

Abstract
The human genome is arguably the most complete mammalian reference assembly1,2,3, yet more than 160 euchromatic gaps remain4,5,6 and aspects of its structural variation remain poorly understood ten years after its completion7,8,9. To identify missing sequence and genetic variation, here we sequence and analyse a haploid human genome (CHM1) using single-molecule, real-time DNA sequencing10. We close or extend 55% of the remaining interstitial gaps in the human GRCh37 reference genome—78% of which carried long runs of degenerate short tandem repeats, often several kilobases in length, embedded within (G+C)-rich genomic regions. We resolve the complete sequence of 26,079 euchromatic structural variants at the base-pair level, including inversions, complex insertions and long tracts of tandem repeats. Most have not been previously reported, with the greatest increases in sensitivity occurring for events less than 5 kilobases in size. Compared to the human reference, we find a significant insertional bias (3:1) in regions corresponding to complex insertions and long short tandem repeats. Our results suggest a greater complexity of the human genome in the form of variation of longer and more complex repetitive DNA that can now be largely resolved with the application of this longer-read sequencing technology.
Similar content being viewed by others


Main
Data generated by single-molecule, real-time (SMRT) sequencing technology differ drastically from most sequencing platforms because native DNA is sequenced without cloning or amplification, and read lengths typically exceed 5 kilobases (kb). Despite overall lower individual read accuracy (∼85%), longer read length facilitates high confidence mapping across a greater percentage of the genome11,12.We generated ∼40-fold sequence coverage from a human CHM1 hydatidiform mole using long-read SMRT sequence technology (average mapped read length = 5.8 kb; Supplementary Table 1). We selected a complete hydatidiform mole to sequence because it is haploid, lacking allelic variation, and provides higher effective sequence coverage. We aligned 93.8% of all sequence reads to the human reference genome (GRCh37) using a modified version of BLASR11 (Supplementary Information) and generated local assemblies of the mapped reads using Celera13 and Quiver14, the latter of which leverages estimates of insertion, deletion and substitution probabilities to determine consensus sequences accurately. We compared the consensus sequences of regions with previously sequenced and assembled large-insert bacterial artificial chromosome (BAC) clones generated from CHM1tert (ref. 15). The comparison shows a consensus sequencing concordance of >99.97% (phred quality = 37.5), with 72% of the errors confined to indels within homopolymer stretches (Supplementary Table 3).
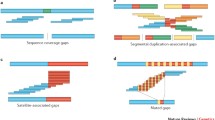
We initially assessed whether the mapped reads could facilitate closure of any of the 164 interstitial euchromatic gaps within the human reference genome (GRCh37). We extended into gap regions using a reiterative map-and-assemble strategy, in which SMRT whole-genome sequencing (WGS) reads mapping to each edge of a gap were assembled into a new high-quality consensus, which, in turn, served as a template for recruiting additional sequence reads for assembly (Supplementary Information). Using this approach, we closed 50 gaps and extended into 40 others (60 boundaries), adding 398 kb and 721 kb of novel sequence to the genome, respectively (Supplementary Table 4). The closed gaps in the human genome were enriched for simple repeats, long tandem repeats, and high (G+C) content (Fig. 1) but also included novel exons (Supplementary Table 20) and putative regulatory sequences based on DNase I hypersensitivity and chromatin immunoprecipitation followed by high-throughput DNA sequencing (ChIP-seq) analysis (Supplementary Information). We identified a significant 15-fold enrichment of short tandem repeats (STRs) when compared to a random sample (P < 0.00001) (Fig. 1a). A total of 78% (39 out of 50) of the closed gap sequences were composed of 10% or more of STRs. The STRs were frequently embedded in longer, more complex, tandem arrays of degenerate repeats reaching up to 8,000 bp in length (Extended Data Fig. 1a–c), some of which bore resemblance to sequences known to be toxic to Escherichia coli16. Because most human reference sequences17,18 have been derived from clones propagated in E. coli, it is perhaps not surprising that the application of a long-read sequence technology to uncloned DNA would resolve such gaps. Moreover, the length and complex degeneracy of these STRs embedded within (G+C)-rich DNA probably thwarted efforts to follow up most of these by PCR amplification and sequencing.

a, Gap closures are enriched for simple repeats compared to equivalently sized regions randomly sampled from GRCh37. b, Human genome gaps typically consist of (G+C)-rich sequence (yellow) flanking complex (A+T)-rich STRs (green) (empirical P value; Supplementary Information). Red line indicates genomic (G+C) content.
Next, we developed a computational pipeline (Extended Data Fig. 2) to characterize structural variation systematically (structural variation defined here as differences ≥50 bp in length, including deletions, duplications, insertions and inversions7). Structural variants were discovered by mapping SMRT sequencing reads to the human reference genome11 and searching for specific mapping signatures (Supplementary Information). At every variant locus, we recruited all uniquely mapping reads, created a local de novo assembly, defined breakpoints compared to the human reference, and classified each structural variant by type and probable mechanism (Table 1). We identified a total of 26,079 insertions/deletions ≥50 bp within the euchromatic portion of the genome. Almost all insertion and deletion breakpoints were resolved at the single-base-pair level, generating one of the most comprehensive catalogues of structural variation (47,238 breakpoint positions). A total of 6,796 of the events map within 3,418 genes with a subset of events (169) corresponding to variation in the spliced transcripts of 140 genes (Supplementary Table 9). From all targeted sequencing experiments combined (Supplementary Information) we estimate an overall validation rate of 97%, of which only a fraction can be detected by application of Illumina next-generation sequencing.
Of all copy number differences found, 85% were novel compared to previous studies of structural variation7,8,19, in large part owing to increased ascertainment of smaller variation (average length 497 bp). The effect was most pronounced for insertions in which 92% of all differences had not been previously reported, in contrast to deletions in which 69% of the events were novel (Fig. 2). When comparing the size distribution of insertions and deletions between the two haplotype references, we found that insertions within CHM1 were longer and more abundant with 5,473 additional insertion events when compared to the human reference (Table 1). This difference contributes to a significant insertional bias of 3.9 megabases (Mb) of additional sequence either missing or expanded when compared to the human reference (Table 1). We find a substantial increase in the amount of long, ≥50 bp STR insertions relative to deletions (P < 2.2 × 10−16), including STRs within genes (Supplementary Table 9). In addition to being 2.80 times more frequent than deletions, the STR insertions ≥50 bp are, on average, 2.87 times longer. This asymmetry becomes more pronounced with increasing STR insertion length (Fig. 2b). The genomic distribution of STR insertions is highly non-random being biased to the last 5 Mb of human chromosomes (Extended Data Fig. 3) correlating with recombination rate20 (r2 = 0.21) and human–chimpanzee divergence (r2 = 0.20). We note that 2,285 of these expanded STRs occur within genes, including 11 within an untranslated region (noting shorter insertions in FMR1 and C9orf72, a common mutated locus for amyotrophic lateral sclerosis; Supplementary Information) and two within the coding sequence of genes (MUC2 and SAMD1). A total of 189 genes have an STR expansion >1 kb, representing potential sites of genomic instability (Supplementary Table 9).

a, Histograms display the distribution of novel insertions (black/grey) and deletions (red/pink) between CHM1 and GRCh37 haplotypes compared to copy number variants identified from other studies for insertions and deletions less than 1 kb (left) and greater than or equal to 1 kb (right). Most of the increased sensitivity occurs below 5 kb. Peaks at ∼300 bp and 6 kb correspond to Alu and L1 insertions, respectively. b, STR insertions in CHM1 (green) are longer than the human genome (blue; GRCh37), and this effect becomes more pronounced with increasing length (x axis). c, The percentage repeat composition (x axis) of 1-kb sequences flanking insertion sites for Alu, L1 and SVA mobile element insertions. Insertion calls from the 1000 Genomes Project (pink)21 compared to calls from CHM1 using SMRT reads (blue) show increased sensitivity for repeat-rich insertions.
The remaining half of the insertional bias (∼1.5 Mb) was accounted for by 1,116 more complex structural variants (which we define as insertions having either several annotated repeat elements, or at least 30% of the remaining sequence not annotated as repeat) (Table 1 and Extended Data Fig. 4). Sequence analyses of these regions of the genome revealed these insertions were frequently embedded within regions already enriched for clusters of mobile element insertions. Complex repetitive regions such as these represent a major challenge in structural variant detection owing to spurious mapping of short-read sequence data. We performed site complexity analysis of annotated mobile element insertion loci by assessing the repeat composition of the 1-kb sequences 5′ and 3′ flanking the retrotransposons AluY, L1 and SVA insertions in both the CHM1 sequencing data and insertion sites from population-scale low-coverage sequencing data21. While we observed a small bias in the repeat complexity of AluY insertions (53% versus 48%; P = 4.8 × 10−6, Kolmogorov–Smirnov test), a much more marked shift is seen for L1 and SVA insertions. We found that human-specific L1Hs insertion sites in CHM1 have a flanking common repeat content of 59% when compared to 39% in the 1000 Genomes Project data set (P = 1.8 × 10−10, Kolmogorov–Smirnov test) (Fig. 2c). The bias for SVA insertions is even greater, with 76% of insertions mapping adjacent to repeats when compared to 50% using Illumina read-pair data (P = 3.84 × 10−14, Kolmogorov–Smirnov test).
The large STR and complex insertions are enriched for regions annotated as having potential clone assembly problems. This enrichment becomes more pronounced the larger and more complex the insertion (for example, the 185-fold enrichment of ‘black tag’ annotations for STR insertions; Supplementary Information). Notably, less than 1% of these variants are present in newer assemblies of the human genome, including GRCh38 and CHM1.1 (ref. 22) (derived primarily by Illumina sequencing technology). Because we find evidence of most of these complex events in additional human or chimpanzee genomes (Supplementary Information), we propose that ∼1,700 sites (3.5 Mb) represent deficiencies or ‘muted’ gaps that can now be accessed as a result of SMRT technology (Supplementary Table 7). We incorporated these inserted sequences as well as gap closures into a patched GRCh37 reference, effectively mapping 0.026% additional Illumina reads and discovering additional single nucleotide polymorphisms (SNPs) (for example, 9,231 SNPs; Supplementary Information).
In addition to insertions and deletions, we also searched for the presence of inversions—a structural variation class that is notoriously difficult to ascertain. We developed a search algorithm that specifically leveraged the increased length of the SMRT sequence reads to search for ‘reversals’ in order when aligned to the reference. Regions with two or more reversals were then locally assembled to define the breakpoints of each event optimally. We identified 34 inversions with an average length of 7.1 kb, corresponding to a total of ∼240 kb of inverted sequence (Supplementary Table 8 and Supplementary Fig. 6). We subcloned and sequenced 15 events using a large-insert BAC library with a validation rate of 100% (15 out of 15) (Extended Data Fig. 5). None of the events disrupted genes, no enrichment was observed on the X chromosome, and 68% (23 out of 34) of the inversions were flanked by inverted repeats (Supplementary Table 8).
A limitation of our approach is its dependence on the local assembly of mapped reads to the human reference genome. Even with an average mapped read length of 5.8 kb, not all reads may be uniquely mapped to a specific location. As a result, gaps (n = 82) adjacent to segmental duplications were largely unresolved, inversions exceeding the read length (>20 kb) could not be detected (for example, 15q13.3 region), and SMRT sequence read synthesis within or flanking long, highly identical repeats could not be reliably assembled. We identified a total of 737 euchromatic regions (12.5 Mb) of our genome, in which large-scale mapping inconsistences (n = 22) or deficiencies (n = 715) were noted but were unresolvable by this approach (Supplementary Tables 26 and 27). We selected one 6.5-Mb region mapping to chromosome 10q11.23 for a more detailed analysis. The region carried seven gaps within the human reference genome (GRCh37), none of which was resolved or extended by SMRT WGS reads. We applied an alternative clone-based hierarchical approach (Supplementary Information) and identified a tiling path of 32 BACs and assembled the clone inserts using SMRT sequencing14. We generated sequence contigs spanning two large clusters of segmental duplication (2.7 and 1.2 Mb), closing six of the seven gaps in this region (Fig. 3 and Extended Data Fig. 6), adding 416 kb of missing reference sequence, correcting the orientation of 1,451 kb, and eliminating 856 kb of redundant sequence that was represented twice within the reference. Two gaps remain, each at the same location within paralogous segmental duplications, corresponding to a nearly perfect 50-kb tandem repeat that cannot be resolved at the level of large-insert clones using existing methods. These results indicate that although it is possible to use reads to close gaps and detect variation missed by other next-generation sequencing methods, the resolution of larger, complex regions of the genome still require targeted efforts that leverage both clones and WGS data. Complete de novo assembly of human genomes will probably require the development of even longer-range sequencing data. The approaches outlined here will have broader application to many of the unfinished and complex regions of mammalian genomes.

The clone-based assembly is composed primarily of BACs from the CH17 library as shown in the tiling path below the internal repeat structure of the region. Coloured arrows indicate large segmental duplications (SDs) with homologous sequences connected by lines generated by Miropeats23.
Methods
SMRT WGS data (41-fold sequence coverage) was generated using a Pacific Biosciences RSII instrument (P5C3 chemistry) from genomic libraries generated from a complete hydatidiform mole DNA (CHM1tert). Sequence reads were mapped to the human reference genome (GRCh37) using a modified version of BLASR (http://www.github.com/EichlerLab/blasr) (Supplementary Methods); a bioinformatics pipeline was developed to identify regions of structural variation and extensions into gaps (http://www.github.com/EichlerLab/chm1_scripts); corresponding sequence reads were de novo assembled and a high-quality consensus sequence generated for each region using Celera v.8.1 (ref. 13) and Quiver v.0.7.6 (ref. 14). Reads are selected for support of a variant if the mapping quality is greater than 20; a minimum of 5 reads are required to trigger an assembly. For the purpose of this analysis, we focused only on the euchromatic portion of the genome excluding pericentromeric regions (5 Mb flanking annotated centromeres), all acrocentric portions of chromosomes, and subtelomeric regions (150 kb from the annotated telomeric sequence). Repeat content of all structural variants was determined using CENSOR24, RepeatMasker25, Miropeats23 and TRF (http://tandem.bu.edu/). The sequence accuracy of the assemblies and structural variant polymorphisms were inferred by comparison to 18 sequenced large-insert BAC (CH17) and 89 fosmid clones8, Sanger-based BAC-end sequence generated for CHM1tert (GenBank accessions in Supplementary Table 35), and comparison to Illumina-based WGS generated for human genomes1. We also generated Illumina WGS data (41-fold) for comparison (SRA SRP044331). For the chromosome 10q11 region, 125 CH17 BACs were identified and sequenced using a Nextera-Illumina protocol26. A minimal tiling path of 35 clones was deeply sequenced (300-fold coverage) using 1 SMRT cell per clone; inserts were assembled and an alternative reference was created using methods described previously15.
Accession codes
Primary accessions
Sequence Read Archive
Data deposits
All underlying SMRT WGS read data have been released within the NCBI Sequence Read Archive (SRA) under accession SRX533609 and may also be accessed as part of all the SMRT data sets (NCBI SRA accession SRP040522). Illumina WGS data for CHM1 are available in the NCBI SRA under accession SRP044331 as well as finished BAC and fosmid clone inserts using SMRT sequence data (GenBank accessions in Supplementary Table 35). For the purpose of mapping and annotation, we developed a patched GRCh37 reference genome including a track hub for upload into the UCSC Genome Browser. A complete list of all inaccessible regions of the human genome and a database of heterochromatic and subtelomeric sequence reads that could not be assembled are available at (http://eichlerlab.gs.washington.edu/publications/chm1-structural-variation).
References
The 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012)
The International HapMap Project Consortium. The International HapMap Project. Nature 426, 789–796 (2003)
International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004)
Kurahashi, H. et al. Molecular cloning of a translocation breakpoint hotspot in 22q11. Genome Res. 17, 461–469 (2007)
Genovese, G. et al. Using population admixture to help complete maps of the human genome. Nature Genet. 45, 406–414 (2013)
Bovee, D. et al. Closing gaps in the human genome with fosmid resources generated from multiple individuals. Nature Genet. 40, 96–101 (2008)
Mills, R. E. et al. Mapping copy number variation by population-scale genome sequencing. Nature 470, 59–65 (2011)
Kidd, J. M. et al. A human genome structural variation sequencing resource reveals insights into mutational mechanisms. Cell 143, 837–847 (2010)
Eichler, E. E., Clark, R. A. & She, X. An assessment of the sequence gaps: unfinished business in a finished human genome. Nature Rev. Genet. 5, 345–354 (2004)
Eid, J. et al. Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138 (2009)
Chaisson, M. J. & Tesler, G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 13, 238 (2012)
Lee, H. & Schatz, M. C. Genomic dark matter: the reliability of short read mapping illustrated by the genome mappability score. Bioinformatics 28, 2097–2105 (2012)
Myers, E. W. et al. A whole-genome assembly of Drosophila. Science 287, 2196–2204 (2000)
Chin, C. S. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nature Methods 10, 563–569 (2013)
Huddleston, J. et al. Reconstructing complex regions of genomes using long-read sequencing technology. Genome Res. 24, 688–696 (2014)
Kimelman, A. et al. A vast collection of microbial genes that are toxic to bacteria. Genome Res. 22, 802–809 (2012)
Lander, E. S. et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001)
Venter, J. C. et al. The sequence of the human genome. Science 291, 1304–1351 (2001)
Conrad, D. F. et al. Origins and functional impact of copy number variation in the human genome. Nature 464, 704–712 (2010)
Kong, A. et al. A high-resolution recombination map of the human genome. Nature Genet. 31, 241–247 (2002)
Stewart, C. et al. A comprehensive map of mobile element insertion polymorphisms in humans. PLoS Genet. 7, e1002236 (2011)
Steinberg, K. M. et al. Single haplotype assembly of the human genome from a hydatidiform mole. Genome Res (in press)
Parsons, J. D. Miropeats: graphical DNA sequence comparisons. Comput. Appl. Biosci. 11, 615–619 (1995)
Jurka, J., Klonowski, P., Dagman, V. & Pelton, P. CENSOR–a program for identification and elimination of repetitive elements from DNA sequences. Comput. Chem. 20, 119–121 (1996)
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-3.0 http://www.repeatmasker.org (1996–2010)
Adey, A. et al. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol. 11, R119 (2010)
Wu, T. & Watanabe GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875 (2005)
Acknowledgements
We thank D. Alexander, D. Church and A. Klammer for discussions, K. Mohajeri and L. Harshman for technical assistance and T. Brown for assistance in manuscript preparation. This work was supported, in part, by US National Institutes of Health (NIH) grant HG002385 and HG007497 to E.E.E. M.Y.D. is supported by the US National Institute of Neurological Disorders and Stroke (award K99NS083627). E.E.E. is an investigator of the Howard Hughes Medical Institute.
Author information
Authors and Affiliations
Contributions
E.E.E., M.J.P.C., M.Y.D., J.H. and J.K. designed experiments; M.M. prepared DNA; M.M. and M.B. prepared libraries and generated sequence data; P.H.S., J.H. and M.Y.D. identified clones for sequencing; J.H., P.H.S., M.Y.D., F.H. and M.J.P.C. performed bioinformatics analyses; M.Y.D., F.A. and M.M. performed targeted sequencing of clones; M.J.P.C. designed algorithms and pipelines for mapping SMRT sequence data and detection of structural variants; M.W.H., U.S., R.S. and J.A.S. provided access to critical resources; J.M.L. deposited SMRT sequence data into SRA; M.J.P.C., J.H. and E.E.E. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
M.B., J.L., M.W.H. and J.K. are employees of Pacific Biosciences, Inc., a company commercializing DNA sequencing technologies; E.E.E. is on the scientific advisory board (SAB) of DNAnexus, Inc. and was formerly an SAB member of Pacific Biosciences, Inc. (2009–2013) and SynapDx Corp. (2011–2013); and M.J.P.C. was a former employee for Pacific Biosciences, Inc.
Extended data figures and tables
Extended Data Figure 1 Sequence content of gap closures.
a–c, Gap closures are enriched for simple repeats compared to equivalently sized regions randomly sampled from GRCh37; examples of the organization of these regions are shown using Miropeats for chromosome 4 (GRCh37, chr4:59724333–59804333) (a), chromosome 11 (GRCh37, chr11:87673378–87753378) (b), and chromosome X (GRCh37, chrX:143492324–143572324) (c). Dotplots show the architecture of the degenerate STRs with the core motif highlighted below. Shared sequence motifs between blocks are indicated by colour.
Extended Data Figure 2 Variant detection pipeline.
At every variant locus, we collected the full-length reads that overlap the locus, performed de novo assembly using the Celera assembler, and called a consensus using Quiver after remapping reads used in the assembly as well as reads flanking the assembly (yellow reads) to increase consensus quality at the boundaries of the assembly. BLASR is used to align the assembly consensus sequences to the reference, and insertions and deletions in the alignments are output as variants. Reads spanning a deletion event within a single alignment are shown as bars connected by a solid line, and double hard-stop reads spanning a larger deletion event and split into two separate alignments of the same read are shown as a dotted line.
Extended Data Figure 3 Genome distribution of closed gaps and insertions.
Chromosome ideogram heatmap depicts the normalized density of inserted CHM1 base pairs per 5-Mb bin with a strong bias noted near the end of most chromosomes. Locations of structural variants and closed gaps are given by coloured diamonds to the left of each chromosome: closed gap sequences (red), inversions (green), and complex events (blue).
Extended Data Figure 4 Confirmation of complex insertions in additional genomes.
Top, genotypes of polymorphic complex regions using read depth of unique k-mers (blue: present; white: absent). Bottom, extended examples of complex insertion events: alignment to chimpanzee panTro4 reference (dark blue); existing human reference hg19 (light teal); inserted sequence (dark teal). The bottom rows show repeat annotations, with darker hues for repeats overlapping the inserted region.
Extended Data Figure 5 Inversion validation by BAC-insert sequencing.
Inversions detected by alignment of single long reads were validated by sequencing clones from the CHM1 BAC library (CHORI17), in which end mappings to GRCh37 spanned the putative inversions. Inversions were validated by aligning the corresponding BAC sequences to GRCh37 with Miropeats. Shared sequence between the BACs and GRCh37 is shown in black; inversion events are indicated in red.
Extended Data Figure 6 CHM1 clone-based assembly of the human 10q11 genomic region.
a, The clone-based assembly is composed primarily of BACs from the CH17 library as shown in the tiling path below the internal repeat structure of the region. Coloured arrows indicate large segmental duplications with homologous sequences connected by coloured lines (Miropeats). Genes annotated from alignment of RefSeq messenger RNA sequences with GMAP27 are shown. b, Miropeats comparisons of the 10q11 clone-based assembly against the corresponding sequence from GRCh37, with gaps shown in red, highlight the degree to which the reference was misassembled.
Supplementary information
Supplementary Information
This file contains Supplementary Methods, Text and Data, Supplementary Figures 1-29, Supplementary Tables 1-35 and additional references. Tables shown in this file represent views of the full tables given in the Supplementary Tables file. (PDF 5107 kb)
Supplementary Tables
This file contains the full table values for the Supplementary Tables 1-35 (see separate Supplementary information file). (XLSX 442 kb)
Rights and permissions
About this article

Cite this article
Chaisson, M., Huddleston, J., Dennis, M. et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature 517, 608–611 (2015). https://doi.org/10.1038/nature13907
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature13907
- Springer Nature Limited
This article is cited by
-
Investigating the dark-side of the genome: a barrier to human disease variant discovery?
Biological Research (2023)
-
A long-read sequencing and SNP haplotype-based novel preimplantation genetic testing method for female ADPKD patient with de novo PKD1 mutation
BMC Genomics (2023)
-
Illuminating the biosynthesis pathway genes involved in bioactive specific monoterpene glycosides in Paeonia veitchii Lynch by a combination of sequencing platforms
BMC Genomics (2023)
-
SPUMONI 2: improved classification using a pangenome index of minimizer digests
Genome Biology (2023)
-
Identification and characterization of two DMD pedigrees with large inversion mutations based on a long-read sequencing pipeline
European Journal of Human Genetics (2023)