

Abstract
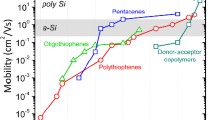
Conjugated polymers enable the production of flexible semiconductor devices that can be processed from solution at low temperatures. Over the past 25 years, device performance has improved greatly as a wide variety of molecular structures have been studied1. However, one major limitation has not been overcome; transport properties in polymer films are still limited by pervasive conformational and energetic disorder2,3,4,5. This not only limits the rational design of materials with higher performance, but also prevents the study of physical phenomena associated with an extended π-electron delocalization along the polymer backbone. Here we report a comparative transport study of several high-mobility conjugated polymers by field-effect-modulated Seebeck, transistor and sub-bandgap optical absorption measurements. We show that in several of these polymers, most notably in a recently reported, indacenodithiophene-based donor–acceptor copolymer with a near-amorphous microstructure6, the charge transport properties approach intrinsic disorder-free limits at which all molecular sites are thermally accessible. Molecular dynamics simulations identify the origin of this long sought-after regime as a planar, torsion-free backbone conformation that is surprisingly resilient to side-chain disorder. Our results provide molecular-design guidelines for ‘disorder-free’ conjugated polymers.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Main
In several donor–acceptor co-polymers7,8,9,10 surprisingly high field-effect mobilities >1 cm 2 V −1 s −1 have recently been found despite the microstructure of these polymers being less ordered than those of crystalline or semicrystalline polymers, such as poly-3-hexylthiophene3 (P3HT) or poly(2,5-bis(3-alkylthiophen-2-yl)thieno(3,2-b)thiophene)5 (PBTTT), and in some cases being near amorphous. The high mobilities have been attributed to a network of tie chains providing interconnecting transport pathways between crystalline domains3, but this does not fully explain how these polymers can exhibit significantly higher mobilities than P3HT or PBTTT. To probe energetic disorder in these systems, we investigate the Seebeck coefficient α, which can be determined experimentally by measuring the electromotive force EMF that develops across a material in response to an applied temperature differential ΔT as follows: α = EMF/ΔT. For small carrier concentration, as in the experiments reported here, the dominant contribution to α is the entropy of mixing associated with adding a carrier into the density of states, which is determined by the density of thermally accessible transport states11,12,13,14. If the energetic dispersion is less than kBT (kB, Boltzmann’s constant) then the density of thermally accessible states will be temperature independent and equal to the density of molecular sites. By contrast, if the energetic dispersion among hopping sites is much greater than kBT then the density of thermally accessible states will increase as the temperature is raised. Thus, we can estimate the energetic disorder relative to kBT associated with transport by measuring the temperature dependence of the Seebeck coefficient of field-effect transistors (FETs) which independently control the carrier density15.
We have investigated a range of state-of-the-art diketopyrrolopyrrole (DPP) and isoindigo copolymers, and here show results for PSeDPPBT16,17 and DPPTTT18,19 with mobilities of 0.3–0.5 cm2 V−1 s−1 and 1.5–2.2 cm2 V−1 s−1, respectively (for the chemical structures of PSeDPPBT and DPPTTT, see Supplementary Fig. 13a). PBTTT serves as a semicrystalline polymer reference system. Among the many polymers we investigated, we find the lowest degree of energetic disorder in indacenodithiophene-co-benzothiadiazole (IDTBT). IDTBT is a highly soluble polymer (Supplementary Information section 1) exhibiting high field-effect mobilities despite a lack of long-range crystalline order6,20. Top-gate IDTBT FETs with films annealed at 100 °C and Cytop gate dielectrics reliably exhibit near-ideal performance: a low threshold voltage of VTh = –3 V, a low contact resistance (Fig. 1a) and a high saturation mobility of 1.5–2.5 cm2 V−1 s−1 extracted from a near-ideal, quadratic current dependence on gate voltage.

a, Room-temperature output characteristics and device architecture of a typical IDTBT organic FET with channel length L = 20 µm and channel width W = 1 mm. b, γ plotted versus 1,000/T for IDTBT (structure shown), PSeDPPBT, DPPTTT and PBTTT organic FETs. c, Temperature evolution of IDTBT transfer curves fitted with a disorder-free MOSFET model (drain voltage, VD = −60 V). d, Gate-voltage dependence of saturation mobility μ at 300 and 240 K for patterned IDTBT (top) and PBTTT (bottom) devices.
These mobility values are lower than the highest values claimed in the literature9,10,19. On the one hand, there is ongoing debate about the possible overestimation of mobilities in polymer FETs owing to deviations from the ideal in their electrical characteristics21. All mobility values reported here were conservatively estimated. Artefacts related to contact resistance make it possible, for example, to extract mobilities up to an order of magnitude higher from non-optimized IDTBT devices with non-ideal electrical characteristics (Supplementary Information section 2). On the other hand, we have restricted ourselves to top-gate FETs with spin-coated films and have not used techniques that may enhance mobilities for certain materials by increasing the interfacial orientation or alignment relative to that present in the bulk10. This enables us to correlate interface-sensitive FET Seebeck measurements with bulk-sensitive optical spectroscopy.
Among the polymers investigated, IDTBT had not only one of the highest mobilities, if not the highest, but also the most-ideal electrical characteristics (Supplementary Information section 2). This is evident in the temperature-dependent dependence of drain current ID on gate voltage VG in the saturation regime, which was fitted to ID ∝ (VG – VTh) γ between 200 and 300 K. For IDTBT, the exponent γ takes the ideal, temperature-independent value 2, (Fig. 1b). By contrast, γ increases with decreasing temperature as γ = T0/T + 1 for PBTTT, PSeDPPBT and DPPTTT, which is commonly observed in polymer FETs and is interpreted in terms of carriers hopping within an exponential density of states with characteristic width kBT0 > kBT (ref. 22). Concomitantly, the mobility rises on increasing the magnitude of the gate voltage as trap states within the band tail are progressively filled (Supplementary Information section 2). Whereas this disorder model fits the other polymers, it does not fit the IDTBT FET data even when T0 is taken to be as small as 330 K. We know of no prior report of such ideal (γ = 2) behaviour for a polymer FET. The IDTBT transfer characteristics are well fitted over the entire temperature range with a disorder-free, metal–oxide–semiconductor FET-like model with a thermally activated, but gate-voltage-independent mobility (Fig. 1c). This was confirmed by directly extracting the gate voltage dependence of the mobility from the transfer characteristics of devices with patterned semiconductor layers to minimize leakage and fringe currents. In IDTBT, the mobility was nearly independent of gate voltage for |VG| > 20 V across the entire temperature range, whereas in PBTTT the mobility strongly increases with gate voltage at lower temperatures (Fig. 1d). These results suggest that energetic disorder is significantly lower in IDTBT than in the other polymers.
To accurately measure the Seebeck coefficients of FETs with 20–50 μm channel lengths as functions of gate voltage and temperature, we developed a microfabricated device architecture with an integrated heater and temperature sensors positioned along the FET’s channel23 (Supplementary Information section 3). The carrier concentrations n in the accumulation layer were estimated from measurements of capacitance versus gate voltage (Supplementary Information section 4). We find Seebeck coefficients (Fig. 2) that are much larger than kB/e ≈ 86 μV K−1 (e, elementary charge), that are decreasing functions of increasing carrier concentration n and that are independent of temperature between 200 and 300 K within the measurement error. Temperature-independent Seebeck coefficients over a similar temperature range have been reported previously only for single crystals of the molecular semiconductors pentacene and rubrene15.

a, Temperature independence of the field-effect-modulated Seebeck coefficients of PBTTT and IDTBT. b, Slopes of the Seebeck coefficients versus the logarithm of carrier concentration in the accumulation region for IDTBT, PBTTT and PSeDPPBT at 300 K. The solid lines in b are plots of α = (kB/e)ln(2N/n). The carrier concentration in IDTBT is slightly lower than in the other polymers because of the Cytop gate dielectric used, which has a low dielectric constant. The measurement error of the Seebeck coefficient is estimated to be ±70 μV K−1 for the IDTBT device (Supplementary Information section 3).
We have attempted to interpret the Seebeck and FET measurements as functions of temperature consistently in terms of the variable-range hopping disorder model used in ref. 24, that is, akin to models used to explain analogous measurements in amorphous silicon25. For PBTTT and PSeDPPBT this may be possible, but the fits depend on several unknown parameters and, as discussed above, the disorder model breaks down for IDTBT (Supplementary Information section 2). A simpler, more consistent interpretation of the three salient Seebeck features that is applicable to all polymers is given by a narrow-band model in which charge carriers experience a small degree of energetic disorder and are able to access a temperature-independent density of thermally accessible sites. The narrowness of the carriers’ energy bands is probably due to polaron formation14, as supported by charge accumulation spectroscopy (Supplementary Information sections 5 and 6). In the simplest narrow-band model, the Seebeck coefficient can be expressed as the sum of three contributions14 (Supplementary Information section 7):

The first contribution is the change of the entropy of mixing when the density of mobile polarons is nc and the density of thermally accessible sites is N. The second contribution is the entropy change arising from the twofold spin degeneracy. The final term is the high-temperature limit of the entropy change produced by a polaron altering the stiffness or frequencies of the molecular vibrations. Only the first contribution depends explicitly on carrier density. Because in our organic FETs nc  N, the primary contribution to the Seebeck coefficient comes from the mixing contribution. Thus, a plot of α versus the logarithm of the mobile carrier density should yield a straight line with slope −(kB/e)ln(10) = −198 μV K−1 decade−1. It is evident from Fig. 2b that the slopes of the near-linear, experimental α–log(n) plots depend on the specific polymer and exceed this value. These discrepancies can be reconciled by taking into account that a fraction f of the n injected carriers are trapped in shallow traps and do not participate in transport. Then nc = n(1 − f) and the slope of the α–log(n) plot is increased to −(kB/e)ln(10)/(1 − f). This procedure is justified if these band-tail-like traps are within ∼kBT of the narrow band of conducting polaron states. We extract values of f = 0.3, 0.5 and 0.7 for IDTBT, PBTTT and PSeDPPBT, respectively. Thus, our Seebeck measurements indicate significantly less trapping in IDTBT than in PBTTT or PSeDPPBT; in IDTBT the majority of charge carriers reside in mobile states.
N, the primary contribution to the Seebeck coefficient comes from the mixing contribution. Thus, a plot of α versus the logarithm of the mobile carrier density should yield a straight line with slope −(kB/e)ln(10) = −198 μV K−1 decade−1. It is evident from Fig. 2b that the slopes of the near-linear, experimental α–log(n) plots depend on the specific polymer and exceed this value. These discrepancies can be reconciled by taking into account that a fraction f of the n injected carriers are trapped in shallow traps and do not participate in transport. Then nc = n(1 − f) and the slope of the α–log(n) plot is increased to −(kB/e)ln(10)/(1 − f). This procedure is justified if these band-tail-like traps are within ∼kBT of the narrow band of conducting polaron states. We extract values of f = 0.3, 0.5 and 0.7 for IDTBT, PBTTT and PSeDPPBT, respectively. Thus, our Seebeck measurements indicate significantly less trapping in IDTBT than in PBTTT or PSeDPPBT; in IDTBT the majority of charge carriers reside in mobile states.
To interpret the magnitude of the Seebeck coefficients, we estimate the number of equivalent sites in our polymers. By assuming there to be one equivalent site on each polymer repeat unit, we obtain N = 7.4 × 1020 cm−3 (IDTBT) and N = 8.9 × 1020 cm−3 (PBTTT) on the basis of reported unit cell parameters20,26. The solid black and red lines in Fig. 2b show the resulting estimates of the Seebeck coefficients for IDTBT and PBTTT, respectively, on ignoring the carrier-induced changes in these molecules’ vibrations. The small discrepancies between the solid lines of Fig. 2b and the experimental data may indicate the vibrational contribution. This interpretation yields 50–100 μV K−1 for the vibrational contribution of IDTBT. This appears reasonable, although smaller than what has been reported for pentacene (265 μV K−1; ref. 27) or boron carbides (200 μV K−1 at 300 K; ref. 28).
The small degree of disorder in IDTBT is also consistent with optical absorption measurements by photothermal deflection spectroscopy (Supplementary Information section 8). This technique provides a bulk-sensitive way of probing energetic disorder manifesting itself as sub-bandgap tail states of the excitonic joint density of states and of estimating their widths in terms of the Urbach energy, Eu, extracted from the optical absorption coefficient in the vicinity of the band gap Eg,  for E < Eg. For more disordered polymers, Eu has previously been found to correlate with the T0 values extracted from fits of device characteristics according to an empirical relationship Eu ≈ kBT0 (ref. 17). Among the ∼20 high-mobility polymers measured in this work (examples in Fig. 3 and Supplementary Fig. 13), IDTBT exhibits the lowest Urbach energy of 24 meV, which is less than kBT at room temperature and, to the best of our knowledge, is the lowest value reported in a conjugated polymer. Notably, the second- and third-lowest values are also measured in high-mobility polymers, naphtalenediimide-based P(NDI2OD-T2)8,17,29 (Eu = 31 meV) and DPPTTT (Eu = 33 meV). This should be compared with PBTTT (Eu = 47 meV).
for E < Eg. For more disordered polymers, Eu has previously been found to correlate with the T0 values extracted from fits of device characteristics according to an empirical relationship Eu ≈ kBT0 (ref. 17). Among the ∼20 high-mobility polymers measured in this work (examples in Fig. 3 and Supplementary Fig. 13), IDTBT exhibits the lowest Urbach energy of 24 meV, which is less than kBT at room temperature and, to the best of our knowledge, is the lowest value reported in a conjugated polymer. Notably, the second- and third-lowest values are also measured in high-mobility polymers, naphtalenediimide-based P(NDI2OD-T2)8,17,29 (Eu = 31 meV) and DPPTTT (Eu = 33 meV). This should be compared with PBTTT (Eu = 47 meV).

Absorption coefficient of IDTBT, DPPTTT, PSeDPPBT and PBTTT films, measured by photothermal deflection spectroscopy. Solid lines represent exponential tail fits for extraction of the Urbach energies Eu (inset). A relative error of 5% in the value of Eu was estimated to result from uncertainty in the fitting procedure.
Our results demonstrate that donor–acceptor copolymers without pronounced crystallinity can exhibit a lower degree of energetic disorder than crystalline or semicrystalline conjugated polymers; it is important to understand the underlying microstructural origin for this. We are also interested in whether IDTBT’s exceptional properties originate in certain unique molecular design features that may not yet be implemented to the same degree in other polymers with comparable mobilities but with otherwise less ideal transport characteristics. IDTBT cannot simply be understood as a classical rigid-rod polymer; its high solubility in a wide range of solvents suggests a degree of chain flexibility that is not common for such polymers (Supplementary Information section 1). To understand these matters better, we have modelled the three-dimensional structures of IDTBT, P(NDI2OD-T2) and PBTTT by combining quantum chemical and molecular dynamics calculations30,31 (Supplementary Information section 9). The conformational search points to interdigitated side chains as the thermodynamic, lowest-energy structures in the three polymers (Supplementary Fig. 14). However, in contrast to PBTTT26, for IDTBT the X-ray pattern simulated for such a dense, ordered, interdigitated side-chain arrangement is not in agreement with experimental data20. Instead, much better agreement with the measured X-ray diffraction is obtained when a less dense, disordered, non-interdigitated side-chain arrangement is built from numerical annealing experiments (Supplementary Fig. 15). A similar protocol was also applied to simulate side-chain disorder in P(NDI2OD-T2) and PBTTT. In relation to their crystalline phases, the backbone conformations in these disordered structures differ significantly between the polymers (Fig. 4a): IDTBT adopts a wavy, yet remarkably planar, largely torsion-free backbone; the deviation from planarity remains exceptionally small (torsion angle of 5.2 ± 4.0°). P(NDI2OD-T2) behaves similarly; although it is not a planar molecule, the torsion-angle distribution between the NDI and thiophene units remains relatively narrow (38.2 ± 10.7°). In contrast, PBTTT chains, while maintaining a linear conformation, explore a broader range of torsion angles (27.2 ± 14.6° between thiophene and thienothiophene).

a, Simulations of the backbone conformation of IDTBT and PBTTT in side-chain-disordered and non-interdigitated structures. The side chains and hydrogen atoms are omitted for clarity. Yellow, sulphur atoms; blue, nitrogen atoms. b, Pressure dependence of the intensity ratio of the Raman transitions at 1,542 cm−1 and 1,613 cm−1 (top) and the Raman spectrum of IDTBT measured using a diamond-anvil cell (bottom). a.u., arbitrary units. c, Simulation of the backbone conformation of IDTBT in the amorphous phase. A single chain from the simulated unit cell has been highlighted in bright yellow (other colours as in a). d, Calculated gas-phase torsion potentials of IDTBT and PBTTT. For PBTTT, the potential for torsion between the thiophene and thienothiophene units is shown.
We have direct experimental evidence for a near-torsion-free backbone in IDTBT from pressure-dependent Raman spectroscopy (Fig. 4b and Supplementary Information section 10). If there was significant torsion in as-deposited films, the backbone could be planarized by applying a hydrostatic pressure of a few gigapascals, as previously observed for structurally related poly-dioctylfluorene-co-benzothiadiazole32 (F8BT), and the Raman intensity ratio between the ring stretching mode of the IDT unit at 1,613 cm−1 and the ring stretching mode of the BT unit at 1,542 cm−1 would be expected to be pressure dependent. However, we find experimentally that this ratio is remarkably pressure independent between 0 and 2.5 GPa, suggesting that the IDTBT backbone is indeed already planar in as-deposited films.
The frontier orbitals of the three theoretically investigated polymers are spread along the backbones (Supplementary Fig. 20), such that conformational disorder is expected to broaden the density of states (DOS). We have calculated the tail width of the DOS of the highest occupied molecular orbital (HOMO) in IDTBT to be the least affected by side-chain disorder; likewise for the DOS of the lowest unoccupied molecular orbital (LUMO) of P(NDI2OD-T2), here partly because of the stronger confinement of the LUMO on the NDI units. In contrast, the HOMO DOS of PBTTT broadens significantly on introducing side-chain disorder (Table 1). Remarkably, even in a completely amorphous phase simulated by cooling low-density systems made of initially highly energetic, randomly distributed oligomers (Supplementary Information section 9), IDTBT accommodates side-chain disorder through bends in the backbone while retaining its near-planar conformation (Fig. 4c); its DOS is not significantly broadened. In contrast, the other two polymers, in particular PBTTT, adopt conformations with larger spans in torsion angles and wider DOSs. The relative trend in disorder resilience evident from Table 1 is remarkably consistent with the measured Urbach energies and transport properties.
Our results provide an explanation for the surprisingly high mobilities in donor–acceptor copolymers with less crystalline microstructures than crystalline or semicrystalline P3HT or PBTTT, in terms of a low degree of energetic disorder originating in a remarkable resilience of the backbone conformation to side-chain disorder, which is inevitable when thin films are solution-deposited by rapid drying techniques. The exceptional properties of IDTBT suggest several cooperating molecular design guidelines for discovering a wider class of such ‘disorder-free’ conjugated polymers: (1) collinear conjugated units with only a single or a minimal number of torsion-susceptible linkages in an extended repeat unit (also, the electronic structure will tend to be less susceptible to residual torsions for larger conjugated units); (2) a relatively steep gas-phase torsion potential with minima ideally (though not necessarily) around 180°, 0° or both (Fig. 4d); and (3) long side-chain substitution on both sides of one of the conjugated units to enable space filling in non-interdigitated structures without introducing backbone torsion and hindering close π−π contacts. Transport in such torsion-free polymers is approaching intrinsic limits, in which all molecular sites along the polymer backbone are thermally accessible; even higher mobilities might be achievable in this regime through closer π–π contacts. The level of energetic disorder as measured by the Urbach energy is comparable to that of certain inorganic crystals, such as GaN (ref. 33). That this is possible in near-amorphous polymers is highly surprising. Our results could lead to a new generation of disorder-free conjugated polymers with improved charge, exciton, spin and other transport properties for a broad range of applications, and to the observation of physical phenomena that have hitherto been prevented by disorder-induced localization.
References
Bruetting, W. Physics of Organic Semiconductors (Wiley-VCH, 2005)
Bässler, H. Localized states and electronic transport in single component organic solids with diagonal disorder. Phys. Stat. Solidi B 107, 9–54 (1981)
Sirringhaus, H. Device physics of solution-processed organic field-effect transistors. Adv. Mater. 17, 2411–2425 (2005)
Rivnay, J. et al. Structural origin of gap states in semicrystalline polymers and the implications for charge transport. Phys. Rev. B 83, 121306 (2011)
Noriega, R. et al. A general relationship between disorder, aggregation and charge transport in conjugated polymers. Nature Mater. 12, 1038–1044 (2013)
Zhang, W. M. et al. Indacenodithiophene semiconducting polymers for high performance air-stable transistors. J. Am. Chem. Soc. 132, 11437–11439 (2010)
Nielsen, C. B., Turbiez, M. & McCulloch, I. Recent advances in the development of semiconducting DPP-containing polymers for transistor applications. Adv. Mater. 25, 1859–1880 (2013)
Kim, N.-K. et al. Solution-processed barium salts as charge injection layers for high performance N-channel organic field-effect transistors. ACS Appl. Mater. Interfaces 6, 9614–9621 (2014)
Kim, G. et al. A thienoisoindigo-naphthalene polymer with ultrahigh mobility of 14.4 cm2/V s that substantially exceeds benchmark values for amorphous silicon semiconductors. J. Am. Chem. Soc. 136, 9477–9483 (2014)
Tseng, H.-R. et al. High-mobility field-effect transistors fabricated with macroscopic aligned semiconducting polymers. Adv. Mater. 26, 2993–2998 (2014)
Callen, H. B. Thermodynamics Ch. 17 (Wiley, 1960)
Emin, D. Seebeck effect. In Wiley Encyclopedia of Electrical and Electronics Engineering Online (ed. Webster, J. G. ) (Wiley, 2013)
Emin, D. Enhanced Seebeck coefficient from carrier-induced vibrational softening. Phys. Rev. B 59, 6205–6210 (1999)
Emin, D. Polarons (Cambridge Univ. Press, 2013)
Pernstich, K. P., Roessner, B. & Batlogg, B. Field-effect-modulated Seebeck coefficient in organic semiconductors. Nature Mater. 7, 321–325 (2008)
Kronemeijer, A. J. et al. A selenophene-based low-bandgap donor–acceptor polymer leading to fast ambipolar logic. Adv. Mater. 24, 1558–1565 (2012)
Kronemeijer, A. J. et al. Two-dimensional carrier distribution in top-gate polymer field-effect transistors: correlation between width of density of localized states and Urbach energy. Adv. Mater. 26, 728–733 (2014)
Chen, Z. et al. High performance ambipolar diketopyrrolopyrrole-thieno[3,2-b]thiophene copolymer field-effect transistors with balanced electron and hole mobilities. Adv. Mater. 24, 647–652 (2012)
Li, J. et al. A stable solution-processed polymer semiconductor with record high mobility for printed transistors. Sci. Rep. 2, 754 (2012)
Zhang, X. et al. Molecular origin of high field-effect mobility in an indacenodithiophene–benzothiadiazole copolymer. Nature Commun. 4, 2238 (2013)
Sirringhaus, H. Organic field-effect transistors — the path beyond amorphous silicon. Adv. Mater. 26, 1319–1335 (2014)
Brondijk, J. J. et al. Two-dimensional charge transport in disordered organic semiconductors. Phys. Rev. Lett. 109, 056601 (2012)
Venkateshvaran, D., Kronemeijer, A. J., Moriarty, J., Emin, D. & Sirringhaus, H. Field-effect modulated Seebeck coefficient measurements in an organic polymer using a microfabricated on-chip architecture. APL Mater. 2, 032102 (2014)
Germs, W. C., Guo, K., Janssen, R. A. J. & Kemerink, M. Unusual thermoelectric behavior indicating hopping to bandlike transition in pentacene. Phys. Rev. Lett. 109, 016601 (2012)
Overhof, H. & Beyer, W. A model for the electronic transport in hydrogenated amorphous silicon. Philos. Mag. B 43, 433–450 (1981)
DeLongchamp, D. M. et al. High carrier mobility polythiophene thin films: structure determination by experiment and theory. Adv. Mater. 19, 833–837 (2007)
von Mühlenen, A., Errien, N., Schaer, M., Bussac, M.-N. & Zuppiroli, L. Thermopower measurements on pentacene transistors. Phys. Rev. B 75, 115338 (2007)
Aselage, T. L., Emin, D., McCready, S. S. & Duncan, R. V. Large enhancement of boron carbides’ Seebeck coefficients through vibrational softening. Phys. Rev. Lett. 81, 2316–2319 (1998)
Yan, H. et al. A high-mobility electron-transporting polymer for printed transistors. Nature 457, 679–686 (2009)
Olivier, Y. et al. High-mobility hole and electron transport conjugated polymers: how structure defines function. Adv. Mater. 26, 2119–2136 (2014)
Cho, E. et al. Three-dimensional packing structure and electronic properties of biaxially oriented poly(2,5-bis(3-alkylthiophene-2-yl)thieno[3,2-b]thiophene) films. J. Am. Chem. Soc. 134, 6177–6190 (2012)
Schmidtke, J. P., Kim, J.-S., Gierschner, J., Silva, C. & Friend, R. H. Optical spectroscopy of a polyfluorene copolymer at high pressure: intra- and intermolecular interactions. Phys. Rev. Lett. 99, 167401 (2007)
Jacobson, M. A., Konstantinov, O. V., Nelson, D. K., Romanovskii, S. O. & Hatzopoulos, Z. Absorption spectra of GaN: film characterization by Urbach spectral tail and the effect of electric field. J. Cryst. Growth 230, 459–461 (2001)
Acknowledgements
We gratefully acknowledge financial support from the Engineering and Physical Sciences Research Council through a programme grant (EP/G060738/1) and the Technology Strategy Board (PORSCHED project). D.V. acknowledges financial support from the Cambridge Commonwealth Trust through a Cambridge International Scholarship. K.B. acknowledges post-doctoral fellowship support from the German Research Foundation. M.Z. acknowledges funding from NanoDTC in Cambridge. The work in Mons was supported by the European Commission/Région Wallonne (FEDER – Smartfilm RF project), the Interuniversity Attraction Pole programme of the Belgian Federal Science Policy Office (PAI 7/05), Programme d’Excellence de la Région Wallonne (OPTI2MAT project) and FNRS-FRFC. D.B. and J.C. are FNRS Research Fellows.
Author information
Authors and Affiliations
Contributions
D.V. designed and fabricated the devices and performed field-effect modulated Seebeck measurements on them. M.N. and A.J.K. optimized the fabrication of IDTBT-based organic FETs and performed transistor measurements. A.S. and M.N. performed photothermal deflection spectroscopy measurements. V.P. optimized the patterning procedure for organic devices. V.L., M.Z., Y.O., J.C. and D.B. performed quantum chemical and molecular dynamic simulations. M.Z. and M.K. acquired the high-pressure induced Raman spectra. K.B. performed measurements on DPPTTT-based devices. I.N. and I.R. performed charge accumulation spectroscopy measurements (Supplementary Information). I.M. and M.H. synthesized IDTBT. D.E. explained the Seebeck measurements on the basis of a narrow-band model. H.S. directed and coordinated the research. D.V., M.N., V.L., Y.O., J.C., D.B., D.E. and H.S. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
This file contains Supplementary Text and Data, Supplementary Figures 1-20, Supplementary Tables 1-2 and Supplementary references. (PDF 8680 kb)
Rights and permissions
About this article

Cite this article
Venkateshvaran, D., Nikolka, M., Sadhanala, A. et al. Approaching disorder-free transport in high-mobility conjugated polymers. Nature 515, 384–388 (2014). https://doi.org/10.1038/nature13854
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature13854
- Springer Nature Limited
This article is cited by
-
Nanoscale doping of polymeric semiconductors with confined electrochemical ion implantation
Nature Nanotechnology (2024)
-
Unraveling the crucial role of trace oxygen in organic semiconductors
Nature Communications (2024)
-
Numerical Simulation and Optimization of Stable Coaxial Jet Formation and Direct-Write Printing Array Nanoarchitectonics
Journal of Electronic Materials (2024)
-
Strain-enhanced electrical performance in stretchable semiconducting polymers
npj Flexible Electronics (2023)
-
Wafer-scale organic-on-III-V monolithic heterogeneous integration for active-matrix micro-LED displays
Nature Communications (2023)