of restorers. Rice Genet Newsl 20:69–71
Nematzadeh Gh, Kiani G (2010) Genetic analysis of fertility restoration genes for WA type cytoplasmic male sterility in Iranian restorer rice line DN-33-18. African J Biotechnology 9(38):6273–6277
Ngangkham U, De Sandip, Singh SK, Parida SK, Raj Kumar KA, Singh AK, Mohapatra T (2010) Genic markers for wild abortive (WA) cytoplasm based male sterility and its fertility restoration in rice. Mol Breed 26(2):275–292
Sarkar CKG, Fu Z, Singh AK (2002) Genetics of fertility restoration of WA based cytoplasmic male sterility system in rice (Oryza sativa L.) using basmati restorer lines. Indian. J Genet 62(4):305–308
Sattari M, Kathiresan A, Gregorio G, Hernandez JE, Nas TM, Virmani SS (2007) Development and use of a two-gene marker-aided selection system for fertility restorer genes in rice. Euphytica 153:35–42
Sattari M, Kathiresan A, Gregorio G, Virmani SS (2008) Comparative genetic analysis and molecular mapping of fertility restoration genes for WA Dissi, and Gambiaca cytoplasmic male sterility systems in rice. Euphytica 160:305–315
Sharma VK, Singh VP, Singh AK, Zaman FU (2001) Inheritance pattern of spikelet fertility restoration in hybrid rice. Indian J Genet 61(2):160–161
Sheeba NK, Viraktamath BC, Sivaramakrishnan S, Gangashetti MG, Pawan K, Sundaram RM (2009) Validation of molecular markers linked to fertility restorer gene(s) for WA-CMS lines of rice. Euphytica 167:217–227
Shen Y, Cai Q, Gao M, Wang X (1996) Isolation and genetic characterization of a fertility restoring revertant induced from cytoplasmic male sterile rice. Euphytica 90:17–23
Shen Y, Guang ZQ, Lu J, Zhuang LY, Zheng KL, Gao MW, Wang XM (1998) Linkage analysis of a fertility restoring mutant generated form CMS rice. Theor Appl Genet 97:261–266
Shinjyo C, Sato S (1994) Chromosomal location of fertility-restoring gene Rf2. Rice Genet Newsl 11:93–95
Singh AK, Mahapatra T, Prabhu KV, Singh VP, Zaman FU, Mishra GP, Nandakumar N, Joseph M, Gopalakrishnan S, Aparajita G, Tyagi NK, Prakash P, Sharma RK, Shab US, Singh SK (2005) Application of molecular markers in rice breeding: progress at IARI. Advances in marker assisted selection workshop. Trainee’s manual, Handouts and references
Sohu VS, Phul PS (1995) Inheritance of fertility restoration of three sources of cytoplasmic male sterility in rice. J Genet Breed 49:93–96
Tan XL, Vanavichit A, Amornsilpa S, Trangoonrung S (1998) Genetic analysis of rice CMS-WA fertility restoration based on QTL mapping. Theor Appl Genet 96:994–999
Virmani SS, Viraktamath BC, Casa CL, Toledo RS, Lopez MT, Manalo JO (1997) Hybrid rice breeding manual. IRRI, Philippines, p 139
Virmani SS, Mao CX, Hardy B (eds) (2003) Hybrid rice for food security, poverty alleviation, and environmental protection. Proceedings of the 4th international symposium on hybrid rice, Hanoi, Vietnam, 14–17 May 2002. Los Banos (Philippines): IRRI, 407 pp
Waghmode BD, Mehta1 HD (2011) Genetics of fertility restoration of diverse cytosterile sources in rice (Oryza sativa L.) Indian J Genet 71(1):1–8
Wang Z, Zou Y, Li X, Zhang Q, Chen L, Wu H, Su D, Chen Y, Guo J, Luo D, Long Y, Zhong Y, Liu YG (2006) Cytoplasmic male sterility of rice with Boro II cytoplasm is caused by a cytotoxic peptide and is restored by two related PPR motif genes via distinct modes of mRNA silencing. Plant Cell 18:676–687
Wang SC, Basten J, Zeng ZB (2010) Windows QTL Cartographer 2.5. Department of Statistics. North Carolina State University, Raleigh, NC. http://statgen.ncsu.edu/qtlcart/WQTL.Cart.htm
Yang J, Hu Ch, Hu H, Rongdong Yu, Xia Zhen, Ye Xiuzi, Zhu Jun (2008) QTLNetwork: mapping and visualizing genetic architecture of complex traits in experimental populations. Bioinformatics 24(5):721–723
Yao FY, Xu CG, Yu SB, Li JX, Gao YJ, Li XH, Zhang Q (1997) Mapping and genetic analysis of two fertility restorer loci in the wild abortive cytoplasmic male sterility system of rice (Oryza sativa L.). Euphytica 98:183–187
Zhang Q, Bharaj TS, Virmani SS, Huang H (1997) Mapping of the Rf3 nuclear fertility restoring gene for WA cytoplasmic male sterility in rice using RAPD and RFLP markers. Theor Appl Genet 94:27–33
Zhang QY, Liu YG, Mei MT (2002) Molecular mapping of the fertility restorer gene Rf4 for WA cytoplasmic male sterility. Acta Genet Sin 29:1001–1004
Zheng KL, Subudhi PK, Dmingo J, Magpantag G, Huang N (1995) Rapid DNA isolation for marker assisted selection in rice breeding. Rice Genet Newsl 12:255–257
Zhuang JY, Fan YY, Wu JL, Xia YW, Zheng KL (2000) Mapping major and minor QTL for rice CMS-WA fertility restoration. Rice Genet Newsl 17:56–58
Acknowledgments
The authors thank Department of Science and Technology, Government of India for the research grant and the two reviewers for suggestions and improvements.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Balaji Suresh, P., Srikanth, B., Hemanth Kishore, V. et al. Fine mapping of Rf3 and Rf4 fertility restorer loci of WA-CMS of rice (Oryza sativa L.) and validation of the developed marker system for identification of restorer lines. Euphytica 187, 421–435 (2012). https://doi.org/10.1007/s10681-012-0737-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-012-0737-6