
Abstract
While therapy-related (t)-myelodysplastic syndromes (MDS) have worse outcomes than de novo MDS (d-MDS), some t-MDS patients have an indolent course. Most MDS prognostic models excluded t-MDS patients during development. The performances of the International Prognostic Scoring System (IPSS), revised IPSS (IPSS-R), MD Anderson Global Prognostic System (MPSS), WHO Prognostic Scoring System (WPSS) and t-MDS Prognostic System (TPSS) were compared among patients with t-MDS. Akaike information criteria (AIC) assessed the relative goodness of fit of the models. We identified 370 t-MDS patients (19%) among 1950 MDS patients. Prior therapy included chemotherapy alone (48%), chemoradiation (31%), and radiation alone in 21%. Median survival for t-MDS patients was significantly shorter than for d-MDS (19 vs 46 months, P<0.005). All models discriminated survival in t-MDS (P<0.005 for each model). Patients with t-MDS had a significantly higher hazard of death relative to d-MDS in every risk model, and had inferior survival compared to patients with d-MDS within all risk group categories. AIC Scores (lower is better) were 2316 (MPSS), 2343 (TPSS), 2343 (IPSS-R), 2361 (WPSS) and 2364 (IPSS). In conclusion, subsets of t-MDS patients with varying clinical outcomes can be identified using conventional risk stratification models. The MPSS, TPSS and IPSS-R provide the best predictive power.
Similar content being viewed by others


Introduction
Myelodysplastic syndromes (MDS) include a number of biologically and clinically heterogeneous clonal hematologic entities that are collectively characterized by varying degrees of bone marrow failure, dysplastic maturation of myeloid elements, and a tendency for progression into more aggressive phenotypes, that is, acute myeloid leukemia (AML).1 Therapy-related (t)-MDS represent a subset of all MDS diagnoses that are descriptively defined when the diagnosis of MDS is made subsequent to DNA-damaging chemotherapy or radiotherapy administered for a prior non-myeloid malignancy or medical condition.2, 3 Although it is difficult to accurately ascertain the prevalence of t-MDS, different reports estimate that 6–20% of all MDS cases meet these criteria.4 Secondary MDS is another classification of myeloid neoplasms that is at times used interchangeably with t-MDS; however, this is not correct as the term secondary MDS is a broader, more inclusive diagnosis that refers to cases of MDS for which the cause is known including cases that evolve from antecedent hematologic disorders, genetic diseases, and environmental/occupational exposures (for example, benzene or radiation) in addition to cases with a history of exposure to prior iatrogenic or occupational chemotherapy or radiotherapy.5 As the number of cancer survivors increases with time, more patients will be diagnosed with t-MDS.5, 6, 7
Patients with MDS have been well recognized to exhibit wide variability in their clinical course and disease aggressiveness. As such, accurate risk stratification has a paramount role in directing appropriate recommendation of risk-adaptive therapies.8, 9 Among the commonly used prognostic tools for de novo (d)-MDS in clinical practice are the International Prognostic Scoring System (IPSS) reported in 1997 and its revised version (IPSS-R), which was published 25 years later.10, 11 Other risk stratification tools used in the clinic include the World Health Organization (WHO)-based Prognostic Scoring System (WPSS) and the MD Anderson Global Prognostic Scoring System (MPSS).12, 13 These tools use standard clinicopathologic and laboratory parameters such as bone marrow blast percentage, number and severity of blood cytopenias, cytogenetic aberrations and/or transfusion dependence to provide prognostic information to be used in the process of clinical decision-making.14 Patients with higher-risk MDS are generally recommended to undergo aggressive interventions such allogeneic hematopoietic stem cell transplantation (alloHSCT) if possible or/and therapy with hypomethylating agents (HMAs) or, rarely, intensive chemotherapy.15 Patients with lower-risk (LR)-MDS are treated generally with supportive measures focused at improving quality of life.
Historically t-MDS were associated with a poor prognosis due to the high prevalence of adverse prognostic factors, such as poor risk karyotypes and higher BM blast percentages.2, 16 One study found that morphologic subclassification and percentage of bone marrow blasts may not be even clinically relevant among patients with t-MDS as such patients experience a uniformly poor outcome.16 There has been a tendency to treat patients with t-MDS as a group with more aggressive interventions when feasible in an attempt to alter the natural history of the disease and prolong survival. However, it has become increasingly apparent that patients with t-MDS exhibit significant heterogeneity in clinical outcomes with a subset of patients having a more indolent disease course and who might not necessarily benefit from aggressive therapeutic interventions.17
Although the previously mentioned prognostic tools may be commonly used in the clinic for risk stratification of patients with t-MDS, the IPSS, IPSS-R and the WPSS were actually developed from databases that excluded patients with t-MDS, whereas the MPSS data set included a small proportion of patients with t-MDS.10, 11, 12, 13 Therefore, the prognostic utility of these tools remains uncertain in this setting. The MD Anderson group recently reported a risk stratification developed specifically for patients with t-MDS, the t-MDS Prognostic Scoring System (TPSS).17 However, the TPSS has only been validated externally in a small single-institution European analysis among azacitidine-treated patients, and its prognostic utility has not been directly compared to the other risk stratification tools.18 To better identify variation in the natural history of t-MDS, we compared the clinical outcomes and assessed the performance of several risk models in patients with t-MDS and d-MDS in one of the largest cohorts reported to date.
Materials and methods
Study population and procedures
We identified patients diagnosed with t-MDS and those with d-MDS in the Moffitt Cancer Center MDS Clinical database. This study was performed under the umbrella of the MDS Clinical Research Consortium. We defined patients with t-MDS as any patient with a pathologically confirmed diagnosis of MDS who had a prior history of exposure to chemotherapy and/or radiotherapy for a malignancy or a medical condition regardless of latency period. The risk stratification models including the IPSS, IPSS-R, MPSS, WPSS and the TPSS were calculated as previously reported at the time of presentation.10, 11, 12, 13, 17 The study protocol was reviewed and approved by the Moffitt Institutional Review Board.
Statistical considerations
Baseline characteristics were summarized for both groups (t-MDS and d-MDS) using the means, medians and proportions, and were compared using analysis of variance (ANOVA), Kruskal–Wallis test, and Pearson’s χ2-test as appropriate. Responses were evaluated using the International Working Group 2006 criteria (IWG-2006).19 Overall survival (OS) was calculated from time of diagnosis. Kaplan–Meier estimates were used to depict OS curves and estimate median OS. Log-rank tests were used to assess survival differences across groups. Cox proportional hazards models were used to estimate adjusted hazard ratios of t-MDS relative to d-MDS while likelihood ratio tests from these models were used to assess interaction effects of MDS type with risk model. Akaike information criteria (AIC) were used to assess the relative goodness fit of the various Cox models where the lower the AIC, the better the fit. Two sided P-values <0.05 defined significance.
Results
Study cohort
We identified 370 patients diagnosed with t-MDS (19%) and 1576 patients diagnosed with d-MDS (81%). Table 1 summarizes the baseline characteristics and demographics of patients in both of these groups. More than three quarters of patients were 60 years old at diagnosis or older (76% vs 81% for t-MDS vs d-MDS, respectively). More than half of the patients had previously been treated for a solid tumor. Prior therapy included chemotherapy alone (48%), chemoradiation (31%) and radiation alone in 21%. More patients with t-MDS had refractory anemia with excess blasts 1 (RAEB-1) and 2 (RAEB-2) than did patients with d-MDS (43% vs 36%, respectively, <0.005). In addition, there was a significantly higher prevalence of adverse prognostic factors among patients with t-MDS than d-MDS (poor risk karyotypes including complex cytogenetics and chromosome 7 abnormalities, and circulating blasts, Table 1). As a result, more patients with t-MDS had higher-risk categories when applying the different prognostic tools than those with d-MDS (Table 1). Nonetheless, a significant subset of patients with t-MDS had LR-MDS by the IPSS (low (14%), intermediate-1 (37%)), the IPSS very low (9%), low (25%)) and the MPSS (low (11%), intermediate-1 (29%)).
Treatment patterns and clinical outcomes
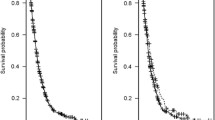
The median OS for patients with t-MDS cases was significantly shorter than those with d-MDS (19 vs 46 months, respectively, P<0.005). The rates of documented progression to AML transformation, though, were not significantly different between t-MDS and d-MDS (30% vs 28%, respectively). Among patients with t-MDS, there was no significant statistical difference in OS based on prior therapy received in unadjusted analysis (median OS for prior chemotherapy alone, 18.5 months (95% confidence interval (CI), 13.7–23.3 months); for prior radiotherapy alone, 27.6 months (95% CI, 19.9–35.3 months); and for prior combined chemo-radiotherapy 17.0 months (95%CI, 14.5–19.5 months; P=0.27, Figure 1). There was also no difference in OS based on type of the prior malignancy (Supplementary Information). When we analyzed the effect of inclusion of alkylating agents in the regimens, we found no overall effect on survival though there was a trend of inferior survival in the first year in landmark analyses that disappeared in subsequent years (Supplementary Information).

Comparison of overall survival among patients with t-MDS (n=370) based on prior therapy received (chemotherapy, radiotherapy, combined chemotherapy/radiotherapy).
Similarly, the rates of use of HMA were not different between t-MDS and d-MDS (63 vs 60%, respectively). Among patients who received HMAs, patients with t-MDS received a significantly lower number of cycles (median, 5 vs 6 cycles, P<0.001) compared to patients with d-MDS (Supplementary Information). Of t-MDS patients treated with HMAs, 34% responded according to IWG-2006 criteria with no difference in the complete remission (CR) from patients with d-MDS who receive HMAs.
Although a higher proportion of patients with t-MDS underwent alloHSCT compared to those with d-MDS (21.5% vs 12.2%, P<0.001), there were no significant differences in survival at 6 months, 1 year and 2 years from time of transplantation (Supplementary Information). However, there was a statistically insignificant trend for inferior 2-year survival probability for patients with t-MDS compared to d-MDS (38.6% vs 47.3%).
Prognostic utility of the risk stratification tools for survival prediction
All prognostic models discriminated OS based on risk group among t-MDS patients (Table 2, Figures 2a–c and 3a and b) with log-rank P-values <0.001 for every risk model. Patients with t-MDS had a significantly higher hazard of death relative to d-MDS in every risk model (Table 3) and had inferior OS compared to patients with d-MDS within all risk group categories (Table 2). No significant evidence was found that the hazard ratio for t-MDS vs d-MDS varied with risk category for any risk model (Table 3). The scores generated using AIC to assess the relative goodness of fit (lower is better) on the subset of patients for which there were observed risk categories across all models (n=323) were 2316 (MDSS), 2343 (TPSS), 2343 (IPSS-R), 2361 (WPSS) and 2364 (IPSS).

Estimates of overall survival among patients with t-MDS (n=370) by the risk group categories of the (a) IPSS-R, (b) IPSS, and (c) WPSS.

Estimates of overall survival among patients with t-MDS (n=370) by the risk group categories of the (a) MPSS and (b) TPSS.
Discussion and conclusions
Formally, t-MDS is recognized within the WHO-defined spectrum of therapy-related myeloid neoplasms (t-MN), which are diagnosed following exposure to DNA damaging agents in patients with a prior non-myeloid malignancy.2, 3, 16 In this study, we have specifically evaluated t-MDS rather than the broader category of t-MN to appropriately apply MDS risk stratification tools.17 We report one of the largest series of patients with t-MDS (n=370) and compare their outcomes to patients with d-MDS (n=1576). We confirm that, as a group, patients with t-MDS have significantly inferior survival than d-MDS (median, 19 vs 46 months, respectively, P<0.005). We also confirm that patients with t-MDS have a higher prevalence of adverse prognostic factors such as poor risk cytogenetics and higher blast percentages.
For every risk category in the different stratification tools, patients with t-MDS had comparatively worse survival than d-MDS (although not reaching statistical significance for IPSS-R very low and MPSS low risk groups). This finding suggests that factors beyond the standard parameters captured by the traditional prognostic tools might be involved in the inferior outcomes of t-MDS (for example, higher prevalence of adverse genetic mutations such as TP53). The lack of difference in rates of progression to AML between t-MDS (30%) and d-MDS (28%) suggests that shorter survival among the t-MDS patients could be related to worse cytopenias, more comorbidities, prior malignancies, or higher toxicity with MDS therapies
It is possible that patients who are diagnosed with t-MDS soon after finishing chemotherapy or radiotherapy for the original malignancy could be dying from reasons related to their original malignancy rather than complications of t-MDS. As we did not have causes of death for individual patients available in our data set, we conducted a multivariable Cox proportional hazards analysis for effect on survival based on time of diagnosis of t-MDS from completing prior cancer-directed therapy and found that a diagnosis of t-MDS within 2 years of completing therapy (whether chemotherapy, radiation or chemoradiation) was not associated with worse survival compared to those who developed t-MDS more than 2 years from finishing prior therapy (Supplementary Information).
Our findings also suggest that worse transplant-related mortality (TRM) and inferior survival after alloHSCT for patients with t-MDS compared to those with d-MDS do not account for the worse survival of patients with t-MDS as a group compared to those with d-MDS (Supplementary Information).
The rates of HMAs use were similar but median number of cycles was lower for t-MDS patients compared to d-MDS. It is unclear whether this was due to higher rates of intolerance or resistance among patients with t-MDS. We confirmed activity of HMAs as a treatment option for patients with t-MDS with a response rate (34%) that is similar to what was previously reported (38–43%)20, 21, 22 and without difference in CR rates from d-MDS.
The highly variable clinical course has long been recognized in MDS. As such, risk stratification tools have played a central role in the management paradigms. Importantly, the risk stratification models were developed from databases that excluded (IPSS, IPSS-R, and WPSS) or had a small proportion of patients with t-MDS (MPSS).10, 11, 13 Furthermore, these tools have not been formally validated in large cohorts of t-MDS patients and therefore the prognostic utility of these risk systems has not been definitely established in the setting of t-MDS.
Due to their worse outcomes, the conventional wisdom has been that all patients with t-MDS should be offered treatments similar to what is offered to higher-risk MDS, including aggressive therapeutic interventions such as alloHSCT. However, it has become been well established that subsets of patients with t-MDS have a more indolent clinical course than expected.17 In our report, we show that patients with t-MDS who belonged to the lowest risk group categories in the different risk stratification tools had reasonably favorable OS (median OS in months: IPSS low 58, IPSS-R very low 58, IPSS-R low 41, MPSS low 97, WPSS low 36 and TPSS low 46) in comparison to the overall median OS for patients with t-MDS (19 months). In fact, the survival estimates for patients with IPSS very low and MPSS low risk groups were not even significantly different for patients with t-MDS than those with d-MDS.
These findings provide further evidence for the significant variation in the natural history of t-MDS and that some patients with t-MDS have less aggressive disease phenotypes and therefore serve as rationale for incorporating risk stratification in the counseling and individualizing the clinical decision-making process. Here we also validated the prognostic utility of the IPSS, IPSS-R and MPSS in this setting and allowed us to compare the performance of several risk models in t-MDS. Each of the IPSS, IPSS-R, and MPSS retains prognostic utility in patients with t-MDS with significantly different median OS between the risk groups of each risk tool. We confirm the very poor survival of patients with the highest risk categories (IPSS intermediate-2 and high, IPSS-R high and very high, MPSS intermediate-2 and high, and WPSS high and very high) in t-MDS supporting use of aggressive therapeutic interventions for these patients.
Furthermore, our results provide t-MDS-specific benchmarks for survival estimation across the risk categories for each prognostic tool that can be used for counseling, risk-adaptive decisions, and clinical trial design for patients with t-MDS. We also externally validated the prognostic utility of the recently described t-MDS-specific TPSS.17 However, the AIC scores suggest that the TPSS does not offer significant prognostic precision beyond that provided by the commonly used IPSS-R and MPSS. In fact, the MPSS provided better, and the IPSS-R provided similar, predictive power compared to the TPSS. All these systems had better predictive power among t-MDS than the WPSS and the IPSS.
Like any retrospective analysis, this study has several limitations. Our results come from a single center and a very small proportion (2%) of t-MDS patients had a prior myeloid malignancy. It is important to note that t-MDS is a descriptive designation and does not necessarily imply causation in relation to prior therapy.3 Recent data suggest that the behavior of MDS might differ based on the exposure with more aggressive phenotypes following combined chemoradiation.7 It is even in doubt whether modern radiation therapy with limited fields in the absence of chemotherapy increases the risk of subsequent development of MDS in some malignancies (for example, localized breast and prostate cancers) and therefore the biology and behavior of MDS that occur in this setting might be closer to d-MDS than t-MDS under which it is currently classified.7, 23, 24, 25 Although examination of radiation intensity effect on outcomes is important, we were unable to conduction this analysis due to lack of availability of the data regarding the different variables of radiation intensity such as dose, field, technique, use of shielding and so on.
In our cohort, we found no difference in OS among t-MDS patients based on type of prior malignancy or modality of prior therapy. However, this analysis was unadjusted and larger number of patients to ensure adequate power for adjusted analyses. It would also be of interest to compare the natural history and the prognostic utility of risk tools among the three subgroups of t-MDS based on prior therapeutic exposure. In addition, the patients in this cohort were treated with different treatment modalities and we did not account for the differential effects of treatments on the survival predictions and risk tool comparisons. We did not evaluate whether the use of azacitidine versus decitabine leads to differential clinical benefit in patients with t-MDS (or even in all patients of MDS),26, 27, 28, 29 and this would be an important question to address in future studies for medical and economic reasons.
Finally, the importance of molecular mutations in driving the pathogenesis and informing the prognostication of MDS has been well established.24, 30, 31 For example, recent data suggest that the presence of TP53 mutations independently predict for significantly inferior OS of patients regardless of the karyotypes.30, 31, 32 We did not have molecular data in the current cohort to inform the evaluation of the survival predictions or the risk tool comparisons. Also, recent data suggested that patients with clonal hematopoiesis of indeterminate potential (CHIP) at time of their first cancer diagnosis are at higher risk for subsequent development of t-MN.33 However, how do the presence of of pre-existing CHIP and specific therapy administered for the primary malignancy interact in the subsequent development and clinical behavior of t-MN is not yet understood. Future investigations adding genomic data at the time of primary malignancy and/or time of t-MN diagnosis may improve the prognostic utility of t-MDS risk stratification tools.
In conclusion, understanding the differences in biology between t-MDS and d-MDS is vital to differentiate ‘true’ t-MDS from d-MDS that is incidentally diagnosed after prior radiation and/or chemotherapy. This in turn will help improve the risk stratification tools by incorporating some of the independently prognostic biomarkers to optimize the risk stratification tools precision and accuracy. Importantly, it could also uncover targets for therapeutics that might improve the poor outcomes of patients with t-MDS and potentially open the door for preventive strategies to reduce the risk of occurrence of t-MDS following therapeutic intervention for the primary malignancy.
References
Zeidan AM, Linhares Y, Gore SD . Current therapy of myelodysplastic syndromes. Blood Rev 2013; 27: 243–259.
Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 2009; 114: 937–951.
Larson RA . Cytogenetics, not just previous therapy, determines the course of therapy-related myeloid neoplasms. J Clin Oncol 2012; 30: 2300–2302.
Churpek JE, Larson RA . The evolving challenge of therapy-related myeloid neoplasms. Best Pract Res Clin Haematol 2013; 26: 309–317.
Granfeldt Ostgard LS, Medeiros BC, Sengelov H, Norgaard M, Andersen MK, Dufva IH et al. Epidemiology and clinical significance of secondary and therapy-related acute myeloid leukemia: a National Population-Based Cohort Study. J Clin Oncol 2015; 33: 3641–3649.
Kaplan H, Malmgren J, De Roos AJ . Risk of myelodysplastic syndrome and acute myeloid leukemia post radiation treatment for breast cancer: a population-based study. Breast Cancer Res Treat 2013; 137: 863–867.
Wolff AC, Blackford AL, Visvanathan K, Rugo HS, Moy B, Goldstein LJ et al. Risk of marrow neoplasms after adjuvant breast cancer therapy: the national comprehensive cancer network experience. J Clin Oncol 2015; 33: 340–348.
Zeidan AM, Komrokji RS . There's risk, and then there's risk: The latest clinical prognostic risk stratification models in myelodysplastic syndromes. Curr Hematol Malig Rep 2013; 8: 351–360.
Faltas B, Zeidan A, Gergis U . Myelodysplastic syndromes: toward a risk-adapted treatment approach. Expert Rev Hematol 2013; 6: 611–624.
Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997; 89: 2079–2088.
Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Sole F et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012; 120: 2454–2465.
Malcovati L, Germing U, Kuendgen A, Della Porta MG, Pascutto C, Invernizzi R et al. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J Clin Oncol 2007; 25: 3503–3510.
Kantarjian H, O'Brien S, Ravandi F, Cortes J, Shan J, Bennett JM et al. Proposal for a new risk model in myelodysplastic syndrome that accounts for events not considered in the original International Prognostic Scoring System. Cancer 2008; 113: 1351–1361.
Lee EJ, Podoltsev N, Gore SD, Zeidan AM . The evolving field of prognostication and risk stratification in MDS: Recent developments and future directions. Blood Rev 2016; 30: 1–10.
Zeidan AM, Stahl M, Komrokji R . Emerging biological therapies for the treatment of myelodysplastic syndromes. Expert Opin Emerg Drugs. 2016; 21: 283–300.
Singh ZN, Huo D, Anastasi J, Smith SM, Karrison T, Le Beau MM et al. Therapy-related myelodysplastic syndrome: morphologic subclassification may not be clinically relevant. Am J Clin Pathol 2007; 127: 197–205.
Quintas-Cardama A, Daver N, Kim H, Dinardo C, Jabbour E, Kadia T et al. A prognostic model of therapy-related myelodysplastic syndrome for predicting survival and transformation to acute myeloid leukemia. Clin Lymphoma Myeloma Leuk 2014; 14: 401–410.
Breccia M, Fianchi L, Lunghi M, Gaidano G, Levis A, Finelli C et al. Newly proposed therapy-related myelodysplastic syndrome prognostic score predicts significant differences in overall survival and leukemia-free survival in patients treated with azacitidine. Leuk Lymphoma 2013; 54: 1786–1787.
Cheson BD, Greenberg PL, Bennett JM, Lowenberg B, Wijermans PW, Nimer SD et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood 2006; 108: 419–425.
Klimek VM, Dolezal EK, Tees MT, Devlin SM, Stein K, Romero A et al. Efficacy of hypomethylating agents in therapy-related myelodysplastic syndromes. Leuk Res 2012; 36: 1093–1097.
Duong VH, Lancet JE, Alrawi E, Al-Ali NH, Perkins J, Field T et al. Outcome of azacitidine treatment in patients with therapy-related myeloid neoplasms with assessment of prognostic risk stratification models. Leuk Res 2013; 37: 510–515.
Bally C, Thepot S, Quesnel B, Vey N, Dreyfus F, Fadlallah J et al. Azacitidine in the treatment of therapy related myelodysplastic syndrome and acute myeloid leukemia (tMDS/AML): a report on 54 patients by the Groupe Francophone Des Myelodysplasies (GFM). Leuk Res 2013; 37: 637–640.
Nardi V, Winkfield KM, Ok CY, Niemierko A, Kluk MJ, Attar EC et al. Acute myeloid leukemia and myelodysplastic syndromes after radiation therapy are similar to de novo disease and differ from other therapy-related myeloid neoplasms. J Clin Oncol 2012; 30: 2340–2347.
Abou Zahr A, Kavi AM, Mukherjee S, Zeidan AM . Therapy-related myelodysplastic syndromes, or are they? Blood Rev, e-pub ahead of print 24 November 2016; doi:10.1016/j.blre.2016.11.002.
Wang R, Zeidan AM, Yu JB, Soulos PR, Davidoff AJ, Gore SD et al. Myelodysplastic syndromes and acute myeloid leukemia after radiotherapy for prostate cancer: a population-based study. Prostate, e-pub ahead of print 21 November 2016; doi:10.1002/pros.23281.
Zeidan A, Davidoff A, Long J, Hu X, Wang R, Ma X et al. Comparative clinical effectiveness of azacitidine versus decitabine in older patients with myelodysplastic syndromes. Br J Haematol 2016; 175: 829–840.
Zeidan A, Sekeres M, Garcia-Manero G, Steensma DP, Zell K, Barnard J et al. Comparison of risk stratification tools in predicting outcomes of patients with higher-risk myelodysplastic syndromes treated with azanucleosides. Leukemia 2016; 30: 649–657.
Zeidan A, Wang R, Gross C, Gore S, Huntington S, Prebet T et al. Modest improvement in survival of patients with refractory anemia with excess blasts in the hypomethylating agents era in the United States. Leuk Lymphoma 2016; 58: 982–985.
Zeidan A, Sekeres M, Wang X, Al Ali N, Garcia-Manero G, Steensma D et al. Comparing the prognostic value of risk stratifying models for patients with lower-risk myelodysplastic syndromes: is one model better? Am J Hematol 2015; 90: 1036–1040.
Abou Zahr A, Bernabe Ramirez C, Wozney J, Prebet T, Zeidan A . New insights into the pathogenesis of MDS and the rational therapeutic opportunities. Expert Rev Hematol 2016; 9: 377–388.
Nazha A, Sekeres M, Gore S, Zeidan A . Molecular testing in myelodysplastic syndromes for the practicing oncologist: will the progress fulfill the promise? Oncologist 2015; 20: 1069–1076.
Bejar R . Implications of molecular genetic diversity in myelodysplastic syndromes. Curr Opin Hematol 2017; 24: 73–78.
Gillis N, Ball M, Zhang Q, Ma Z, Zhao Y, Yoder S et al. Clonal haemopoiesis and therapy-related myeloid malignancies in elderly patients: a proof-of-concept, case-control study. Lancet Oncol 2016; 18: 112–121.
Acknowledgements
Funding for this research has been provided, in part from the Edward P. Evans Foundation to EP, MAS, DPS, AD, GR, GGM and RK.
Author contributions
AMZ designed the research, interpreted the data and wrote the manuscript. RK designed the research, supervised data analysis, interpreted the data, and critically revised the manuscript. JB analyzed the data and contributed to manuscript writing. NAA, EP, JEL, MAS, DPS, AD, GR, EJ, GGM, and AL reviewed the data and critically reviewed and contributed to the manuscript significantly. All authors approved the final manuscript for submission.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Leukemia website
Supplementary information
Rights and permissions
About this article

Cite this article
Zeidan, A., Al Ali, N., Barnard, J. et al. Comparison of clinical outcomes and prognostic utility of risk stratification tools in patients with therapy-related vs de novo myelodysplastic syndromes: a report on behalf of the MDS Clinical Research Consortium. Leukemia 31, 1391–1397 (2017). https://doi.org/10.1038/leu.2017.33
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2017.33
- Springer Nature Limited
This article is cited by
-
Therapy-related myeloid neoplasms: a real-world pharmacovigilance analysis (1970–2023)
Annals of Hematology (2024)
-
Therapy-related MDS dissected based on primary disease and treatment—a nationwide perspective
Leukemia (2023)
-
Therapy-related myelodysplastic syndromes deserve specific diagnostic sub-classification and risk-stratification—an approach to classification of patients with t-MDS
Leukemia (2021)
-
Which lower risk myelodysplastic syndromes should be treated with allogeneic hematopoietic stem cell transplantation?
Leukemia (2020)
-
The mutational burden of therapy-related myeloid neoplasms is similar to primary myelodysplastic syndrome but has a distinctive distribution
Leukemia (2019)