

Abstract
The interaction of myosin with actin filaments is the central feature of muscle contraction1 and cargo movement along actin filaments of the cytoskeleton2. The energy for these movements is generated during a complex mechanochemical reaction cycle3,4. Crystal structures of myosin in different states have provided important structural insights into the myosin motor cycle when myosin is detached from F-actin5,6,7. The difficulty of obtaining diffracting crystals, however, has prevented structure determination by crystallography of actomyosin complexes. Thus, although structural models exist of F-actin in complex with various myosins8,9,10,11, a high-resolution structure of the F-actin–myosin complex is missing. Here, using electron cryomicroscopy, we present the structure of a human rigor actomyosin complex at an average resolution of 3.9 Å. The structure reveals details of the actomyosin interface, which is mainly stabilized by hydrophobic interactions. The negatively charged amino (N) terminus of actin interacts with a conserved basic motif in loop 2 of myosin, promoting cleft closure in myosin. Surprisingly, the overall structure of myosin is similar to rigor-like myosin structures in the absence of F-actin, indicating that F-actin binding induces only minimal conformational changes in myosin. A comparison with pre-powerstroke and intermediate (Pi-release)7 states of myosin allows us to discuss the general mechanism of myosin binding to F-actin. Our results serve as a strong foundation for the molecular understanding of cytoskeletal diseases, such as autosomal dominant hearing loss and diseases affecting skeletal and cardiac muscles, in particular nemaline myopathy and hypertrophic cardiomyopathy.
Similar content being viewed by others
Main
Using electron cryomicroscopy (cryo-EM) and single-particle-based analysis of helical specimens (Methods), we determined the structure a human actomyosin–tropomyosin (ATM) complex, composed of the motor domain of non-muscular myosin-2C (NM-2C), cytoplasmic γ1-F-actin and cytoplasmic tropomyosin 3.1 (Fig. 1, Extended Data Figs 1a–g and 2 and Supplementary Video 1). We also reprocessed our previous F-actin–tropomyosin data set12 and obtained an improved reconstruction at 3.6 Å resolution (Extended Data Figs 1h–k and 2a). The density of tropomyosin did not improve in both data sets and is limited to ~7 Å as described previously12.

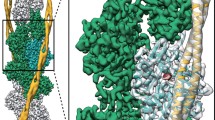
a, Cryo-EM reconstruction of F-actin (five central subunits in green and cyan) decorated with tropomyosin (blue) and myosin (central molecules in red). The peripheral densities (shown in grey) and tropomyosin were low-pass filtered and symmetrized for better visualization. b, Subdomain organization of F-actin and myosin head region. The closed actin-binding cleft between L50 (red) and U50 (dark red) domains is indicated with a dotted line. c, d, Front and back views of the F-actin–myosin interface. Involved structural parts of myosin are highlighted in red. For all figures and videos, we use a general colour code for each protein and state, if not labelled differently. The central F-actin subunit is shown in cyan (M-state) and yellow (A-state); surrounding F-actin subunits are depicted in green (M-state) and in darker yellow (A-state); rigor state myosin (red), Pi-release state myosin (blue) and PPS state myosin (purple). Less relevant parts of models or densities in respective figures are depicted in grey or faded out.
The ATM structure reveals that myosin interacts intimately with F-actin (Fig. 1a). The overall organization of the ATM complex is similar to that described in our previous structure of the complex between Dictyostelium discoideum myosin-IE, skeletal α-F-actin and α-tropomyosin10 (Supplementary Videos 2 and 3). However, given the superior resolution of our present structure, we could clearly identify large side chains and therefore reveal most intermolecular interactions between F-actin and myosin in detail.
The large interface between the two proteins is formed mainly between the helix–loop–helix (HLH) motif and surface loops of myosin (CM-loop, loop 2, loop 3, loop 4, and ‘activation’ loop13) and the subdomain (SD) 1 and 2 of one actin subunit and SD2 (D-loop) of the adjacent actin subunit as previously predicted9,10,11 (Fig. 1b–d and Supplementary Video 3).
Tpm3.1, which is resolved to ~7 Å, is in the same position as skeletal muscle tropomyosin 1.1 (Tpm1.1) in our previous structure, namely the M-state10, interacting with loop 4 of myosin and SD3 of actin (Figs 1b, c and 2a). Interestingly, although Tpm3.1 is shorter (stretching over six actin subunits) than Tpm1.1 (stretching over seven actin subunits), the pitch of the coiled-coil structure is equivalent. Tpm3.1, which is mainly negatively charged on its surface, interacts with arginine 384 of loop 4, indicating electrostatic interactions (Fig. 2a). In addition, the negatively charged residue D387 on loop 4 (N377–D393) interacts with a positively charged region on F-actin (K325, K327) (Fig. 2b). Notably, in the absence of myosin, these actin residues directly interact with tropomyosin (A-state)12.

a, b, Interface of loop 4 with tropomyosin (blue) and SD3 of one F-actin subunit (cyan). c, d, Interaction of the HLH motif of myosin (red) with hydrophobic groove formed by the D-loop of one F-actin subunit (green ribbon in c and as surface in d) and SD1 and SD3 of the adjacent subunit (surfaces depicted by low (white) to high (yellow) hydrophobicity). Hydrophobic residues at the interface (c, d) and a possible electrostatic interaction (dotted lines (c)) are highlighted. e, f, The CM-loop (red) binds to a region formed by SD1 and SD3 of F-actin. Charged (e) and hydrophobic (f) residues of myosin. g, Loop 3 (red) interacts with SD1 of an adjacent F-actin subunit forming the Milligan contact9,21. The F-actin surface is coloured either by hydrophobicity (f) or electrostatic Coulomb potential from −10 kcal mol−1 (red) to +10 kcal mol−1 (blue) (e, g). In all panels, coloured residue labels depict F-actin residues.
The HLH motif (L550–E575) in the lower 50-kDa (L50) domain of NM-2C plays an essential role in strong binding of myosin to F-actin (Figs 1c, d and 2c, d). It enters a hydrophobic groove on actin that is formed between two adjacent actin subunits comprising SD1 and SD3 of one and the D-loop (R38–V53) of the adjacent subunit. In particular, the hydrophobic loop of the HLH motif interacts with the hydrophobic groove and F560 is completely immersed into a hydrophobic cavity resembling a lock-and-key interaction (Fig. 2d, Extended Data Fig. 3a–d and Supplementary Video 4). The key role of F560 for the actomyosin interaction has also been shown by mutational analyses in which a F560A mutation resulted in a complete disruption of motility, whereas alanine mutants of the directly adjacent W559 and P561 showed only one-tenth the motility compared with wild type14. Interestingly, compared with its position in the pre-powerstroke (PPS) state the loop is the only part of the HLH motif that alters its position upon actin binding, stressing its important role in the actin–myosin interaction (Extended Data Fig. 3a, c).
In addition to the hydrophobic contacts, there are also two electrostatic interactions that stabilize the HLH motif binding to F-actin. E570 probably forms a salt bridge with K49 of the D-loop and E556 interacts with the backbone of S349 and T350 in SD1 of F-actin (Fig. 2c, Extended Data Fig. 3b, e and Supplementary Video 4). Both residues are part of a highly conserved acidic patch in several myosin classes (Extended Data Fig. 3e, f), and a single point mutation (E556Q) in myosin results in a tenfold reduced F-actin binding affinity15.
The cardiomyopathy loop (CM-loop), forming one antiparallel β-strand pair (T417–T432), is the major site of the myosin upper 50-kDa (U50) domain that interacts with actin (Figs 1c and 2e, f and Supplementary Video 4). The CM-loop is fully ordered and the interface with actin is mainly stabilized by hydrophobic interactions (Fig. 2f), supported by weak electrostatic interactions at the tip and the base of the CM-loop (Fig. 2e). K429, which is found in all myosin-II isoforms (Extended Data Fig. 4a, b), interacts with a negatively charged patch on F-actin formed mainly by D24 and E333 in SD1 and SD3, respectively (Fig. 2e). In addition, the positively charged tip (R424) interacts with the negatively charged region around E55 and D92 of F-actin (Fig. 2e). However, residue 424 is only positively charged in smooth and non-muscular isoforms of myosin (Extended Data Fig. 4b) and therefore does not play a role in skeletal and cardiac muscles. Importantly, we did not find any prominent possible salt bridges that would stabilize the contacts between the CM-loop and F-actin, supporting previous mutagenesis studies suggesting that charged residues, in particular the highly conserved residue D425, play a minor role in this interface14,16.
As speculated previously8, the highly conserved and disease-related residue R419 (R403 in β-cardiac myosin) indeed does not directly interact with F-actin (Fig. 2e, Extended Data Fig. 4b–d and Supplementary Video 4). In our structure, R419 clearly interacts with Y426 on the opposing strand of the CM-loop, thereby bridging and stabilizing the conformation of the loop (Fig. 2e). Both residues are highly conserved, suggesting that this bridge is present in all myosin-II isoforms (Extended Data Fig. 4b). Many disease-causing mutations are found in the region of the CM-loop, demonstrating the high importance of the CM-loop for the strong binding between actin and myosin (Extended Data Fig. 4b–e).
Several studies showed that loop 2, connecting the L50 and U50 domains in myosin (W638–T669), plays a major role in the initial binding to F-actin14,15,17,18,19. We see clear density for loop 2 in the ATM structure. However, whereas the base of the loop is ordered, the rest of the loop is more flexible (Fig. 3a, b). It occupies a large predominantly hydrophobic surface of the actin SD1 domain (Fig. 3c and Extended Data Fig. 5a, b).

a, Myosin densities of lower resolution (grey) are shown together with more highly resolved regions of myosin (red) and F-actin (cyan). The densities can be assigned to more flexible parts of loop 1, loop 2 and the outer lever arm (Methods), respectively. b, Close-up view onto region of the density map corresponding to loop 2. Loop 3 of the adjacent myosin does not interact with loop 2 (red). c, d, Interaction of the stabilized base of loop 2 with SD1 of F-actin (coloured by hydrophobicity (c) or by electrostatic Coulomb potential (d)). The flexible part of loop 2 and possible electrostatic interactions are indicated by different dotted lines.
A conserved hydrophobic patch at the carboxy (C)-terminal base of loop 2 (G665–F667) and the tip of helix-R (W559) (Fig. 3c, Extended Data Fig. 3f and Supplementary Video 4) interacts with a hydrophobic groove of actin SD1 (Fig. 3c and Extended Data Fig. 3d). Mutagenesis studies showed that especially W559 is essential for forming the F-actin–myosin interface and obtaining motility14,20. Compared with their position in the PPS state, these residues orient towards actin to stabilize the newly formed interface (Fig. 4a).

a, Involved residues of loop 2 and helix-R undergo changes in rotamer orientation during base stabilization of loop 2 (PPS state (purple), PDB accession number 5I4E; rigor state (red)). The F-actin surface is shown in grey. Inset shows a top view of rotating residues of loop 2. F667 rotates to SD1, R668 rotates outwards to the U50 of myosin. b, Density comparison of the N terminus between myosin-unbound F-actin (yellow, A-state) and myosin-bound F-actin (cyan, M-state) illustrates the pulled conformation of the terminus induced by loop 2 interaction. The flexible N terminus (1–4 in α-actin) in the A-state is depicted as dotted lines. c, Superposition of SD1 and SD2 of F-actin in the A-state (yellow) and M-state (cyan) visualizing the myosin-induced changes in F-actin (indicated by arrows).
Interestingly, the adjacent conserved positively charged region (R661–R664) interacts with the acidic N terminus of actin and a negatively charged area (D23, D24) of SD1 (Fig. 3d, Extended Data Fig. 5b and Supplementary Video 4). While one of the conserved arginines (R663) forms a possible salt bridge with actin E3 (Fig. 3d), the other one (R664) interacts with D640 and D642 at the other end of loop 2, forming an electrostatic belt that stabilizes the base of loop 2 (Fig. 3c). Notably, D640 and D642 are only conserved in myosins with a long loop 2 (Extended Data Fig. 5c), suggesting that the electrostatic belt is not required for myosins with a shorter loop 2.
In line with our previous observation10, loop 3 (Q576–D593) forms the so-called Milligan-contact9,21 connecting the L50 domain to SD1 of the adjacent actin subunit (Figs 1c and 2g and Extended Data Fig. 5d, e). The small interface is only formed by complementary charged surfaces and not by specific salt bridges as previously expected (Fig. 2g). The weak nature of the interactions, and the fact that not all myosin isoforms have a long loop 3 that can form this contact22, suggest that, for most myosin proteins, loop 3 plays only an ancillary role in strong F-actin binding.
Several studies suggested that a proline-rich loop in the L50 domain, a so-called activation loop (I541–G549), is directly involved in activation of myosin by interacting with the negatively charged N terminus of actin13,23. Our ATM structure confirms that this loop is part of the actomyosin interface. Together with helix-W and the base of loop 2, it forms a positively charged basin that interacts with the negatively charged N terminus of actin (Fig. 3d, Extended Data Fig. 6a, b and Supplementary Video 4). However, R543, which is the only positively charged residue of the loop and conserved in myosin-II (Extended Data Fig. 6c), points away from the interface and therefore cannot be involved in a direct interaction with the glutamates of the actin N terminus (Fig. 4b and Extended Data Fig. 6d). In other myosins with shorter activation loops, namely myosin-V, myosin-VI or Dictyostelium myosin-II, the position of the positively charged residue is shifted by one position and would therefore allow a direct interaction with the N terminus (Extended Data Fig. 6c, e, f). Compared with the PPS conformation, the proline-rich loop orients slightly closer to F-actin (Extended Data Fig. 6b). Since the actin-induced conformational changes in the proline-rich loop and the adjacent relay helix are minor, we conclude that they are probably not responsible for a direct activation of myosin and therefore suggest using the term supporting loop rather than activation loop.
To identify myosin-induced conformational changes in F-actin, we compared the ATM structure with our reprocessed F-actin–tropomyosin structure. As expected from our previous observations10, the overall changes are minimal. Whereas the D-loop and other interface regions orient slightly towards myosin (Fig. 4c and Extended Data Fig. 3a), areas interacting with the CM-loop and more distal regions of actin only move marginally away from the interface (Extended Data Fig. 7a–c).
The most prominent changes occur at the N and C termini of actin (Fig. 4b, c and Extended Data Fig. 7d–k). The highly conserved and negatively charged N terminus (Extended Data Fig. 7c), which is only partly resolved in F-actin12, but essential for myosin binding24,25,26, is completely ordered in the ATM structure and pulled into a positively charged basin on the myosin structure (Fig. 4b, c and Extended Data Fig. 6a). Two glutamates at its tip (E2, E3) are potentially involved in salt bridges with positively charged residues on helix-W and loop 2 (Fig. 3d). The conformational change of the N terminus is partly transmitted to the nucleotide-binding pocket (Extended Data Fig. 7e, f). Actin residues D10 and K17 slightly change their position (Extended Data Fig. 7f, g); however, this does not considerably alter the position of ADP and Mg2+ (Extended Data Fig. 7f, h).
Interestingly, although the actin C terminus does not participate in forming the actomyosin interface, it is completely ordered in the ATM structure and orients towards the SD1 domain (Fig. 4c and Extended Data Fig. 7i–k). This is in line with previous studies showing that myosin binding to F-actin results in quenching of fluorescence of pyrene-labelled actin at C373 (C374 in α-actin)14. The higher myosin-induced stability of the D-loop, N terminus and surrounding regions is probably responsible for the stabilization of the C terminus, which is not well ordered in the F-actin–tropomyosin structure12 (Extended Data Fig. 7j). We believe that the minimal but substantial myosin-induced conformational changes are exemplary for most actin–myosin interactions. We therefore suggest using the term M-state for F-actin bound to myosin in contrast to A-state for bare F-actin (Figs 1a and 4c).
To understand which conformational changes F-actin binding induces in myosin, we compared our rigor ATM structure with rigor-like crystal structures (Extended Data Fig. 8). Surprisingly, the overall structures of the different rigor-like states are very similar compared with that of the rigor state (Extended Data Fig. 8a, b), indicating that F-actin induces only minimal conformational changes in myosin as previously predicted11,27. Differences are found at actin-interacting loops that are partly ordered in the crystal structures and are stabilized by actin in the ATM structure (Extended Data Fig. 8c). In addition, we found that the converter and lever arm regions differ in their position relative to the rest of the protein (Extended Data Fig. 8d). Importantly, the comparison between rigor and rigor-like structures shows that F-actin stabilizes the closed conformation of myosin, but does not induce major additional conformational changes.
To gain further insight into how actin accommodates first a weak and then a strong myosin-binding state, we compared our rigor NM-2C structure with the crystal structure of the same protein in the PPS state. On the basis of recently determined crystal structures of the motor domain of myosin-VI in the Pi-release state, an important intermediate state, a detailed mechanism of myosin binding to F-actin has been proposed7. To obtain the best possible approximation for an F-actin–NM-2C complex in the Pi-release state, we used a two-step approach. First, we calculated a homology model of the Pi-release state of NM-2C on the basis of the atomic model of myosin-VI (ref. 7). On the basis of the positions of actin and myosin in our rigor actomyosin structure and the myosin PPS structure, we then performed different alignments of the Pi-release state model to obtain a model for the actomyosin complex in its Pi-release state (Fig. 5 and Extended Data Fig. 9a–c). Exclusive alignment to the U50 domain in all states would cause steric clashes with F-actin in the Pi-release state (Extended Data Fig. 9a). Alignment to the L50 domain would result in a rotation of the U50 away from F-actin before the final strong binding (Extended Data Fig. 9b). We therefore chose a combined alignment instead (Extended Data Fig. 9c and Supplementary Video 5) to describe the possible global conformational changes during myosin binding (Extended Data Fig. 9d–f, Fig. 5 and Supplementary Video 6). We aligned the Pi-release state model first to the L50 domain of the rigor state. The PPS state was then aligned to the U50 domain of the Pi-release state.

a–c, Cartoon representation of the myosin–F-actin-binding mechanism. Numbers refer to different steps of the actomyosin interaction and are directly described in the lower panel. U50 and L50 domains are coloured according to their respective myosin state: PPS (purple, PDB accession number 5I4E), Pi-release (blue, homology model of PDB accession number 4PFO (ref. 7)) and rigor state (red). Other domains are coloured as in Fig. 1b. F-actin subunit colours give their current state (A-state, yellow; M-state, cyan and green). Loop 2 (grey) and strut (dark blue) are shown as lines. Flexibility in loop 2 is indicated by dotted lines. Arrows highlight rotations and binding of domains.
There is general agreement that loop 2 initiates weak binding of myosin-ADP-Pi to F-actin by interacting with the SD1 and SD3 of one actin subunit14,15,17,18,19 (Extended Data Fig. 9d). This brings both the L50 and U50 close to F-actin (Fig. 5a). As proposed in ref. 7, the L50 domain rotates and binds to actin, resulting in the Pi-release state that represents the initial strong binding state of myosin. The interface is mainly mediated by hydrophobic interactions of the HLH domain with SD1, SD3 and the D-loop and hydrophilic interactions with the N terminus of actin, in line with our previous prediction10 (Fig. 5b). On the basis of our results, we propose that this process stabilizes the base of loop 2 (Extended Data Fig. 9e, Fig. 5b and Supplementary Video 6), creating a positively charged patch to which the negatively charged strut (bridge between L50 and U50) is attracted (Extended Data Fig. 9f and Fig. 5b). Thus, the base of loop 2 acts as a key region that shifts the equilibrium between the open and closed actin-binding cleft28 towards the closed conformation, in which F-actin directly interacts with the CM-loop and loop 4 of the U50 domain (Fig. 5c). Notably, both the strut and the highly conserved basic region at the base of loop 2 have previously been shown to be essential for strong binding17,27,29. In our model, the closure of the cleft is mediated by a rotation of the U50 domain towards F-actin and not by a back-rotation of the L50 domain (Fig. 5c).
As described before (reviewed in ref. 4), cleft closure results in the strong binding of myosin to actin, providing the necessary anchoring of the motor domain for the subsequent powerstroke (Fig. 5c). The time point and effect of Pi and ADP release during this process is highly debated7,30. Because we lack high-resolution structures of intermediate states of myosin bound to F-actin, we cannot determine whether Pi is released before or after the powerstroke. However, our observation that the rigor state is very similar to the rigor-like state suggests that actin promotes and stabilizes the closed conformation of myosin, ultimately resulting in the release of phosphate and ADP.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Protein expression and purification
G-actin (γ1-actin, ACTG1 from Homo sapiens) was recombinantly expressed using the baculovirus/Sf9-cell system and purified as described previously31,32. Afterwards, the sample was polymerized to F-actin by increasing the salt concentration to 100 mM KCl and 2 mM MgCl2. Tropomyosin 3.1 (TPM3 from H. sapiens, isoform Tpm3.1) was expressed and purified from Escherichia coli on the basis of the protocol of ref. 33 with no additional modifications. The motor domain of non-muscular myosin-2C (MYH14, isoform 2 from H. sapiens) consisting of amino acids 1–799 was directly fused to an artificial lever arm (spectrin repeats 1 and 2 from α-actinin)34 and a C-terminal Flag-tag. The protein was recombinantly overproduced in the Sf9/baculovirus system as previously described34 and purified via Flag capture and size-exclusion chromatography on a Superdex 26/60–200 prep grade column. Before grid preparation for electron microscopy studies, the F-actin sample was spun down (100,000g) and carefully suspended in nucleotide-free F-actin buffer (5 mM Tris-HCl pH 7.5, 1 mM DTT, 100 mM KCl, and 2 mM MgCl2). The actomyosin complex was prepared by mixing F-actin with tropomyosin initially at a molar ratio of 7:1. The final concentration of tropomyosin for frozen specimens was then adjusted empirically to obtain complete decoration and only little unbound tropomyosin in the background by standard negatively stained studies as described previously12. The F-actin–tropomyosin filaments were decorated with myosin during preparation of vitrified sample grids (see below).
Optimized grid preparation and image acquisition for cryo-EM
Best conditions for reconstituting the F-actin–tropomyosin complex were screened by using the negatively staining protocol as described above. When myosin was incubated in solution with the optimized F-actin–tropomyosin sample before applying the sample to the grid, we always obtained only bundles of fully myosin-decorated actin filaments. Because of this, we optimized the protocol to reconstitute the full actomyosin filaments with reduced bundling on a grid. Therefore, the normal cryo-preparation protocol was changed. First we applied 2 μl of F-actin–tropomyosin solution to a glow-discharged holey carbon grid (C-flats 2/1, Protochips), incubated for 20 s and manually blotted from the backside for less than a second with filter paper. A thin layer of solution stayed on the grid and the filaments where pre-straightened in the holes. Afterwards, 1.5 μl of myosin solution (3 μM in F-actin buffer without nucleotide) were added directly on the grid, incubated for 10 s and then manually blotted for 5 s from the backside with filter paper (Whatman no. 5), before vitrification by plunging the grid into liquid ethane using a Cp3 plunger (Gatan).
Screening for the best sample and blotting conditions was performed on a JEOL JEM 3200FSC electron microscope equipped with a field emission gun and operated at a voltage of 200 kV. The omega in-column energy filter of the microscope was used to estimate best ice conditions (~70–100 nm thickness). Finally, a data set was taken with a spherical aberration-corrected FEI Titan Krios transmission electron microscope equipped with an extra-high brightness field emission gun (X-FEG) and operated at a voltage of 300 kV. Although the sample preparation protocol was optimized, we had to screen and choose usable grid squares extensively. Images were recorded with a back-thinned 4k × 4k FEI Falcon 2 direct detection camera under minimal dose conditions using the automatic data collection software EPU (FEI). Within each selected grid hole, three different positions were imaged, each with a total exposure of 1 s and a frame recording time of 55 ms. Seven frames from 85 to 475 ms with a total dose of ~16 electrons per square ångström and one total average (integrated image) with an electron dose of ~35 electrons per square ångström were used for image processing. The used magnification of 125,000 (nominal magnification of 59,000) corresponds to a pixel size of 1.1 Å. The defocus range of the data set was 0.7–2.8 μm (Extended Data Table 1).
Image processing of the cryo-EM data set
In total, we collected ~6,300 images in two sessions. Despite the extensive pre-screening of grid squares before starting automatic data collection (see above), we deleted ~68% of the recorded images because of bundled filaments, contaminations or bad ice quality. Resulting frames were aligned and afterwards summed up using motion correction35. The drift-corrected averages were used for determination of defocus and astigmatism values with CTFFIND336. Filaments were manually selected (Extended Data Fig. 1a) and exported from the 1 s integrated images using sxhelixboxer in SPARX37 without changing the orientation of the filaments to the y axis. A total of ~138,000 segments were extracted with a box size of 256 pixels and a boxing distance of 29 pixels (overlap ~90%). Thus, the approximate distance between them (~32 Å) slightly exceeded the rise of the helical assembly of actin (~27–28 Å). The same procedure was applied to the drift-corrected average and all individual frames. Afterwards all segments were transformed to RELION38-readable image stack formats and initial metadata files were created for further refinement steps in RELION.
First, two-dimensional reference-free classification and sorting of bad classes from the integrated images led to a resulting data set of 126,000 particles (Extended Data Fig. 1b). The resulting data set was used in several rounds of three-dimensional auto-refinements with particles from the integrated images and local three-dimensional auto-refinements with the particles from the drift-corrected averages. The refinement showed an expected Gaussian distribution of projection direction around the filament axis (Extended Data Fig. 1d, e). To improve processing time, we limited the tilt angle afterwards (Extended Data Fig. 1d). Finally, we applied a particle-based movie refinement and frame weighting39 and continued three-dimensional auto-refinements with the resulting contrast-enhanced particles. We did not make use of helical symmetry during refinement but masked F-actin and myosin in the outer regions to focus the refinement on the central parts. Initially, we applied a standard spherical mask (diameter 270 Å, Extended Data Fig. 1d) to the reconstruction. After a global refinement with the spherical mask, we continued with local refinement and a mask at the size of seven F-actin subunits and six myosin molecules. We used a preliminary model and the ‘Colour zone’ and ‘Split Map’ options in CHIMERA40,41 to extract the central part of the map from the current density map. From this part of the map we calculated a smoothed mask with the ‘relion_mask_create’ function in RELION. To evaluate the results, we again performed a two-dimensional classification with projection parameters derived from three-dimensional refinement (Extended Data Fig. 1c). In single class averages, we could already detect secondary structure elements (Extended Data Fig. 1f, g). As a final sorting step, we deleted particles, which were outliers with respect to their neighbouring segments coming from the same filament. Therefore, we kept track throughout the whole processing steps to know to which filament each particle belongs. This resulted in 118,000 particles, which we used for a local three-dimensional auto-refinement. In the final iteration, we applied a mask of the central five F-actin subunits and two central myosin molecules (coloured region in Fig. 1a). This mask was created as described above for the mask of seven F-actin subunits and six myosin molecules. Outer regions of the lever arm were also excluded.
Fourier shell correlation (FSC) analysis was performed within the central area (five F-actin subunits and two myosin molecules) of the volume, resulting in an average resolution of 3.9 Å (FSC0.143 criterion42) for the F-actin–myosin electron density map (Extended Data Fig. 2a). The density map of F-actin–myosin was then sharpened using a negative b factor of –200 Å2 and filtered to its nominal resolution. Because tropomyosin was masked out during refinements as we did before12, we filtered the tropomyosin density map to ~7 Å and merged it with the final F-actin–myosin map to obtain a map of the entire F-actin–myosin–tropomyosin complex.
Local resolution was estimated using ResMap43 on the full density map without masking (Extended Data Fig. 2b), revealing a resolution gradient from higher (~3.5 Å, F-actin core) to lower (4.5 Å - 5 Å, outer myosin domains) values, which could be a result of induced forces from the protruded lever arm (Extended Data Fig. 2c–e). We converted the local resolution map to absolute frequencies and applied a local filtering algorithm on the final map with sxfilterlocal in SPARX. To estimate and confirm the helical symmetry of the actomyosin complex, we used the symmetry search function of sxhelicon_utils in SPARX.
We reprocessed our previous F-actin–tropomyosin12 data set with the same protocol as described above (Extended Data Fig. 1h–k) and obtained an improved reconstruction at an average resolution of 3.6 Å for the central five F-actin subunits (Extended Data Fig. 2a). During the helical reconstruction approach, several asymmetrical units are averaged by symmetrization. This results in a decrease of resolution in flexible and bent regions. However, using the single particle approach, we refine only on the central region, thereby probably decreasing the influence of flexibility induced by the bending of the filament. This probably improves the resolution of the resulting reconstruction.
For each data set, we provide two density maps in the Electron Microscopy Data Bank (EMDB) databank. One entry contains the map after ‘post-processing’ in RELION without masking, the other one the same map locally filtered, masked and merged with the filtered tropomyosin density map at its respective position (as described above).
Atomic model building and refinement
The central F-actin subunit (chain A) and the central myosin molecules (chain F, G) show all available contacts to adjacent chains (B, C, D, E) and were therefore used for further structural analysis. We used our previous F-actin model12 as a starting model for F-actin and the highly homologous model of rigor-like NM-2B (PDB accession number 4PD344) for myosin. We performed homology modelling with the respective sequences of both proteins using MODELLER45. Derived models were rigid-body fitted into the density map, using ‘Fit in Map’ and the map was segmented with ‘Split Map’ in CHIMERA40,41. Additionally, models were flexibly fitted with iMODFIT46 into the respective density maps. Finally, the flexibly fitted models were used to create a starting F-actin–myosin model. The electron density was converted to structure factors with the CCP4 program suite47. The model and the density map were then used for real space refinement and model building in COOT48. Flexible parts of the N terminus (residues 1–49), loop 1 (residues 222–232), loop 2 (residues 640–661) and lever arm (residues 817–1039) of myosin were deleted from the model. Some parts (especially surface loops and termini) of F-actin and myosin were built de novo. Domains showing resolution lower than 4.5 Å (outer myosin regions) were not manually changed and only the backbone trace was optimized.
The resulting model was refined in REFMAC, using the modified version of the program for cryo-EM maps49 and applying secondary structure and reference restraints derived by ProSMART50. In addition, we added non-crystallographic symmetry restraints for F-actin (chain A–E) and myosin (chain F and G). To prevent overfitting, we first determined refinement settings by refining the model (atoms randomly displaced by 0.5 Å) only versus a density map belonging to one half of the data set and compared the FSC curves of the refined model with both half maps (Extended Data Fig. 2f). Finally, the model was refined versus the full map with the derived refinement parameters, which did not show significant differences in the two FSC curves (Extended Data Fig. 2f). We used MOLPROBITY51 to evaluate the resulting atomic model. The data statistics are given in Extended Data Table 1.
For the reprocessed F-actin–tropomyosin data set, we used the same refinement strategies as for the actomyosin data set. The final data statistics are given in Extended Data Table 1.
Fit of the tropomyosin models
To describe the interaction between F-actin–myosin and tropomyosin, we used the tropomyosin model from our previous structure10. As already described in the main text, the pitch of the coiled-coil structure is equivalent. We directly rigid-body fitted the tropomyosin model (PDB accession number 4A7F) into the density map, using ‘Fit in Map’ in CHIMERA40. We reduced the model to the five central pseudo-repeats as before12 and shifted the residue numbering regarding the differences from long Tpm1.1 to the shorter Tpm3.1. Owing to the limited resolution of the cryo-EM density in the region of tropomyosin, we avoided interpretation of tropomyosin at the single amino-acid level.
For the reprocessed F-actin–tropomyosin model, we could use the tropomyosin model from our previous model (PDB accession number 3JA8), as the structure and resolution in that region did not differ.
Structure analysis and visualization
For visualization of models and density maps in all figures and videos, we used CHIMERA40. The actomyosin complex was protonated using H++ (ref. 52) at pH 7.5, and the electrostatic Coulomb potential of the filament surface was calculated ranging from −10 to +10 kcal mol−1. For visualization of the hydrophobicity per amino-acid residue, we used ‘Define attribute’ in CHIMERA and generated amino-acid-specific scores53. The densities of flexible parts of myosin were detected by using the ‘Colour Zone’ function on a low-pass-filtered density map in CHIMERA41. For comparison of differences in density of A-state and M-state F-actin, we low-pass filtered both density maps to the same resolution of 3.9 Å. Sequence alignments were performed using the ClustalOmega online server54. The HGMD55 library and UNIPROT56 were browsed to find mutations in regions of interest. For creating a homology model of NM-2C in the Pi-release state, we used MODELLER in the CHIMERA ‘Multalign viewer’-interface with NM-2C as target sequence and the coordinates of the previously determined crystal structure of the Pi-release state of myosin-VI (PDB accession number 4PFO)7 as reference structure.
Accession codes
Primary accessions
Electron Microscopy Data Bank
Protein Data Bank
Data deposits
The coordinates and electron microscopy density maps have been deposited in the Protein Data Bank (PDB) under accession numbers 5JLF and 5JLH and the Electron Microscopy Data Bank (EMDB) under accession numbers EMD-8162 to EMD-8165.
References
Huxley, H. E. The mechanism of muscular contraction. Science 164, 1356–1366 (1969)
Heissler, S. M. & Manstein, D. J. Nonmuscle myosin-2: mix and match. Cell. Mol. Life Sci. 70, 1–21 (2013)
Lymn, R. W. & Taylor, E. W. Mechanism of adenosine triphosphate hydrolysis by actomyosin. Biochemistry 10, 4617–4624 (1971)
Sweeney, H. L. & Houdusse, A. Structural and functional insights into the myosin motor mechanism. Annu. Rev. Biophys. 39, 539–557 (2010)
Coureux, P.-D., Sweeney, H. L. & Houdusse, A. Three myosin V structures delineate essential features of chemo-mechanical transduction. EMBO J. 23, 4527–4537 (2004)
Gulick, A. M., Bauer, C. B., Thoden, J. B. & Rayment, I. X-ray structures of the MgADP, MgATPγS, and MgAMPPNP complexes of the Dictyostelium discoideum myosin motor domain. Biochemistry 36, 11619–11628 (1997)
Llinas, P. et al. How actin initiates the motor activity of myosin. Dev. Cell 33, 401–412 (2015)
Lorenz, M. & Holmes, K. C. The actin-myosin interface. Proc. Natl Acad. Sci. USA 107, 12529–12534 (2010)
Rayment, I. et al. Structure of the actin-myosin complex and its implications for muscle contraction. Science 261, 58–65 (1993)
Behrmann, E. et al. Structure of the rigor actin-tropomyosin-myosin complex. Cell 150, 327–338 (2012)
Holmes, K. C., Angert, I., Kull, F. J., Jahn, W. & Schröder, R. R. Electron cryo-microscopy shows how strong binding of myosin to actin releases nucleotide. Nature 425, 423–427 (2003)
von der Ecken, J. et al. Structure of the F-actin–tropomyosin complex. Nature 519, 114–117 (2015)
Várkuti, B. H. et al. A novel actin binding site of myosin required for effective muscle contraction. Nature Struct. Mol. Biol. 19, 299–306 (2012)
Onishi, H., Mikhailenko, S. V. & Morales, M. F. Toward understanding actin activation of myosin ATPase: the role of myosin surface loops. Proc. Natl Acad. Sci. USA 103, 6136–6141 (2006)
Furch, M., Remmel, B., Geeves, M. A. & Manstein, D. J. Stabilization of the actomyosin complex by negative charges on myosin. Biochemistry 39, 11602–11608 (2000)
Sasaki, N., Asukagawa, H., Yasuda, R., Hiratsuka, T. & Sutoh, K. Deletion of the myopathy loop of Dictyostelium myosin II and its impact on motor functions. J. Biol. Chem. 274, 37840–37844 (1999)
Joel, P. B., Trybus, K. M. & Sweeney, H. L. Two conserved lysines at the 50/20-kDa junction of myosin are necessary for triggering actin activation. J. Biol. Chem. 276, 2998–3003 (2001)
Murphy, C. T. & Spudich, J. A. The sequence of the myosin 50–20K loop affects myosin’s affinity for actin throughout the actin–myosin ATPase cycle and its maximum ATPase activity. Biochemistry 38, 3785–3792 (1999)
Uyeda, T. Q. P., Ruppel, K. M. & Spudich, J. A. Enzymatic activities correlate with chimaeric substitutions at the actin-binding face of myosin. Nature 368, 567–569 (1994)
Kojima, S. et al. Functional roles of ionic and hydrophobic surface loops in smooth muscle myosin: their interactions with actin. Biochemistry 40, 657–664 (2001)
Milligan, R. A., Whittaker, M. & Safer, D. Molecular structure of F-actin and location of surface binding sites. Nature 348, 217–221 (1990)
Van Dijk, J., Furch, M., Lafont, C., Manstein, D. J. & Chaussepied, P. Functional characterization of the secondary actin binding site of myosin II. Biochemistry 38, 15078–15085 (1999)
Várkuti, B. H., Yang, Z. & Malnasi-Csizmadia, A. Structural model of weak binding actomyosin in the prepowerstroke state. J. Biol. Chem. 290, 1679–1688 (2015)
Cook, R. K., Root, D., Miller, C., Reisler, E. & Rubenstein, P. A. Enhanced stimulation of myosin subfragment 1 ATPase activity by addition of negatively charged residues to the yeast actin NH2 terminus. J. Biol. Chem. 268, 2410–2415 (1993)
Miller, C. J., Wong, W. W., Bobkova, E., Rubenstein, P. A. & Reisler, E. Mutational analysis of the role of the N terminus of actin in actomyosin interactions. Comparison with other mutant actins and implications for the cross-bridge cycle. Biochemistry 35, 16557–16565 (1996)
Sutoh, K., Ando, M., Sutoh, K. & Toyoshima, Y. Y. Site-directed mutations of Dictyostelium actin: disruption of a negative charge cluster at the N terminus. Proc. Natl Acad. Sci. USA 88, 7711–7714 (1991)
Coureux, P.-D. et al. A structural state of the myosin V motor without bound nucleotide. Nature 425, 419–423 (2003)
Klein, J. C. et al. Actin-binding cleft closure in myosin II probed by site-directed spin labeling and pulsed EPR. Proc. Natl Acad. Sci. USA 105, 12867–12872 (2008)
Sasaki, N., Ohkura, R. & Sutoh, K. Insertion or deletion of a single residue in the strut sequence of Dictyostelium myosin II abolishes strong binding to actin. J. Biol. Chem. 275, 38705–38709 (2000)
Muretta, J. M., Rohde, J. A., Johnsrud, D. O., Cornea, S. & Thomas, D. D. Direct real-time detection of the structural and biochemical events in the myosin power stroke. Proc. Natl Acad. Sci. USA 112, 14272–14277 (2015)
Müller, M. et al. Functional characterization of the human α-cardiac actin mutations Y166C and M305L involved in hypertrophic cardiomyopathy. Cell. Mol. Life Sci. 69, 3457–3479 (2012)
Ohki, T., Ohno, C., Oyama, K., Mikhailenko, S. V. & Ishiwata, S. Purification of cytoplasmic actin by affinity chromatography using the C-terminal half of gelsolin. Biochem. Biophys. Res. Commun. 383, 146–150 (2009)
Coulton, A., Lehrer, S. S. & Geeves, M. A. Functional homodimers and heterodimers of recombinant smooth muscle tropomyosin. Biochemistry 45, 12853–12858 (2006)
Heissler, S. M. & Manstein, D. J. Comparative kinetic and functional characterization of the motor domains of human nonmuscle myosin-2C isoforms. J. Biol. Chem. 286, 21191–21202 (2011)
Li, X. et al. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nature Methods 10, 584–590 (2013)
Mindell, J. A. & Grigorieff, N. Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol. 142, 334–347 (2003)
Hohn, M. et al. SPARX, a new environment for cryo-EM image processing. J. Struct. Biol. 157, 47–55 (2007)
Scheres, S. H. W. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012)
Scheres, S. H. W. Beam-induced motion correction for sub-megadalton cryo-EM particles. eLife 3, e03665 (2014)
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004)
Pintilie, G. D., Zhang, J., Goddard, T. D., Chiu, W. & Gossard, D. C. Quantitative analysis of cryo-EM density map segmentation by watershed and scale-space filtering, and fitting of structures by alignment to regions. J. Struct. Biol. 170, 427–438 (2010)
Scheres, S. H. W. & Chen, S. Prevention of overfitting in cryo-EM structure determination. Nature Methods 9, 853–854 (2012)
Kucukelbir, A., Sigworth, F. J. & Tagare, H. D. Quantifying the local resolution of cryo-EM density maps. Nature Methods 11, 63–65 (2014)
Münnich, S., Pathan-Chhatbar, S. & Manstein, D. J. Crystal structure of the rigor-like human non-muscle myosin-2 motor domain. FEBS Lett. 588, 4754–4760 (2014)
Šali, A. & Blundell, T. L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 (1993)
Lopéz-Blanco, J. R. & Chacón, P. iMODFIT: efficient and robust flexible fitting based on vibrational analysis in internal coordinates. J. Struct. Biol. 184, 261–270 (2013)
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D 67, 235–242 (2011)
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010)
Brown, A. et al. Tools for macromolecular model building and refinement into electron cryo-microscopy reconstructions. Acta Crystallogr. D 71, 136–153 (2015)
Nicholls, R. A., Fischer, M., McNicholas, S. & Murshudov, G. N. Conformation-independent structural comparison of macromolecules with ProSMART. Acta Crystallogr. D 70, 2487–2499 (2014)
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 (2010)
Anandakrishnan, R., Aguilar, B. & Onufriev, A. V. H. H++ 3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 40, W537–W541 (2012)
Hessa, T. et al. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature 433, 377–381 (2005)
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011)
Stenson, P. D. et al. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 133, 1–9 (2014)
UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res. 43, D204–D212 (2015)
Dausse, E. et al. Familial hypertrophic cardiomyopathy. Microsatellite haplotyping and identification of a hot spot for mutations in the beta-myosin heavy chain gene. J. Clin. Invest. 92, 2807–2813 (1993)
Richard, P. et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 107, 2227–2232 (2003)
Yu, B. et al. Denaturing high performance liquid chromatography: high throughput mutation screening in familial hypertrophic cardiomyopathy and SNP genotyping in motor neurone disease. J. Clin. Pathol. 58, 479–485 (2005)
Epstein, N. D., Cohn, G. M., Cyran, F. & Fananapazir, L. Differences in clinical expression of hypertrophic cardiomyopathy associated with two distinct mutations in the β-myosin heavy chain gene. A 908Leu→Val mutation and a 403Arg→Gln mutation. Circulation 86, 345–352 (1992)
Geisterfer-Lowrance, A. A. et al. A molecular basis for familial hypertrophic cardiomyopathy: a β cardiac myosin heavy chain gene missense mutation. Cell 62, 999–1006 (1990)
Blanchard, E., Seidman, C., Seidman, J. G., LeWinter, M. & Maughan, D. Altered crossbridge kinetics in the αMHC403/+ mouse model of familial hypertrophic cardiomyopathy. Circ. Res. 84, 475–483 (1999)
Cuda, G., Fananapazir, L., Zhu, W. S., Sellers, J. R. & Epstein, N. D. Skeletal muscle expression and abnormal function of beta-myosin in hypertrophic cardiomyopathy. J. Clin. Invest. 91, 2861–2865 (1993)
Woo, A. et al. Mutations of the beta-myosin heavy chain gene in hypertrophic cardiomyopathy: critical functional sites determine prognosis. Heart 89, 1179–1185 (2003)
Van Driest, S. L. et al. Comprehensive analysis of the beta-myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 44, 602–610 (2004)
Mohiddin, S. A. et al. Utility of genetic screening in hypertrophic cardiomyopathy: prevalence and significance of novel and double (homozygous and heterozygous) β-myosin mutations. Genet. Test. 7, 21–27 (2003)
Marian, A. J. et al. A transgenic rabbit model for human hypertrophic cardiomyopathy. J. Clin. Invest. 104, 1683–1692 (1999)
Lankford, E. B., Epstein, N. D., Fananapazir, L. & Sweeney, H. L. Abnormal contractile properties of muscle fibers expressing beta-myosin heavy chain gene mutations in patients with hypertrophic cardiomyopathy. J. Clin. Invest. 95, 1409–1414 (1995)
Perrot, A. et al. Prevalence of cardiac beta-myosin heavy chain gene mutations in patients with hypertrophic cardiomyopathy. J. Mol. Med. 83, 468–477 (2005)
Erdmann, J. et al. Mutation spectrum in a large cohort of unrelated consecutive patients with hypertrophic cardiomyopathy. Clin. Genet. 64, 339–349 (2003)
Moolman, J. C., Brink, P. A. & Corfield, V. A. Identification of a new missense mutation at Arg403, a CpG mutation hotspot, in exon 13 of the β-myosin heavy chain gene in hypertrophic cardiomyopathy. Hum. Mol. Genet. 2, 1731–1732 (1993)
Moolman-Smook, J. C., De Lange, W. J., Bruwer, E. C., Brink, P. A. & Corfield, V. A. The origins of hypertrophic cardiomyopathy-causing mutations in two South African subpopulations: a unique profile of both independent and founder events. Am. J. Hum. Genet. 65, 1308–1320 (1999)
Greber-Platzer, S. et al. Beta-myosin heavy chain gene mutations and hypertrophic cardiomyopathy in Austrian children. J. Mol. Cell. Cardiol. 33, 141–148 (2001)
Villard, E. et al. Mutation screening in dilated cardiomyopathy: prominent role of the beta myosin heavy chain gene. Eur. Heart J. 26, 794–803 (2005)
Ley, T. J. et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 456, 66–72 (2008)
Toydemir, R. M. et al. Mutations in embryonic myosin heavy chain (MYH3) cause Freeman-Sheldon syndrome and Sheldon-Hall syndrome. Nature Genet. 38, 561–565 (2006)
Kamisago, M. et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N. Engl. J. Med. 343, 1688–1696 (2000)
Daehmlow, S. et al. Novel mutations in sarcomeric protein genes in dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 298, 116–120 (2002)
Acknowledgements
We thank O. Hofnagel for assistance in cryo sample preparation. We acknowledge R. Matadeen and S. de Carlo for image acquisition at the Netherlands Centre for Nanoscopy in Leiden. We thank R. S. Goody for reading the manuscript. This work was supported by the Max Planck Society, the European Research Council under the European Union’s Seventh Framework Program (FP7/2007-2013) (grant number 615984) (to S.R.), the Behrens-Weise foundation (to S.R.) and German Research Foundation (DFG) grant MA 1081/21-1 (to D.J.M.). J.v.d.E. is a fellow of Studienstiftung des deutschen Volkes.
Author information
Authors and Affiliations
Contributions
D.J.M. and S.R. designed the project. S.M.H. and S.P.-C. purified actin, tropomyosin, and myosin constructs. D.J.M. supervised protein work. J.v.d.E. prepared specimens, recorded, analysed and processed the data, and prepared figures. S.R. managed the project. J.v.d.E. and S.R. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information Nature thanks E. Nogales, J. Löwe and A. Houdusse for their contribution to the peer review of this work.
Extended data figures and tables
Extended Data Figure 1 Micrographs, two-dimensional classifications and three-dimensional refinement.
a–c, Representative of ~2,000 digital micrographs (a) and of 200 two-dimensional class averages of the F-actin–tropomyosin–myosin data set before (b) and after (c) three-dimensional refinement, respectively. Lower part of the micrograph is band-pass filtered to allow a better visualization of the filaments. Only filaments in rectangular boxes were chosen for refinement and bundled filaments were sorted out. d, e, Box dimension and angular distribution during three-dimensional refinement in side (d) and top (e) views. Histogram (few in blue to many in red) shows distribution of projection direction of each boxed segment relative to the three-dimensional reconstruction (grey). f, Example of two class averages out of c that show secondary structure elements. g, Fit of F-actin–myosin model to assign characteristic domains of myosin (see coloured circles and boxes). h, i, Representative of 200 class averages of the reprocessed F-actin–tropomyosin data set before (h) and after (i) three-dimensional refinement. j, k, In class averages, secondary structure elements in F-actin (green) and the coiled-coil structure of tropomyosin (yellow) are visible. Scale bars in micrograph and class averages are 50 nm and 10 nm, respectively.
Extended Data Figure 2 Resolution and model refinement of the actomyosin complex.
a, FSC curves of the cryo-EM reconstruction of the F-actin–tropomyosin–myosin data set (blue) and the reprocessed data set of F-actin–tropomyosin (red). The average resolution (FSC0.143) of the final electron density maps (central parts, green in subfigures) is estimated at 3.9 Å and 3.6 Å, respectively. Next subfigures illustrate only the actomyosin data set. b, Colour-coded local resolution of the full map and only finally refined part of the map (see Methods) estimated by ResMap43. c–e, Representative regions with higher than the average resolution in the F-actin filament core (c), the average resolution at the interface (d) and lower resolution in outer myosin parts (e). f, FSC curves of the model to each half map to check for overfitting, when the model was only refined versus the first half map. Black curve shows FSC between refined model and full map, when the model was refined against the full map (see Methods). g, h, B-factor distribution of final model from low (blue) to high (red) values. The absolute value strongly depends on the sharpening factor of the map, while the distribution shows the same gradient as the local resolution in b.
Extended Data Figure 3 HLH motif bound to F-actin.
a, Front view of F-actin and the HLH motif of the L50 domain of myosin show only small changes in loop regions while helices do not alter between weak (PPS state in purple, PDB accession number 5I4E) and strong binding (rigor state in red). The D-loop is moved towards the binding interface and is stabilized (A-state in yellow, M-state in green). Arrows indicate changes and scale bar is given. b, Same view as before shows the interface of myosin and F-actin in the rigor state. One possible salt bridge is highlighted with dotted lines. Surfaces are coloured from low (white) to high (yellow) hydrophobicity. c–e, Back view of the HLH motif and the base of loop 2 bound to central (SD1, SD3) and adjacent (D-loop) F-actin subunits. Comparison of rigor (red) and PPS state (purple, PDB accession number 5I4E) shows main differences (c). Final interaction of fully bound myosin is given in d, e. Possible electrostatic interactions are indicated by dotted lines. F-actin surface is depicted per subunit colour (c), by hydrophobicity (d) or electrostatic Coulomb potential (e, −10 kcal mol−1 in red to +10 kcal mol−1 in blue). In all subfigures, coloured residue labels belong to F-actin. f, Sequence alignment of myosin (H. sapiens myosin-II, -I, -III, -V, -VI) in the region of the HLH (helix-R–loop–helix-S) motif. Important functions at the F-actin–myosin interface and roles in stabilizing these regions themselves are highlighted and labelled. Residue numbering refers to our published structure belonging to the sequence of NM-2C (depicted in bold type). Tissue localization of myosin-II is written in parentheses. We refer to the different myosin isoforms according to the nomenclature for the genes encoding the respective myosin heavy chains.
Extended Data Figure 4 Cardiomyopathy loop and disease-causing mutations.
a, Conservation of the CM-loop in the human myosin-II class is visualized as a model on F-actin (cyan) from low (white) to high (purple) conservation. b, Sequence alignment of the CM-loop region of the human myosin-II class. Important functions at the F-actin–myosin interface are highlighted and labelled. Residue numbering refers to our published structure belonging to the sequence of NM-2C (depicted in bold type). Tissue localization of myosin-II is written in parentheses. We refer to the different myosin isoforms according to the nomenclature for the genes encoding the respective myosin heavy chains. c, d, Mutations in β-cardiac myosin (MYH7) can lead to cardiomyopathies. Corresponding residues in β-cardiac myosin are illustrated with our rigor state model (c) and known mutations57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74 are listed (d). e, Table of known disease-causing mutations at the actomyosin interface56,66,69,74,75,76,77,78. Numbers in parentheses give respective residue position in our published structure of NM-2C. Localization is described in parentheses.
Extended Data Figure 5 Loop 2 and loop 3 on F-actin.
a, b, Density map (grey) corresponding to the flexible part (residues 641–661) of loop 2 (red). F-actin is shown as surface model coloured from low (white) to high (yellow) hydrophobicity (a) or electrostatic Coulomb potential (b, −10 kcal mol−1 in red to +10 kcal mol−1 in blue). Residue labels belonging to F-actin are coloured as surface colours. c, Sequence alignment of myosin (H. sapiens myosin-II, -I, -III, -V, -VI) in the region of loop 2 and helix-R of the HLH region. Important functions at the F-actin–myosin interface and in stabilizing these regions are highlighted and labelled. Residue numbering refers to our published structure belonging to the sequence of NM-2C (depicted in bold). Tissue localization of myosin-II is written in parentheses. We refer to the different myosin isoforms according to the nomenclature for the genes encoding the respective myosin heavy chains. d, e, Changes between rigor (red) and PPS state (purple) in the loop 3 region relative to the rest of lower 50-kDa domain when bound to F-actin. Movements are indicated by black arrows and scale bars are given.
Extended Data Figure 6 Sequence-dependent interaction of supporting loop with the N terminus of F-actin.
a, Surface of myosin and N terminus is depicted by electrostatic Coulomb potential (−10 kcal mol−1 in red to +10 kcal mol−1 in blue). Involved charged residues are labelled. b, Position of the proline-rich loop (supporting loop) located between relay helix and helix-R slightly differs between the PPS (purple, PDB accession number 5I4E) and rigor state (red) and shows no direct interaction with the N terminus of F-actin. Regions at the surface of SD1 are pulled to the actomyosin interface indicated by an arrow and a scale bar is given (F-actin in A-state is depicted in yellow; F-actin in M-state is depicted in cyan). c, Sequence alignment of myosin (H. sapiens myosin-II, -I, -III, -V, -VI) in the region of the supporting loop. Different lengths of the loop and a possible supporting function are given in the last column. Residue numbering refers to our published structure belonging to the sequence of NM-2C (depicted in bold). Tissue localization of myosin-II is written in parentheses. We refer to the different myosin isoforms according to the nomenclature for the genes encoding the respective myosin heavy chains. d–g, Comparison of prominent properties in the supporting loop of different myosin classes (comparative models in purple) and their ability to undergo a direct interaction with the N terminus. Main differences are length of loop (numbers give absent amino acids relative to long loop) and position of the prominent positive-charged amino acid (R or K). Only an arginine or lysine sitting on the top would allow a direct interaction (e–g), while a sideward-oriented arginine (d) or a short loop (e) disables or reduces a possible interaction, respectively. In addition, respective densities (d) of the cryo-EM map are displayed.
Extended Data Figure 7 Myosin-induced conformational changes in F-actin.
a–c, Comparison of bare F-actin (A-state, yellow) with myosin-bound F-actin (M-state, cyan). Myosin is depicted in red. Either models (a) or representative parts of the electron density maps (b) illustrate conformational changes in F-actin (c). d, Sequence alignment of the N terminus region of human actin isoforms. Residue numbering refers to our published structure belonging to the sequence of non-muscular γ1-actin (ACTG1, depicted in bold type). To prevent confusion, the gene names instead of protein names are given. Localization is written in parentheses. e, N terminus and nucleotide binding region of F-actin undergo small changes (highlighted with arrows) through transmitted force of N terminus pulling. f, g, Close-ups of structural changes at the nucleotide binding site. h, Coordination of ADP and Mg2+ in the nucleotide binding cleft in M-state F-actin. i–k, Myosin binding induces a stabilization and shifting of the C terminus towards SD1 of F-actin. C373, which was used for pyrene labelling of F-actin, is part of the C-terminal region. Scale bars are given in the subfigures.
Extended Data Figure 8 Comparison of rigor and rigor-like myosin structures.
a–d, Close-ups of superimposed models from rigor-like structures (nucleotide-free myosin crystal structures in light green, PDB accession number 4PD3 (ref. 44) and 1OE9 (ref. 27)) with our rigor cryo-EM structure (red). F-actin is shown as surface model (green, cyan). Illustrated domains are labelled and coloured, while the rest of myosin is shown in grey from the rigor state model. Most regions do not show conformational differences (a, b), but the surface loops of myosin (CM-loop, loop 3 and loop 4) interacting with F-actin differ slightly in the rigor from the rigor-like structures (a, c). In contrast to the cryo-EM structure, loops at the interface (a, c) between F-actin and myosin are not always resolved in crystal structures. Major structural differences in the lever arm and converter regions are indicated by arrows and a scale bar is given (d).
Extended Data Figure 9 Different alignments of models for weak to strong binding of myosin and strut attraction to the base of loop2 promotes cleft closure.
a–c, Three possible alignments of myosin in the PPS (first column, purple, PDB accession number 5I4E), Pi-release (second column, blue) and rigor (third column, red) states are illustrated with respect to F-actin. For better visualization, differences in F-actin are not shown and F-actin is only depicted in the M-state (green, cyan). The Pi-release state represents a homology model of NM-2C based on a crystal structure of myosin in the Pi-release state7 (PBD accession number 4PFO, see Methods). All models are either aligned to the U50 domain (a) or the L50 domain (b) of the rigor state. In c, the model of the Pi-release state was first aligned to the L50 domain of the rigor state. The PPS state was then aligned to the U50 domain of model of the Pi-release state. The first row in each subfigure shows changes in the U50 domain from the top (for a better visualization L50 was deleted). The second row shows the L50 domain from the bottom (U50 is transparent). Possible clashes are indicated by a yellow star (a). d–f, Binding mechanism of the strut, connecting L50 and U50 domains, to the stabilized base of loop 2. To illustrate the conformational changes, the respective regions in the PPS state (PPS, purple, PDB accession number 5I4E) and rigor state (RS, red) of myosin have been partly overlaid. The rest of myosin is shown in grey. L50 binds to F-actin (A-state, yellow) (d, e). The base of loop 2 is stabilized by F-actin (e) and attracts the negatively charged strut with its positive patch. This promotes the binding of the strut, shifting the equilibrium to a closed conformational state of myosin (f). Flexible parts of loop 2 are indicated as dotted lines. Lower panels show surfaces of the same regions as in the upper panels coloured by electrostatic Coulomb potential. For better visualization, the upper parts of the strut were removed. Surface of F-actin is depicted in transparent grey.
Supplementary information
Cryo-EM structure of the ATM complex in detail
a, Cryo-EM reconstruction of F-actin (five central subunits in green and one subunit in cyan) decorated with tropomyosin (blue) and myosin in rigor state (central molecules highlighted in red). Close-up ends with the central part of the map. b, Representative part of the central core region of the F-actin filament shows better than average resolution. c, Interface between D-loop, SD1 of F-actin and HLH motif of myosin. d, Myosin CM-loop bound to SD1 and SD3 of F-actin. (MP4 9614 kb)
Model of the rigor ATM complex and subdomains in F-actin
F-actin (green, cyan) decorated with tropomyosin (blue) and myosin (red). Subdomain organisation of F-actin is shown. (MP4 8246 kb)
Domain organisation of myosin on F-actin
Domain organisation of the NM-2C head region (depicted in different colours and labelled as in Fig. 1b-d). Close-ups on HLH motif and surface loops (CM-loop, loop 3, loop 4) at the F-actin-myosin interface. (MP4 9341 kb)
F-actin-myosin interfaces in detail
a-c, Close-ups on the HLH motif (a), CM-loop (b) and loop 2 (c) of myosin bound to F-actin in rigor state. d, e, A positively charged basin is formed by loop 2, helix-W and the supporting loop of myosin to stabilize the pulled N-terminus conformation of F-actin. Myosin is shown as ribbon with residues (d) or as surface coloured by the electrostatic Coulomb potential ranging from -10 kcal mol-1 in red to +10 kcal mol-1 in blue. (MP4 7622 kb)
Model of weak to strong binding of myosin to F-actin
a, Myosin binds in a weak PPS state (purple, PDB: 5I4E) to F-actin. b, L50 rotates and binds to F-actin resulting in a stronger bound myosin (Pi-release state in blue). c, d, Finally, U50 rotates and binds to F-actin (c) obtaining the full interface of strongly bound myosin (rigor state in red) on F-actin (d). Models were aligned as shown in Extended Data Fig. 9f. For better visualization, differences in F-actin are not shown and F-actin is depicted only in the M-state (green, cyan). (MP4 5344 kb)
Model of myosin-binding to F-actin in detail
a, Initial binding of myosin (purple) to F-actin (yellow). b, L50 rotates and binds to F-actin. c, The base of loop 2 (red) is stabilized by hydrophobic interactions with F-actin. d, It also interacts with the negatively charged N-terminus of F-actin inducing its pulling and ordering (A-state in yellow, M-state in cyan). L50 is in its final rigor state conformation. e, Negatively charged strut gets attracted to the positively charged patch of the base of loop 2. U50 rotates and binds to F-actin resulting in the interface observed in the rigor state (red). (MP4 6017 kb)
Rights and permissions
About this article

Cite this article
Ecken, J., Heissler, S., Pathan-Chhatbar, S. et al. Cryo-EM structure of a human cytoplasmic actomyosin complex at near-atomic resolution. Nature 534, 724–728 (2016). https://doi.org/10.1038/nature18295
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature18295
- Springer Nature Limited
This article is cited by
-
The pig as an optimal animal model for cardiovascular research
Lab Animal (2024)
-
Electron microscopy of cardiac 3D nanodynamics: form, function, future
Nature Reviews Cardiology (2022)
-
The actomyosin interface contains an evolutionary conserved core and an ancillary interface involved in specificity
Nature Communications (2021)
-
Alpha and beta myosin isoforms and human atrial and ventricular contraction
Cellular and Molecular Life Sciences (2021)
-
Actin–tropomyosin distribution in non-muscle cells
Journal of Muscle Research and Cell Motility (2020)