
Abstract
Hypertrophic cardiomyopathy (HCM) is a frequent, autosomal-dominant cardiac disease and manifests predominantly as left ventricular hypertrophy. Mutations in the cardiac beta-myosin heavy chain gene (MYH7) are responsible for the disease in about 30% of cases where mutations were identified. We clinically evaluated a large group of 147 consecutive HCM patients from three cardiology centers in Germany, Poland, and Kyrgyzstan according to the same protocol. The DNA of the patients was systematically analyzed in the whole coding region of the MYH7 gene using PCR, single-strand conformation polymorphism analysis, and automated sequencing. Eleven different missense mutations (including seven novel ones) in 11 unrelated patients were identified, showing a mutation frequency of 7.5% in the study population. We further examined the families of five patients (three of German, one of Polish, and one of Kyrgyz origin) with 32 individuals in total. We observed a clear, age-dependent penetrance with onset of disease symptoms in the fourth decade of life. Genotype–phenotype correlations were different for each mutation, whereas the majority was associated with an intermediate/malign phenotype. In conclusion, we report a systematic molecular screening of the complete MYH7 gene in a large group of consecutive HCM patients, leading to a genetic diagnosis in 38 individuals. Information about the genotype in an individual from one family could be very useful for the clinician, especially when dealing with healthy relatives in doubt of their risk about developing HCM. The increasing application of genetic screening and the increasing knowledge about genotype–phenotype correlations will hopefully lead to an improved clinical management of HCM patients.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Hypertrophic cardiomyopathy [(HCM) OMIM no. 192600)] is characterized predominantly by left ventricular hypertrophy mainly affecting the interventricular septum, with or without involvement of the anterior or posterior wall [1, 2]. Often, the prognosis is benign, but a certain number of patients die of sudden cardiac death or heart failure [3]. HCM is transmitted as an autosomal dominant trait and is the most frequent genetic disorder of the myocardium. The clinical phenotype is as heterogeneous as the genotype (reviewed by Maron [4]).
Twelve disease genes causing HCM were identified to date. Nearly all of them encode for sarcomeric proteins, which led to the hypothesis that HCM is a disease of the cardiac sarcomere. In addition to this locus heterogeneity, there is a wide allelic heterogeneity. More than 250 different mutations have been described in these disease genes (reviewed by Seidman and Seidman [5] and Marian and Roberts [6]).
Mutations in the cardiac beta-myosin heavy chain gene [(MYH7) OMIM no. 160760)] are responsible for the disease in about 30% of cases where mutations were identified as described in the literature [6]. The MYH7 gene is located on chromosome 14q11.2–q13. It is 23 kb long, and 38 exons encode for a protein of 1,935 amino acids [7]. Muscle contraction and cell motility depend on myosin as the essential component of the sarcomere thick filament. Generation of force is mostly done by the globular head of the molecule possessing the important enzymatic activities. Nearly all of the published mutations were found in exons encoding for this part of the protein. Although MYH7 was the first disease gene identified for HCM, prospective genetic screening of different HCM populations revealed many novel mutations and only few frequent ones. Because of this wide allelic heterogeneity and the rarity of each individual mutation [8], it is important to identify all individuals bearing MYH7 mutations and perform extensive genotype–phenotype correlations. The wide range in clinical manifestation indicates a need to establish whether and to what extent genotype influences phenotype. Additionally, examination of patients and families with novel MYH7 mutations will extend our knowledge about the manifestation of this common cardiac disease.
Therefore, we systematically analyzed a well-characterized group of HCM patients from three cardiology centers in the whole coding region of the MYH7 gene. We identified 11 different missense mutations (including seven novel ones) and describe here the disease-associated phenotype in five families and six index patients.
Materials and methods
Phenotyping
After obtaining informed consent, clinical evaluation was performed and blood samples were drawn from the patients and family members at the National Center for Cardiology and Internal Medicine Bishkek, at the Silesian School of Medicine, Katowice, and at the Charité, Berlin.
One hundred forty-seven consecutive HCM patients (six from Kyrgyzstan, 10 from Poland, and 131 from Germany) were evaluated according to the same examination protocol on the basis of medical history, physical examination, two-dimensional and M-mode echocardiography, 12-lead electrocardiogram (ECG) and, in some cases, by Holter ECG and heart catheterization.
The diagnosis of HCM was based on criteria recommended by the WHO [9] and the criteria proposed by McKenna et al. [10]. Briefly, the major inclusion criteria were the presence of a interventricular septal thickness (IVS) ≥13 mm in the absence of other known causes of hypertrophy (e.g., hypertension or aortic valve disease) and major ECG abnormalities like negative T waves, pathological Q waves, and heart blocks. The echocardiographic measurements were performed according to the standards of the American Society of Echocardiography [11], without knowledge of the genetic status.
Ninety-six samples from adult anonymous blood donors were used as controls. They were sex-matched and of matched ethnic origin to the study population. All underwent routine physical examination, and blood tests and were without known cardiovascular disease.
The study protocol was approved by the respective local institutional review board and was in accordance with the Helsinki Declaration.
Genotyping
Genomic DNA was extracted from EDTA blood using standard techniques [12].
All mutations were detected by PCR amplification, followed by single-strand conformation polymorphism (SSCP) analysis and direct sequencing. Primer pairs flanking each of the 38 coding exons were designed using the genomic sequence of the human MYH7 gene according to Hoffmann et al. [19] (GenBank/EMBL accession numbers M30603, M30604, M30605, M58018, NM_000257) and are available on request.
The final PCR reaction mix (25 μl) contained about 20 ng genomic DNA, 200 μM dNTP, 0.1 μM each primer, 0.625 U AmpliTaq Gold DNA polymerase (Applied Biosystems, Darmstadt, Germany), and GeneAmp 10× PCR Buffer [100 mM Tris-HCl, pH 8.3, 500 mM KCl, 15 mM MgCl2, 0.01% (w/v) gelatin, Applied Biosystems]. The reaction mix was denatured initially at 95°C for 10 min, and amplified for 40 cycles in a thermal cycler (Biometra, Göttingen, Germany). Each cycle was composed of denaturing for 15 s at 95°C, annealing for 30 s at the exon-specific temperature, and extension for 20 s at 72°C. Subsequent to the last cycle, a final extension of 3 min at 72°C was performed before cooling the samples to 4°C. SSCP analysis of the PCR products was used for mutation detection. Briefly, 1 μl of a 25-μl reaction mixture was diluted with 5 μl denaturing solution (95% formamide, 20 mM EDTA, 0.05% bromphenol blue, 0.05% xylene cyanol), heated to 95°C, plunged into an ice bath, and resolved on an MDE gel (FMC BioProducts, Philadelphia, Pa., USA) run at 1,500 V, 100 mA, and 60 W (upper limits) for 30 min at 15°C. The DNA banding patterns were visualized by silver staining.
All PCR products exhibiting an aberrant electrophoretic mobility in the SSCP analysis were directly sequenced in both directions using either the forward or the reverse PCR primer in two independent runs. Nucleotide sequence determination was performed by cycle sequencing using the Big Dye Terminator Cycle Sequencing Kit on an ABI 373 fluorescence sequencer (Applied Biosystems).
In order to test the sensitivity of the SSCP, we sequenced all 147 samples in the randomly chosen exons 22 and 23 and compared the SSCP patterns with the sequencing electropherograms.
Results
Genotyping in the MYH7 gene
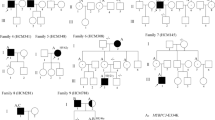
A group of 147 unrelated patients with HCM was examined, and the complete coding region of the MYH7 gene was screened for DNA alterations in these patients. In total, we identified 11 different mutations leading to an amino acid replacement in 11 unrelated HCM patients (see Table 1), showing that 7.5% of the analyzed patients had a mutation in the MYH7 gene. We were able to further examine the families of five of these patients (see Fig. 1), while for six patients there were no families available for examinations.

Pedigrees of hypertrophic cardiomyopathy (HCM) families with respective silver stained single-strand conformation polymorphism analysis gel showing aberrant band patterns. Squares indicate males; circles, females. Open symbols represent unaffected subjects; solid symbols, affected individuals; shaded symbols, probably affected individuals; and slanted bars, deceased individuals. Question marks denote individuals with unknown disease status, arrows indicate the index patient (proband) of each family. Plus symbols represent the presence of a beta-myosin heavy chain (MYH7) mutation, and minus symbols the absence of the MYH7 mutation
All mutations identified were missense mutations showing a clear pattern of heterozygosity in the electropherograms (see Fig. 2). Seven of these were novel mutations not described in the literature before. The mutations were widely distributed over the coding region of the gene in nine different exons (from exon 7 to exon 32). Most of them were located in the globular head (subfragment 1) of the N-terminal heavy meromyosin domain (HMM), while two were located in the neck region (subfragment 2) of the HMM (see Table 1). Of note was that two mutations, Glu1356Lys and Ala1454Thr, occurred in the C-terminal rod termed light meromyosin domain (LMM).

Sequence electropherograms representing the 11 heterozygous missense mutations in the MYH7 gene. The panel represents the mutant sequence. Mutated amino acids are shown above the normal amino acid sequence
An alignment of the corresponding sequences revealed that all seven novel mutations affect highly conserved residues (see Table 2). The residues show full conservation across species including rat, golden hamster, and pig.
None of the 11 described mutations was present in 192 chromosomes from the unrelated control population of matched ethnic origin.
Further, we detected ten genetic polymorphisms that occurred with different frequencies in the patient and control population (unpublished data).
Sequencing of the exons 22 and 23 revealed only the two mutations at codons 901 and 928 detected by SSCP and no further genetic alterations in the 294 PCR products. This shows a sensitivity of 100% in the two exons using our screening method.
Clinical phenotype in families
We evaluated five families (three of German, one of Polish, and one of Kyrgyz origin) with 32 individuals in total. Detailed pedigrees are shown in Fig. 1. Seventeen family members carried a mutation in the MYH7 gene, of whom 11 fulfilled diagnostic criteria of HCM. Detailed clinical characteristics of all examined individuals are presented in Table 3.
In family E of German origin, we identified the Gly741Trp mutation in two individuals (II-1, II-2). The proband II-1 developed dyspnea and angina pectoris at the age of 38 years. The ECG showed preterminal negative T waves in the lateral leads. The diagnosis of HCM is based on a moderate eccentric hypertrophy of the whole IVS. The affected brother II-2 became symptomatic at the age of 36 years, with palpitations and dyspnoea. ECG revealed sinus rhythm and left bundle branch block. Echocardiography showed massive asymmetric hypertrophy of the IVS without outflow tract gradient at rest. Heart catheterization confirmed this finding and showed an outflow tract gradient only after provocation with orciprenaline. The third brother II-3 was genetically and clinically unaffected.
In family D of German origin, we identified the Tyr501Cys mutation in three individuals (I-2, II-1, II-2), of whom only the male proband I-2 fulfilled diagnostic criteria of HCM. Beginning at the age of 18 years, proband I-2 complained of premature fatigue after strong physical exercise. The diagnosis of hypertrophic obstructive cardiomyopathy (HOCM) was established at the age of 38 years. ECG showed negative T waves in precordial leads. Echocardiography revealed asymmetric hypertrophy of the entire IVS, with maximal diastolic thickness of 18 mm and an outflow tract gradient of about 40 mm Hg at rest. Left heart catheterization confirmed this finding and excluded coronary artery disease. His children, individuals II-1 and II-2, carry the mutation but were asymptomatic and had normal ECG and echocardiography findings. The mother, individual I-1, was genetically and clinically unaffected.
In family C of German origin, we identified the Arg403Trp mutation in two individuals (II-2, III-4). The proband II-2 developed symptomatic and intermittent atrial fibrillation and flutter at the age of 53 years. Successful electrophysiologic ablation of atrial flutter was performed at the age of 60 years. ECG revealed right bundle branch block. Echocardiography showed asymmetric septal hypertrophy of 18 mm without outflow tract obstruction. Individual III-4 had dyspnea with greater exertion (NYHA II). ECG revealed preterminal negative T waves in V5–6. Echocardiography showed severe asymmetric septal hypertrophy without outflow tract obstruction. The remaining family members (individuals II-1, II-4, III-1, III-2, III-3, III-5, IV-1) were genetically and clinically unaffected. There are no data available about the death of individual II-3.
In family B, we identified the Ala901Gly mutation in four individuals (III-1, III-2, III-3, IV-1). The family is of Polish origin. The proband III-1 became symptomatic at the age of 40 years with dyspnea at strong exercise. ECG was normal. The left ventricle showed asymmetric hypertrophy with an IVS thickness of 18 mm but without outflow tract obstruction. Individual III-2 had already dyspnea during mild exertion. ECG revealed right bundle branch block and pathological Q and T waves in lateral leads. On echocardiography moderate symmetric left ventricular (LV) hypertrophy with a small outflow tract gradient at rest could be shown. Individual III-3 developed exertional dyspnea at the age of 45 years. She had negative T waves on ECG, and echocardiography showed mild symmetric LV hypertrophy. The asymptomatic individual IV-1 carrying the mutation is 22 years of age. Her ECG and echocardiography findings were normal. There are no data available about the death of individual III-4.
In family A, we identified the Ile736Thr mutation in six individuals (II-2, II-4, II-6, II-7, III-7, III-10), of whom three met the diagnostic criteria of HCM. This family is of Kyrgyz (Central Asian) origin. Among 15 examined family members, we detected the Ile736Thr mutation in six individuals while only three of them were clinically affected. All clinically affected individuals from this family are characterized by asymmetric septal hypertrophy without outflow tract obstruction and pathological Q waves. Individual II-2 had NYHA II, palpitations, and atypical chest pain unrelated to exercise. ECG showed pathological Q wave in aVL lead. Asymmetric LV hypertrophy without outflow tract obstruction was shown by echocardiography. The proband II-4 as well as individual II-7 had NYHA class II. Left anterior hemiblock, pathological Q waves, and negative T waves were registered on ECG of the proband II-4. Echocardiography revealed pronounced asymmetric LV hypertrophy (Maron type III) without outflow tract obstruction. Individual II-7 showed pathological Q waves in I, aVL, V5–6 leads and negative T wave in aVL on ECG. A marked asymmetric LV hypertrophy without outflow tract obstruction was found on echocardiography of this individual. The remaining individuals (II-6, III-7, III-10) had no complaints and showed no abnormalities on ECG and echocardiography.
Clinical phenotype in index patients
Detailed clinical data of the six patients bearing a MYH7 mutation are shown in Table 4. The families of these patients were not available for examination. All patients were of German origin.
The female proband H1 (Pro211Leu) was diagnosed with HOCM at the age of 72 years. Symptoms worsened in the following years. Outflow tract gradient determined by LV heart catheterization was 122 mmHg at rest. Embolization of the first septal branch was successfully performed, leading to an immediate decrease of the gradient to 23 mmHg. Four years later, a DDD pacemaker was implanted after a successful reanimation because of asystoly.
The male proband H2 (Arg453Cys) developed HOCM at the age of 17 years. He suffered from palpitations and dyspnea on exertion and reported several syncopes. On the Holter recording, ventricular premature beats (Lown class IVa) were present. Echocardiography revealed massive hypertrophy of the proximal septum with a outflow tract gradient of 51 mmHg. He was treated with low doses of beta-blockers and with calcium antagonists.
The male proband H3 (His576Arg) presented with atypical angina and dyspnea on exertion. Echocardiography revealed hypertrophy of the basal septum and outflow tract obstruction. Invasive examination confirmed a maximal pressure gradient of 40 mmHg and severe mitral insufficiency. Treatment with a calcium antagonist improved symptoms.
The female proband H4 (Asp928Asn) developed severe dyspnea at the age of 44 years. Physical examination revealed a systolic murmur, and ECG showed signs of myocardial hypertrophy. She had a asymmetrical hypertrophy with outflow tract obstruction of 100 mmHg at rest. Therefore, she underwent septal myectomy, which improved symptoms significantly.
The male proband H5 (Glu1356Lys) developed dyspnea at the age of 34 years. HOCM with a gradient of 70 mmHg was diagnosed by echocardiography and catheterization. Hypertrophy was asymmetrical and confined to the septum. Holter ECG revealed multiple ventricular extrasystolic beats (Lown class IIIb). He died suddenly at the age of 40 years.
The female proband H6 (Ala1454Thr) reported palpitations, angina, and dyspnea on exertion at the age of 61 years. Cardiac examination revealed asymmetrical hypertrophy with a maximal outflow tract gradient of 80 mm Hg. Holter ECG showed ventricular and supraventricular extrasystolic beats (Lown class IVa). Treatment with a calcium antagonist improved symptoms.
Genotype–phenotype correlations
Table 1 shows the genotype–phenotype correlation for each mutation. The classification in “benign/intermediate/malign” was assessed according to the severity of symptoms, degree of hypertrophy, and course/prognosis of the disease in the respective patient and examined family members. There was no phenotype difference between families with different ethnic origin.
We observed a clear, age-dependent penetrance as demonstrated in Fig. 3: mutation carriers with no signs of hypertrophy were all under the age of 30 years (with one exception, individual II-6 from family A). This is underlined by the mean age of all patients of 52.4±10.4 years and by the mean age at disease onset of 34.8±14.1 years. Furthermore, Fig. 3 clearly shows that individuals over 30 years bearing a MYH7 mutation can be diagnosed very well using echocardiography, since 95% of them develop septal hypertrophy with IVS ≥13 mm.

Genotype–phenotype correlation showing age (years) related to interventricular septum (millimeters). Included are all individuals (index patients and family members) bearing a MYH7 mutation (n=23) composed of 17 clinically affecteds (showing manifest HCM) and six clinically nonaffecteds
Discussion
We identified 11 different missense mutations in the MYH7 gene in five families of different ethnic origin and in six index patients, underlining the strong genetic heterogeneity in HCM. Seven of these mutations have not been described before, showing that most of HCM cases bear rare mutations. Further, it emphasizes that a screening only for known genetic variants in HCM is not appropriate. Three of the known mutations we detected (Arg403Trp, Arg453Cys, and Ile736Thr) were localized in well-known hot spot regions; especially in codon 403 were different mutations often detected. The mutations were distributed over the complete MYH7 gene, which is not a typical finding. This underlines the importance to screen the whole coding region of the gene. In the past, MYH7 screening often included only the first half of the gene encoding for the globular head. All 11 detected sequence alterations were missense mutations. They probably lead to “poison peptides” as previously suggested by experimental studies and may act in a dominant-negative manner [5].
We consider the detected genetic alterations as disease-causing mutations rather than rare polymorphisms for the following reasons. First, MYH7 is a proven disease gene for HCM as demonstrated in various patient studies and animal models. Additionally, there is a clear cosegregation of the respective mutation with the disease phenotype in the five examined families: all relatives with normal cardiac findings did not have the mutation, whereas all clinically affected relatives carried the mutation. Further evidence is the alignment of the altered sequences showing that all mutations predicted amino acid changes of highly conserved residues implying functional importance. Finally, none of the MYH7 mutations was present in 192 control alleles.
The frequency of MYH7 mutations in our study population was 7.5% (11 in 147). Previous studies have reported frequencies in MYH7 mutations ranging from 3% [13, 14] up to a frequency of 25% [15]. The largest study included 389 patients with HCM and showed a prevalence of 15% [16]. This value was supported by similar data from a German study and a Danish study, with about 100 patients each [17, 18]. The wide range may be probably caused by differences in genotyping methods, differences in clinical evaluation, and most importantly differences in the composition of the study population. We could not exclude that we have missed a mutation. Altough this seems rather unlikely, because our genotyping method is highly sensitive and accurate, as shown by the high number of detected sequence alterations in MYH7 (pathogenic mutations and frequent polymorphisms). We tested the sensitivity of the applied SSCP method by sequencing all 147 samples in the two randomly chosen exons 22 and 23. Only the mutations in codon 901 and 928 (detected by SSCP before) and no further variants were identified, showing a sensitivity of 100% in these two exons. However, it is rather difficult to predict the sensitivity for the remaining 36 exons, because the sensitivity is exon dependent and therefore variable. Under the assumption that the sensitivity is lower in some other exons (which may lead to a mean sensitivity of about 90%), we would have missed only a few mutations, resulting in a prevalence most likely below 10%. Further, we successfully applied the same method to identify mutations in other genes [19, 20]. SSCP in general is described as a method with a high sensitivity of about 84–89% as comparative tests have shown [21, 22]. Additionally, our screening method and our clinical study protocols are similar to those used by the other groups.
The higher prevalence in a German cohort of 108 HCM patients [17] most likely results from the different inclusion criteria. Erdmann et al. included only patients with septal wall thickness of more than 15 mm, whereas we included patients with 13 mm or more. Including patients with a greater IVS favors the inclusion of patients with MYH7 mutations that usually cause a more pronounced hypertrophy.
The distribution of MYH7 mutations in the different ethnic groups is as follows: one patient with a mutation in six Kyrgyz patients, one patient with a mutation in ten Polish patients, and nine patients with mutations in 131 German patients. Because of the small numbers, it is difficult to draw valid conclusions about general differences in the prevalence of MYH7 mutations in the three ethnic groups.
We observed different phenotypes (severity of symptoms, degree of hypertrophy, and course/prognosis of the disease) related to each mutation. The majority of mutations were associated with an intermediate/malign phenotype. Our findings seem to be in agreement with the published data as shown in Table 1 [3, 23, 24]. Further, we found a clear, age-dependent penetrance with onset of disease symptoms in the fourth decade of life. This is in contrast to previous findings, which showed a penetrance of 90% or more from 10 to 60 years of age in HCM patients with MYH7 mutations [25]. One reason for that may be that mainly patients with a more obvious and severe hypertrophy were included in the former studies. Additionally, a further difficulty of genotype–phenotype correlations in HCM is that clinical diversity is also influenced by genetic background, environment, gender, and acquired conditions [26].
Myosin plays a central role in generating active force and movement in muscle fibers. Most of the mutations detected by us were located in the globular head of the protein, HMM(S1), in or close to the important domains like the ATP- and actin-binding sites, the “active thiols” (SH1 and SH2), the binding sites for the essential and regulatory myosin light chains, and the converter subdomain. They may therefore alter the myosin structure and the functional properties of the molecule like ATPase activity and sliding velocity. The two mutations found in the HMM(S2) neck domain may influence neck flexibility during contraction, while the two mutations in the myosin rod (LMM) may disturb thick filament assembly and binding of accessory proteins [27]. There is no doubt that mutations in beta-myosin have a strong impact on the structure and function of the sarcomere as shown in many studies. However, it remains rather difficult to predict functional consequences (gain or loss of activity) of an individual MYH7 mutation, as shown by contradictory results concerning the Arg403Gln mutation (reviewed by Lowey) [28].
There are some limitations of our study. The evidence of the pathogenicity of the detected mutations is not without difficulties because of the unavailability of the respective family in six of the patients bearing a mutation. Further, we could not rule out double heterozygotes in the 11 cases, i.e., harboring two mutations in different sarcomere genes. Because the sample size for each mutations is small, the genotype–phenotype correlations shall be used with caution for classification of patients with the same mutations.
In conclusion, we report a systematic molecular screening of the complete MYH7 in a large group of consecutive HCM patients, leading to a genetic diagnosis in 38 individuals. Information about the genotype in an individual from one family could be very useful for the clinician, especially when dealing with healthy relatives, in doubt of their risk about developing HCM. Predictions about the disease course in affected patients based on the genotype remains difficult. When assessing HCM subjects, screening of the whole MYH7 gene is necessary and feasible as shown by us, but it requires considerable effort and time.
The increasing application of genetic screening and the increasing knowledge about genotype–phenotype correlations will hopefully lead to better genetic counseling and improved clinical management of HCM patients and their relatives in the future.
References
Ciro E (1983) Heterogeneous morphologic expression of genetically transmitted hypertrophic cardiomyopathy: two-dimensional echocardiographic analysis. Circulation 67:1227–1233
Maron BJ, Bonow RO, Seshagiri TNR, Roberts WC, Epstein SE (1981) Hypertrophic cardiomyopathy with ventricular septal hypertrophy localized to the apical region of the left ventricle. Am J Cardiol 49:1838–1848
Roberts R, Sigwart U (2001) New concepts in hypertrophic cardiomyopathy, part I. Circulation 104:2113–2116
Maron BJ (2002) Hypertrophic cardiomyopathy: a systematic review. JAMA 287:1308–1320
Seidman JG, Seidman C (2001) The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell 104:557–567
Marian AJ, Roberts R (2001) The molecular genetic basis for hypertrophic cardiomyopathy. J Mol Cell Cardiol 33:655–670
Jaenicke T, Diederich KW, Haas W, Schleich J, Lichter P, Pfordt M, Bach A, Vosberg HP (1990) The complete sequence of the human b-myosin heavy chain gene and a comparative analysis of its product. Genomics 8:194–206
Ackerman MJ, Van Driest SL, Ommen SR, Will ML, Nishimura RA, Tajik AJ, Gersh BJ (2002) Prevalence and age-dependence of malignant mutations in the beta-myosin heavy chain and troponin t genes in hypertrophic cardiomyopathy. J Am Coll Cardiol 39:2042–2048
Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O’Connel J, Olsen EGJ, Thiene G, Guyarfas I (1996) Report of the 1995 WHO/ISFC Task Force on the definition and classification of cardiomyopathies. Circulation 93:841–842
McKenna WJ, Spirito P, Desnos M, Dubourg O, Komajda M (1997) Experience from clinical genetics in hypertrophic cardiomyopathy: proposal for new diagnostic criteria in adult members of affected families. Heart 77:130–132
Sahn DJ, DeMaria A, Kisslo J, Weyman A (1978) The Committee on M-mode standardization of the American Society of Echocardiography: recommendations regarding quantitation in M-mode echocardiography: results of a survey of echocardiographic measurements. Circulation 58:1072–1083
Lahiri DK, Nurnberger JI (1991) A rapid non-enzymatic method for the preparation of HMW DNA from blood for RFLP studies. Nucleic Acids Res 19:5444
Jääskeläinen P, Soranta M, Miettinen R, Saarinen L, Pihlajamäki J, Silvennoinen K, Tikanoja T, Laakso M, Kuusisto J (1998) The cardiac beta-myosin heavy chain gene is not the predominant gene for hypertrophic cardiomyopathy in the Finnish population. J Am Coll Cardiol 32:1709–1716
Mörner S, Richard P, Kazzam E, Hellman U, Hainque B, Schwartz K, Waldenström A (2003) Identification of the genotypes causing hypertrophic cardiomyopathy in northern Sweden. J Mol Cell Cardiol 35:841–849
Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichreau C, Benaiche A, Isnard R, Dubourg O, Burban M, Gueffet JP, Millaire A, Desnos M, Schwartz K, Hainque B, Komajda M (2003) Hypertrophic cardiomyopathy—distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 107:2227–2232
Van Driest SL, Jaeger MA, Ommen SR, Will ML, Gersh BJ, Tajik AJ, Ackerman MJ (2004) Comprehensive analysis of the beta-myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 44:602–610
Erdmann J, Daehmlow S, Wischke S, Senyuwa M, Werner U, Raible J, Tanis N, Dyachenko S, Hummel M, Hetzer R, Regitz-Zagrosek V (2003) Mutation spectrum in a large cohort of unrelated consecutive patients with hypertrophic cardiomyopathy. Clin Genet 64:339–349
Hougs L, Havndrup O, Bundgaard H, Kober L, Vuust J, Larsen LA, Christiansen M, Andersen PS (2005) One third of Danish hypertrophic cardiomyopathy patients have mutations in MYH7 rod region. Eur J Hum Genet 13:161–165
Hoffmann B, Schmidt-Traub H, Perrot A, Osterziel KJ, Geßner R (2001) First mutation in cardiac troponin C, L29Q, in a patient with hypertrophic cardiomyopathy. Human Mutat 17:524
Geier C, Perrot A, Özcelik C, Binner P, Counsell D, Hoffmann K, Pilz B, Martiniak Y, Gehmlich K, van der Ven PFM, Fürst DO, Vornwald A, von Hodenberg E, Nürnberg P, Scheffold T, Dietz R, Osterziel KJ (2003) Mutations in the human muscle LIM protein gene in families with hypertrophic cardiomyopathy. Circulation 107:1390–1395
Vidal-Puig A, Moller DE (1994) Comparative sensitivity of alternative single-strand conformation polymorphism (SSCP) methods. Biotechniques 17:490–496
Jordanova A, Kalaydjieva L, Savov A, Claustres M, Schwarz M, Estivill X, Angelicheva D, Haworth A, Casals T, Kremensky I (1997) SSCP analysis: a blind sensitivity trail. Human Mutat 10:65–70
Fananapazir L, Dalakas MC, Cyran F, Cohn G, Epstein ND (1993) Missense mutations in the beta-myosin heavy chain gene cause central core disease in hypertrophic cardiomyopathy. Proc Natl Acad Sci USA 90:3993–3997
Woo A, Rakowski H, Liew JC, Zhao MS, Liew CC, Parker TG, Zeller M, Wigle ED, Sole MJ (2003) Mutations of the beta myosin heavy chain gene in hypertrophic cardiomyopathy: critical functional sites determine prognosis. Heart 89:1179–1185
Niimura H, Bachinski LL, Sangwatanaroj S, Watkins H, Chudley AE, McKenna W, Kristinsson A, Roberts R, Sole M, Maron BJ, Seidman JG, Seidman CE (1998) Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N Engl J Med 338:1248–1257
Arad M, Seidman JG, Seidman CE (2002) Phenotypic diversity in hypertrophic cardiomyopathy. Hum Mol Genet 11:2499–2506
Blair E, Redwood C, Oliveira M, Moolman-Smook JC, Brink P, Corfield VA, Östman-Smith I, Watkins H (2002) Mutations in the light meromyosin domain of the beta-myosin heavy chain rod in hypertrophic cardiomyopathy. Circ Res 90:263–269
Lowey S (2002) Functional consequences of mutations in the myosin heavy chain at sites implicated in familial hypertrophic cardiomyopathy. Trends Cardiovasc Med 12:348–354
Acknowledgements
The authors declare that there are no conflicts of interest. We thank the families for their cooperation in this study. Further, we thank all of our colleagues in the three hospitals for invaluable help in the recruitment of patients. Especially, we thank U. Weiher and A. Köstner for excellent technical assistance. This study was supported by a grant-in-aid from Charité-Universitätsmedizin, Berlin.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article can be found at http://dx.doi.org/10.1007/s00109-005-0722-9
Andreas Perrot and Hajo Schmidt-Traub contributed equally to this work
Rights and permissions
About this article
Cite this article
Perrot, A., Schmidt-Traub, H., Hoffmann, B. et al. Prevalence of cardiac beta-myosin heavy chain gene mutations in patients with hypertrophic cardiomyopathy. J Mol Med 83, 468–477 (2005). https://doi.org/10.1007/s00109-005-0635-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-005-0635-7