
Abstract
Although temozolomide (TMZ) is the first-line chemotherapeutic agent for glioblastoma, it is often non-curative due to drug resistance. To overcome the resistance of glioblastoma cells to TMZ, it is imperative to identify prognostic markers for outcome prediction and to develop chemo-sensitizing agents. Here, the gene expression profiles of TMZ-resistant and TMZ-sensitive samples were compared by microarray analysis, and mitogen-activated protein kinase kinase 2 (MEK2) was upregulated specifically in resistant glioma cells but not in sensitive tumor cells or non-tumor tissues. Moreover, a comprehensive analysis of patient data revealed that the increased level of MEK2 expression correlated well with the advancement of glioma grade and worse prognosis in response to TMZ treatment. Furthermore, reducing the level of MEK2 in U251 glioma cell lines or xenografted glioma models through shRNA-mediated gene knockdown inhibited cell proliferation and enhanced the sensitivity of cells toward TMZ treatment. Further analysis of tumor samples from glioma patients by real-time PCR indicated that an increased MEK2 expression level was closely associated with the activation of many drug resistance genes. Finally, these resistance genes were downregulated after MEK2 was silenced in vitro, suggesting that the mechanism of MEK2-induced chemo-resistance could be mediated by the transcriptional activation of these resistance genes. Collectively, our data indicated that the expression level of MEK2 could serve as a prognostic marker for glioma chemotherapy and that MEK2 antagonists can be used as chemo-sensitizers to enhance the treatment efficacy of TMZ.
Similar content being viewed by others


Introduction
Glioblastoma is one of the most common malignant central nervous system (CNS) tumors1. The prognosis of patients with glioma remains poor despite improved treatment modalities, including maximum surgical resection, chemotherapy with anti-neoplastic drugs and radiotherapy2,3. Temozolomide (TMZ), an alkylating agent, has shown promising results in the treatment of newly diagnosed patients with glioblastoma multiforme or refractory anaplastic astrocytoma when used as single agent as well as in combination with radiation therapy4. TMZ was approved by the Food and Drug Administration5 because it readily passes through the blood–brain barrier due to its lipophilic properties and causes little damage to the liver with its metabolites excreted by the kidneys. TMZ in combination with radiation therapy is recommended by NCCNand NICE6,7, thus making it a standardized treatment protocol for such diseases8. However, many glioma patients have been reported to be insensitive to TMZ treatment, and recent studies have demonstrated that several genes contribute to TMZ resistance, such as the base excision repair enzyme alkylpurine-DNA-N-glycosylate (APNG)9, methylguanine-DNA methyltransferase (MGMT)10 and signal transducers and activators of transcription 3 (STAT3)11. Other articles have also indicated that TMZ resistance is associated with the expression of vascular endothelial growth factor (VEGF)12 and epidermal growth factor receptor (EGFR)13. Changes in EGFR expression by itself activates both MEK and PI3 kinase pathways14,15.
Recently, a number of frequent genetic alterations have been identified in human glioblastoma16. Among all of these altered genes, the Ras-Raf-MAP kinase signaling pathway has demonstrated to be important in the control of glioma cell proliferation17. The importance of Raf in oncogenic signaling was confirmed by the discovery of activating BRAF mutations in a variety of human tumors, including 11% of glioma cell lines18. Additionally, the loss of NF1, a GAP protein that inactivates Ras, has also been detected in ∼20% of human GBM samples from the TCGA studies19. Although the relationship between aberrant activation of the mitogen-activated protein kinase (MAPK) pathway in gliomagenesis has been well established20, the role of this pathway in the drug resistance of gliomas remains to be elucidated.
In this paper, we found that the expression of MEK2 in primary glioma was upregulated and was correlated with glioma grades and the outcome of TMZ treatment. We further revealed that downregulation of MEK2 inhibited the ERK1/2 pathway and sensitized glioma cells to TMZ treatment.
Materials and methods
Cell lines and materials
The human glioma cell lines U251 and U87 were obtained from ATCC (Manassas, VA) and maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, Carlsbad, CA, USA) with 10% fetal bovine serum (Thermo Fisher Scientific, Inc., Waltham, MA, USA) and 50 U/mL penicillin and streptomycin (Invitrogen) in a 5% CO2 humidified atmosphere at 37 °C. TMZ was purchased from Sigma-Aldrich. MEK inhibitors, including PD184352 (CI-1040) and trametinib (GSK1120212), were purchased from Selleck.
Human tissues
All human glioma tissues were obtained from the Changzheng Hospital (China Secondary Military Medical University, Shanghai, China) in compliance with the laws and institutional guidelines approved by the Hospital Ethics Committee. Gliomas were graded by the Pathology Department of Changzheng Hospital according to the World Health Organization grading system. Human normal brain tissues (mostly from the cortex) were acquired from patients with physical injuries to the brain and were authorized by patients for further experiments.
Microarray analysis
RNA was extracted from three TMZ-sensitive and seven TMZ-resistant glioma patients and hybridized to a whole human genome oligo microarray (Agilent Technologies). Quantile normalization was used to equalize the distribution of probe intensities for each array, and two-sample t-statistics were calculated for each gene. The genes of differentiated expression were designated based on the criteria of P ≤ 0.05 and a fold change of ≥2 in their expression values between the two groups. The array clustering was performed by Cluster 3.0.
Real-time PCR
Real-time PCR was performed as previously described21. The following primers were used for each gene (from 5′ to 3′): MEK2 forward, GCGGTCACGGGATGGA TAG, MEK2 reverse, GCTCCGCTGGGTTCTTGAT; ABCB1 forward, GCTCCTGACTATGCCAAAGC, ABCB1 reverse, CTTCACCTCCAGGC TCAGTC; ABCC1 forward, AGGACACGTCGGAACAA GTC, ABCC1 reverse, AGGGGTTCCACTCCTTCTGT; ABCC2 forward, TCATTCAGACGACCATCCAA, ABCC2 reverse, TTCTCAATGCCAGCTTCCTT; ABCG2 forward, CACAGGTGGAGGCAAATCTT, ABCG2 reverse, CCGAAGAGCTGCTGAGAACT; BCL2 forward, TCCATGTCTTTGGACAACCA, BCL2 reverse, CTCCACCAGTGTTCCCATCT; MGMT forward, ATGGATGTTTG-AGCGACACA, MGMT reverse ATAGAGCAAGGGCAGCGTTA; GAPDH forward, ACAACTTTGGTATCGTGGAAGG, GAPDH reverse, GCCATCACGC-CACAGTTTC.
The PCR procedure was as follows: an initial denaturing step at 95 °C for 3 minutes, then 95 °C for 30 seconds, 60 °C for 30 seconds, and 72 °C for 30 seconds for a total of 40 cycles. The results are presented as the relative mRNA level of interest analyzed by normalizing the threshold cycle (Ct) value to that of the internal control (GAPDH) and the external control. The results are representative of at least three independent experiments.
Immunoblot and immunohistochemistry
The experiment was performed as previously described21. The MEK2 antibody was purchased from Abcam (ab28834). In the immunoblot, the relative amount of target protein was determined by normalizing the density value to an internal control (GAPDH) via Image Quant 5.2 (Molecule Dynamic). Immunohistochemistry was performed with paraffin-embedded tissue sections of 5 µm thickness mounted on pre-coated glass slides. Nuclei were stained with hematoxylin. The images were collected by a light microscope (Olympus).
MEK2 RNAi
U251 cells were transfected by a lentivirus carrying the siRNA fragments against MEK2, which was synthesized and packaged by Genechem Inc. The knockdown efficiency was examined 48 hrs after transfection by immunoblot. The RNAi target sequence was 5′-GATCCCCGAAGGAGAGCCTCACAGCATTCAAGAGATGCTG-TGAGGCTCTCCTTCTTTTTA-3′.
Cell growth and viability assay
Cell growth and viability assays of glioma cells transfected with different constructs or treated with different drugs were performed by the (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)) assay as previously described22. Cells were plated in a 96-well plate at a density of 5000 cells/well. After incubation for 2 hrs with the cell-counting kit-8 reagent (10 μl/well), the reaction was terminated by adding SDS solution (0.1% final concentration). Absorbance at the 450 nm wavelength was measured by an enzyme-labeling instrument (ELX-800 type; Bio-Tek, Shanghai, China). The results are representative of at least three independent experiments.
Flow cytometric analysis
Cell cycle and apoptosis were measured by flow cytometry as previously described21. The Cell Cycle Analysis Kit (GenScrip) and Annexin V PE Apoptosis Detection Kit (BD Biosciences) were used for cell cycle and apoptosis staining according to the manufacturer’s instructions. Then, the cells were analyzed on a FACSCalibur (BD Bioscience). Typically, signals from 10 000 cells were acquired and analyzed for each sample using FlowJo 7.6. The results of flow cytometry are representative of at least three independent experiments.
In vivo studies
The in vivo studies were performed as previously described21. In the subcutaneous glioma xenograft model, U87 cells were infected with lentivirus at an MOI of 10 (si-CRTL 2, si-MEK2), and 24 hrs after infection, 2 × 106 cells were subcutaneously implanted into the flank of BALB/c athymic nude male mice. All of the mice were 3–4 weeks old (n = 8 per group). After 2 weeks, TMZ was given orally to the mice (66 mg/kg daily for 2 days), and the mice were killed when signs of sickness were found. We evaluated the tumor burden by the length (L) and width (W) of xenografted tumors every 5 days. The tumor burden was calculated as follows: L*(w)2/2.
In the intracranial glioma model, U87 cells were infected with adenovirus at an MOI of 10 (si-CRTL 2, si-MEK2), and 24 hrs after infection, 5 × 105 cells were injected into the right caudate putamen of athymic nude male mice at 6 weeks of age (n = 14 per group). At 2 weeks, mice were given TMZ by oral gavage (66 mg/kg daily for 2 days) and killed upon signs of sickness.
At 1 and 3 weeks after the transplantation of tumor cells, mouse brains were scanned to detect tumor formation and size by MRI3. After tail vein injection of gadopentetate dimeglumine, the entire mouse brain was covered, and both T1- and T2-weighted images were obtained.
Specific pathogen-free conditions were used to keep the athymic nude mice, and we followed the animal welfare guidelines of Second Military Medical University to take care of all mice.
Statistical analysis
Paired groups were analyzed by Student’s t-test, whereas three or more groups were compared by analysis of variance followed by Dunnett’s t-test for multiple pair-wise comparisons. Kaplan–Meier survival analysis was utilized to compare overall survival times in glioma patients. Univariate survival analysis was conducted using the Kaplan–Meier method and analyzed by the log-rank test to assess survival differences between groups. A two-tailed P value < 0.05 was considered to be statistically significant. The data are presented as the mean ± SD. All statistical analyses were performed using Office Excel 2007 (Microsoft Corporation, Redmond, WA, USA) or SPSS software (version 11.5; SPSS, Inc., Chicago, IL, USA).
Results
Significant upregulation of MEK2 in gliomas
We used bioinformatics methods to analyze the microarray data in the previous experiments, and 25 core genes involved in the chemosensitivity of GBM to Me-CCNU were obtained23. MEK2 was widely expressed in gliomas. To study the genes involved in TMZ resistance, we used microarray analysis to compare the mRNA expression profiles of three glioma patients sensitive to TMZ treatment and seven glioma patients resistant to TMZ (Figure 1a). We found that MEK2 was one of the most upregulated genes in glioma patients with TMZ resistance in comparison with glioma patients sensitive to TMZ (Figure 1a).

MEK2 expression in glioma tissues. (a) The hierarchical cluster of differentially expressed probes in three temozolomide-sensitive (TS) and seven temozolomide-resistant (TR) patients based on cDNA microarray analysis (left). The detailed MEK2 probe (MAPK2K) expression pattern was enlarged (right). (b) The statistical results of MEK2 mRNA expression measured by real-time PCR in the normal brain (Normal, n = 8) and glioma tissues (Tumor, n = 26). **P < 0.01 vs. Normal. (c) The representative results and statistical analysis of MEK2 protein expression measured by immunoblot in the normal brain (Normal, n = 8) and glioma tissues (Tumor, n = 26). **P < 0.01 vs. Normal. (d) The representative images of MEK2 immunostaining in normal brain and glioma tissues (×400). All results are expressed as the mean ± SD from at least three independent experiments.
Next, MEK2 expression was examined in human glioma tissues. As shown in Figure 1b, the MEK2 mRNA level was increased specifically in glioma tissues compared with normal brain tissues as quantified by real-time PCR. Consistently, the results of immunoblotting demonstrated that the MEK2 protein level was also upregulated in glioma tissues versus normal brains (Figure 1c). Immunohistochemistry staining results of MEK2 demonstrated that in glioma cells from all types of patient samples, but not stromal cells, MEK2 was over-expressed, whereas in neurons and normal glia cells of normal brain slices, the expression level of MEK2 was low (Figure 1d). We therefore concluded that MEK2 was over-expressed in tumor tissues.
The level of MEK2 expression is positively correlated with glioma grades and negatively correlated with disease outcome
To determine the contribution of MEK2 to glioma progression, we investigated MEK2 expression levels in glioma samples of different grades. As shown in Figure 2a, stronger MEK2 staining was detected in high grades of glioma (WHO III–IV) (n = 42) compared with low grades of glioma (WHO I–II) (n = 23).

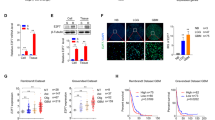
The association of MEK2 expression and clinical outcome in glioma. (a) The representative images of MEK2 immunostaining in the low-grade (left, a case of WHO-I grade) and high-grade (right, a case of WHO-IV grade) glioma tissues (×400). (b) The statistical results of MEK2 mRNA expression measured by real-time PCR in normal brain (Normal, n = 8), low-grade glioma tissues (Low Grade, n = 13) and high-grade glioma tissues (High Grade, n = 13). **P < 0.01 vs. Normal, #P < 0.01 vs. Low Grade. (c) The statistical results of MEK2 protein expression measured by immunoblot in normal brain (Normal, n = 8), low-grade glioma tissues (Low Grade, n = 13) and high-grade glioma tissues (High Grade, n = 13). **P < 0.01 vs. Normal, #P < 0.01 vs. Low Grade. (d) Kaplan–Meier curve of overall survival in 87 Grade IV patients with low (blue curve) and high (green curve) MEK2 expression. (e) Kaplan–Meier curve of overall survival in 50 Grade IV patients receiving temozolomide treatment with low (blue curve) and high (green curve) MEK2 expression. All results are expressed as the mean ± SD from at least three independent experiments.
The expression change of MEK2 mRNA was also studied using real-time PCR, and a similar upregulation pattern was observed in glioma samples. Consistently, the increasing MEK2 mRNA level was closely associated with higher tumor grades (normal tissue, 1.145 ± 0.517; low grade, 1.96 ± 0.890; high grade, 3.57 ± 0.979, respectively) (Figure 2b), which was also true for MEK2 protein levels (normal tissue, 0.17 ± 0.137; low grade, 0.55 ± 0.253; high grade, 1.02 ± 0.216, respectively) (Figure 2c).
Then, we analyzed patient data for the role of MEK2 in general prognosis, and in tumor responses to TMZ-treatment, to determine whether MEK2 could serve as a predictive factor for disease outcome. A total of 87 glioma patients with WHO-IV who underwent glioma surgical resection from October 2008 to December 2010 were included in this study. As shown in Figure 2d, patients with a low protein expression level of MEK2 based on immunohistochemistry showed prolonged overall survival time compared with those with high expression of MEK2. Within this cohort, we further analyzed MEK2 expression in 50 patients who post-operatively received TMZ treatment. We found that higher MEK2 expression was associated with shorter overall survival time in patients receiving TMZ treatment, suggesting that higher expression of MEK2 could confer resistance in glioma cells to TMZ treatment (Figure 2e).
MEK2 is necessary to promote proliferation of glioma cells
To explore the biological function of MEK2 in glioma cells, we selectively knocked down MEK2 using siRNA in U87 and U251 cell lines (Figure 3a). As shown in Figure 3b and 3c, the proliferation of U251 and U87 cells decreased greatly after MEK2 silencing as determined by MTT assays. However, results of 7AAD and Annexin V staining by fluorescence-activated cell sorter analysis indicated that MEK2-silenced glioma cells underwent a similar apoptotic rate to that of cells in the control group (Figure 3d).

The function of MEK2 in glioma cell growth. (a) The immunoblotting results of protein lysates extracted from U251 and U87 cells with MEK2 silenced (si-MEK2) or its control group (si-ctl). (b) The growth curve of U251 cells with MEK2 silenced (si-MEK2) or its control group (si-ctl) measured by MTT assay. **P < 0.01 vs. si-ctl at the same time point. (c) The growth curve of U87 cells with MEK2 silenced (si-MEK2) and its control group (si-ctl) measured by MTT assay. **P < 0.01 vs. si-ctl at the same time point. (d) The apoptotic rates of U251 and U87 cells with MEK2 silenced (si-MEK2) or its control group (si-ctl) measured by Annexin V staining. (e) The cell cycle profile of U87 cells with MEK2 silenced (si-MEK2) and its control group (si-ctl). (f) The percentages of cells at G1, S, and G2/M phases in U251 and U87 cells with MEK2 silenced (si-MEK2) or its control group (si-ctl). All results are expressed as the mean ± SD from at least three independent experiments.
The cell cycle progression was analyzed by flow cytometry to test whether MEK2 played a role in regulating glioma proliferation. The results of a cell cycle analysis indicated that MEK2-silenced cells displayed an increased accumulation in G2/M phase compared with those in the control group (16.3 ± 1.4% vs. 9.7 ± 0.5% for U251 cells, P < 0.01; 20.0 ± 2.4% vs. 13.8 ± 2.8% for U87 cells, P < 0.05, respectively) (Figure 3e and f). Consistently, we did not observe an obvious increase in cell death, in terms of cells in sub-G1 phase, after MEK2 silencing in U251 and U87 cell lines (data not shown).
Enhanced chemotherapy sensitivity of TMZ in MEK2-knockdown glioma cells in vitro
To test whether MEK2 is necessary for glioma cells to resist TMZ-induced cell death, we treated glioma cells in the control and MEK2-silenced groups with TMZ and assessed the apoptotic rate of each group. We found that MEK2-deficient cells displayed an increased cell death rate compared with those in the control group (Control vs. MEK2-silenced group: 15.0 ± 2.5% vs. 42.8 ± 6.2% for U251 cells, P < 0.01; 18.0 ± 6.4% vs. 37.2 ± 3.9% for U87 cells, P < 0.05, respectively), suggesting that MEK2 knockdown in tumor cells enhanced their sensitivity to TMZ treatment (Figure 4a and b).

The effect of MEK2 expression on the temozolomide sensitivity of glioma cells. The representative plots (a) and statistical results (b) of the apoptotic rates of U251 and U87 cells with MEK2 silenced (si-MEK2) or its control group (si-ctl) treated with 100 μM temozolomide (TMZ) for 48 hrs as measured by Annexin V staining. **P < 0.01, *P < 0.05 vs si-ctl. (c) The inhibition curve of U251 with MEK2 silenced (si-MEK2) or its control group (si-ctl) treated with 1, 10, 25, 50, 100, 200, 400, or 800 μM TMZ and measured by MTT assay. The representative plots (d) and statistical results of the apoptotic rates (e) of U251 and U87 cells with MEK2 silenced (si-MEK2) or its control group (si-ctl) treated with 100 μM TMZ and MEK inhibitors (GSK: GSK1120212 at 10 nM, PD: PD184352 at 50 nM) for 48 hrs as measured by Annexin V staining. **P < 0.01, *P < 0.05 vs. DMSO + TMZ group; ##P < 0.01 vs. DMSO within each treatment. All results are expressed as the mean ± SD from at least three independent experiments.
We further determined whether MEK2 silencing increases TMZ sensitivity in glioma cells. As shown in Figure 4c, U251 normally exhibited an IC50 of TMZ at approximately 150 μM by MTT assays. After selective knockdown of MEK2, U251 showed a decrease in IC50 to less than 50 μM. Consistently, administration of the MEK1/2 inhibitors trametinib and PD184352 resulted in more apoptosis in glioma cells upon TMZ administration compared with DMSO (Figure 4d). These results indicated that MEK2 silencing increased the sensitivity of glioma cells to TMZ treatment.
The downstream genes in relation to drug resistance with MEK2 expression
To explore the mechanisms underlying MEK2-dependent TMZ resistance, we analyzed several genes related to drug resistance in human malignancy. As shown in Figure 5a and b, the mRNA levels of ABCB1, ABCC1, ABCC2, ABCG2, BCL2, and MGMT were significantly downregulated in U251 and U87 cell lines when MEK2 was silenced. Furthermore, the MEK2 expression level was associated closely with the ABCG2 (r = 0.790, P < 0.01) and MGMT (r = 0.803, P < 0.01) expression levels in 34 tested human samples, including 8 normal tissues and 26 glioma tissues (Figure 5c and d). Therefore, MEK2 most likely confers chemoresistance by promoting the expression of multiple drug resistance genes.

The downstream genes of MEK2 in regulation of temozolomide sensitivity. ABCB1, ABCC1, ABCC2, ABCG2, BCL2, or MGMT mRNA expression levels in U251 (a) and U87 (b) cells with MEK2 silenced (si-MEK2) or its control group (si-ctl) as measured by real-time PCR.**P < 0.01 vs. si-ctl. The correlation coefficients of MEK2 mRNA expression and ABCG2 (c) or MGMT (d) mRNA expression in 34 human tissues (including normal brain and glioma tissues) as measured by real-time PCR. All results are expressed as the mean ± SD from at least three independent experiments.
Effect of MEK2 inhibition on the chemotherapy sensitivity of TMZ in vivo
To prove that MEK2 is important for the development of glioma resistance in vivo, we generated both subcutaneously and intracranially xenografted glioma models using nude mice and treated them with TMZ.
In the intracranial model (Figure 6a), Si-MEK2-infected U87 cell glioma burden mice (n = 8) had longer survival times (mean = 48 days, 95% CI = 41.9 to 54.1 days, P < 0.001 vs Si-ctl) compared with Si-ctl-infected glioma burden mice (mean = 30.4 days, 95% CI = 25.1 to 35.7 days) (n = 8 in each group). At 21-day post-implantation, an MRI was used to stain the coronal sections of the brains, and the results showed that tumors from Si-MEK2-infected cells were clearly smaller compared with those from Si-ctl-infected cells (Figure 6b and c).

Effect of the Inhibition of MEK2 on chemotherapy sensitivity to TMZ in vivo. (a) Nude mice were intracranially transplanted with 5 × 105 glioma cells infected with Si-MEK2 or Si-ctl (control). The survival curve was plotted by Kaplan–Meier analysis. Mice-bearing gliomas from Si-MEK2-infected U87 cells treated with TMZ survived longer than those in the control group (n = 8 per group). Each curve was compared using the two-sided log-rank test. P < 0.001, Si-MEK2 vs. Si-ctl. The number of mice at risk at each time point is also shown. (b) Representative tumor images were obtained by MRI 1 or 3 weeks after glioma transplantation. (c) Tumor size was quantified and analyzed. (d) In the subcutaneous model, the tumor volumes in Si-MEK2- and Si-ctl-infected groups treated with TMZ (n = 8 per group) were determined every 5 days for 30 days after implantation of U87 cells. The means and upper 95% confidence intervals are shown, Si-MEK2 vs. Si-ctl. P values were calculated using the two-sided student’s t-test. (E) The volume of xenografted tumors on day 30 of implantation. Error bars represent the interquartile range. *P = 0.013, Si-ctl vs. Si-MEK2, P values were calculated using the two-sided student’s t-test.
These results indicated that MEK2 was important for the chemotherapy sensitivity of gliomas to TMZ.
In the subcutaneous group, after injection, the tumor burden was determined every 5 days (Figure 6d). The tumors still grew after 30 days, and tumors from Si-MEK2-infected cells were smaller than those from Si-ctl-infected cells. The mean tumor burden of Si-ctl-infected cells was 125.75 ± 114 mm3 (n = 8) and of Si-MEK2-infected cells was 10.32 ± 2.65 mm3 (n = 8, P = 0.013 vs. Si-ctl) (Figure 6e).
Discussion
In this study, MEK2 was identified to be a gene differentially expressed in glioma patients who were sensitive or resistant to TMZ treatment. It has been reported that the MAPK pathway is highly activated in many cancers, including glioma17,24. Our study further revealed the potential function of MEK2 in the chemotherapy resistance of gliomas.
MEK2 is involved in many important cellular processes, such as cell proliferation, cell apoptosis, and cell differentiation25,26,27. Moreover, it is also implicated in the initiation and progression of many human malignancies28,29,30. We observed that MEK2 was expressed highly in glioma cells, and its expression level increased as tumor grade advanced. These results strongly suggested MEK2 is a gene closely associated with glioma formation and progression.
Additionally, we provided multiple pieces of evidence that MEK2 also plays an important role in TMZ resistance. MEK2 was identified as a differentially expressed gene by comparative study of cDNA microarray from patients insensitive or sensitive to TMZ treatment. In addition, the intracranial glioma model and the subcutaneous model presented convincing in vivo data and reinforced the important role of MEK2 in TMZ resistance of gliomas. Thus, the expression level of MEK2 could serve as a predictor of disease outcome for patients receiving TMZ treatment.
The mechanisms underlying MEK2-dependent TMZ resistance were also investigated in our study. Through silencing MEK2 by siRNA in glioma cell lines, cell growth was blocked significantly due to cell cycle arrest at G2/M phase, and cell apoptosis was largely unaffected in the absence of other treatments. Therefore, the increased sensitivity of MEK2-silenced glioma cells to TMZ was not caused by cell apoptosis. Additionally, TMZ is an alkylating agent that exerts its antineoplastic activity by interfering with DNA replication31. Thus, the cell cycle arrest at S phase, but not G2/M phase, caused by MEK2 silencing could lead to enhanced sensitivity of glioma cells to TMZ treatment. Given that there is a DNA damage checkpoint that acts at the G2/M transition to ensure that cells do not initiate mitosis immediately after replication before they repair damaged DNA, our data might suggest that MEK2 either promotes DNA repair or helps glioma cells bypass the checkpoint and enter M phase. Aberrant activation of the MAPK/ERK pathway has been reported to upregulate ABCB1 expression in acute lymphoblastic leukemia32. Additionally, administration of MAPK pathway inhibitors can reverse the multiple drug resistance phenotype mediated through P-glycoprotein in many different types of tumor cells33,34,35,36. The enhanced ERK kinase activity would affect the nuclear translocation of Y-box binding protein-1, which is capable of binding to the promoter region of multidrug resistance genes and regulating the mRNA expression levels of those genes. The role of MEK2 in regulating the TMZ sensitivity of glioma cells is consistent with previous findings on the function of ERK signaling in the multidrug resistance of human malignancies.
It has been reported that APNG and MGMT were involved in the TMZ resistance of gliomas37. We here found that the mRNA level of MGMT was closely associated with that of MEK2, consistent with a previously identified connection between MEK2-ERK-MDM2-p53 signaling and MGMT expression. In addition, many genes involved in drug transport (ABCB1, ABCC1, ABCC2, ABCG2)38, anti-apoptosis (BCL2)39 and DNA repair (MGMT)40 are upregulated by MEK2 in glioma cell lines, so it is highly likely that MEK2 simultaneously regulates multiple signal pathways to promote TMZ resistance. As for upstream signaling leading to aberrant MEK2 activation, the recurrent mutation of important genes in the RAF-MAPK signaling cascade such as KRAS and BRAF might be a trigger17,18 . Alternatively, abnormal over-expression of VEGF and EGFR, which was reported to be associated with enhanced resistance to TMZ treatment, could activate MAPK signaling28.
Another interesting finding of our study is that MEK2 inhibitors could be used as a sensitizing strategy for TMZ treatment. Here, the administration of trametinib (GSK1120212) and PD184352 (CI-1040), two MEK1/2 inhibitors undergoing clinical trials41, greatly altered the apoptotic rate of glioma cells upon TMZ treatment. Notably, these inhibitors are non-selective inhibitors to MEK1 and MEK2, and the contribution of MEK1 inhibition in improved TMZ sensitivity should be further investigated in future studies. It has been shown that although MEK1 and MEK2 share many similarities in structure and kinase properties, they play distinct roles in many physiological processes42. However, the MEK1/2 inhibitors conferred an increase in the sensitivity of glioma cells to TMZ treatment compared with untreated MEK2 silenced glioma cells, which suggested that MEK1 might also be involved in the regulation of TMZ sensitivity in glioma cells.
Collectively, our study provided clear evidence that MEK2 expression levels were closely related to the outcome of TMZ treatment. Moreover, our study revealed a tremendous potential for MEK2 blockade to be utilized as a useful sensitizing strategy for the improvement of the therapeutic effects of TMZ.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Feili Liu, Shu Li, Yicheng Meng, Yuxin Li helped to supplement experiments. All authors have read and approved the final manuscript.
References
Friedmann-Morvinski D, Bushong EA, Ke E, Soda Y, Marumoto T, Singer O et al . Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science 2012 ; 338 : 1080 – 1084 .
Chen S, Tanaka S, Giannini C, Morris J, Yan ES, Buckner J et al . Gliomatosis cerebri: clinical characteristics, management, and outcomes. J Neurooncol 2013 ; 112 : 267 – 275 .
Zhao J, He H, Zhou K, Ren Y, Shi Z, Wu Z et al . Neuronal transcription factors induce conversion of human glioma cells to neurons and inhibit tumorigenesis. PLoS One 2012 ; 7 : e41506 .
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ et al . Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005 ; 352 : 987 – 996 .
Corsa P, Parisi S, Raguso A, Troiano M, Perrone A, Cossa S et al . Temozolomide and radiotherapy as first-line treatment of high-grade gliomas. Tumori 2006 ; 92 : 299 – 305 .
National Cancer Comprehensive Network . NCCN clinical practice guidelines in oncology: central nervous system cancers, v.1.2008 . Available at http://www.nccn.org/professionals/physician_gls/PDF/cns.pdf ( accessed 7 March 2008 ).
National Institute for Health and Clinical Excellence (NICE) . Carmustine implants and temozolomide for the treatment of newly diagnosed high-grade glioma. Technology Appraisal Guidance No. 121 . London: National Institute for Health and Clinical Excellence (NICE) ; 2007, p . 45 .
Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ . Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin 2010 ; 60 : 166 – 193 .
Agnihotri S, Gajadhar AS, Ternamian C, Gorlia T, Diefes KL, Mischel PS et al . Alkylpurine-DNA-N-glycosylase confers resistance to temozolomide in xenograft models of glioblastoma multiforme and is associated with poor survival in patients. J Clin Invest 2012 ; 122 : 253 – 266 .
Pan Q, Yang XJ, Wang HM, Dong XT, Wang W, Li Y, Li JM . Chemoresistance to temozolomide in human glioma cell line U251 is associated with increased activity of O6-methylguanine-DNA methyltransferase and can be overcome by metronomic temozolomide regimen. Cell Biochem Biophys 2012 ; 62 : 185 – 191 .
Lee ES, Ko KK, Joe YA, Kang SG, Hong YK . Inhibition of STAT3 reverses drug resistance acquired in temozolomide-resistant human glioma cells. Oncol Lett 2011 ; 2 : 115 – 121 .
Lu XY, Cao K, Li QY, Yuan ZC, Lu PS . The synergistic therapeutic effect of temozolomide and hyperbaric oxygen on glioma U251 cell lines is accompanied by alterations in vascular endothelial growth factor and multidrug resistance-associated protein-1 levels. J Int Med Res 2012 ; 40 : 995 – 1004 .
Munoz JL, Rodriguez-Cruz V, Greco SJ, Ramkissoon SH, Ligon KL, Rameshwar P . Temozolomide resistance in glioblastoma cells occurs partly through epidermal growth factor receptor-mediated induction of connexin 43. Cell Death Dis 2014 ; 5 : e1145 .
Poteet E, Choudhury GR, Winters A, Li W, Ryou MG, Liu R et al . Reversing the Warburg effect as a treatment for glioblastoma. J Biol Chem 2013 ; 288 : 9153 – 9164 .
Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M et al . MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005 ; 352 : 997 – 1003 .
Tao BB, He H, Shi XH, Wang CL, Li WQ, Li B et al . Up-regulation of USP2a and FASN in gliomas correlates strongly with glioma grade. J Clin Neurosci 2013 ; 20 : 717 – 720 .
Okabe H, Lee SH, Phuchareon J, Albertson DG, McCormick F, Tetsu O . A critical role for FBXW8 and MAPK in cyclin D1 degradation and cancer cell proliferation. PLoS One 2006 ; 1 : e128 .
Basto D, Trovisco V, Lopes JM, Martins A, Pardal F, Soares P, Reis RM . Mutation analysis of B-RAF gene in human gliomas. Acta Neuropathol 2005 ; 109 : 207 – 210 .
Cancer Genome Atlas Research Network . Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008 ; 455 : 1061 – 1068 .
MacDonald TJ, Brown KM, LaFleur B, Peterson K, Lawlor C, Chen Y et al . Expression profiling of medulloblastoma: PDGFRA and the RAS/MAPK pathway as therapeutic targets for metastatic disease. Nat Genet 2001 ; 29 : 143 – 152 .
Ding X, He Z, Zhou K, Cheng J, Yao H, Lu D et al . Essential role of TRPC6 channels in G2/M phase transition and development of human glioma. J Natl Cancer Inst 2010 ; 102 : 1052 – 1068 .
Li B, He H, Tao BB, Zhao ZY, Hu GH, Luo C et al . Knockdown of CDK6 enhances glioma sensitivity to chemotherapy. Oncol Rep 2012 ; 28 : 909 – 914 .
Zhao Z, Liu Y, He H, Chen X, Chen J, Lu YC . Candidate genes influencing sensitivity and resistance of human glioblastoma to Semustine. Brain Res Bull 2011 ; 86 : 189 – 194 .
Guo G, Yao W, Zhang Q, Bo Y . Oleanolic acid suppresses migration and invasion of malignant glioma cells by inactivating MAPK/ERK signaling pathway. PLoS One 2013 ; 8 : e72079 .
Roskoski R Jr, MEK1/2 dual-specificity protein kinases: structure and regulation. Biochem Biophys Res Commun 2012 ; 417 : 5 – 10 .
Zhang L, Shi R, He C, Cheng C, Song B, Cui H et al . Oncogenic B-Raf(V600E) abrogates the AKT/B-Raf/Mps1 interaction in melanoma cells. Cancer Lett 2013 ; 337 : 125 – 132 .
Chen Z, Zhang L, Xia L, Jin Y, Wu Q, Guo H et al . Genomic analysis of drug resistant gastric cancer cell lines by combining mRNA and microRNA expression profiling. Cancer Lett 2014 ; 350 : 43 – 51 .
Infante JR, Fecher LA, Falchook GS, Nallapareddy S, Gordon MS, Becerra C et al . Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol 2012 ; 13 : 773 – 781 .
Hui K, Yang Y, Shi K, Luo H, Duan J, An J et al . The p38 MAPK-regulated PKD1/CREB/Bcl-2 pathway contributes to selenite-induced colorectal cancer cell apoptosis in vitro and in vivo. Cancer Lett 2014 ; 354 : 189 – 199 .
Boeckx C, Op de Beeck K, Wouters A, Deschoolmeester V, Limame R, Zwaenepoel K et al . Overcoming cetuximab resistance in HNSCC: The role of AURKB and DUSP proteins. Cancer Lett 2014 ; 354 : 365 – 377 .
Ito M, Ohba S, Gaensler K, Ronen SM, Mukherjee J, Pieper RO . Early Chk1 phosphorylation is driven by temozolomide-induced, DNA double strand break- and mismatch repair-independent DNA damage. PLoS One 2013 ; 8 : e62351 .
Tomiyasu H, Watanabe M, Sugita K, Goto-Koshino Y, Fujino Y, Ohno K et al . Regulations of ABCB1 and ABCG2 expression through MAPK pathways in acute lymphoblastic leukemia cell lines. Anticancer Res 2013 ; 33 : 5317 – 5323 .
Barancik M, Bohacova V, Kvackajova J, Hudecova S, Krizanova O, Breier A . SB203580, a specific inhibitor of p38-MAPK pathway, is a new reversal agent of P-glycoprotein-mediated multidrug resistance. Eur J Pharm Sci 2001 ; 14 : 29 – 36 .
Graziosi L, Mencarelli A, Santorelli C, Renga B, Cipriani S, Cavazzoni E et al . Mechanistic role of p38 MAPK in gastric cancer dissemination in a rodent model peritoneal metastasis. Eur J Pharmacol 2012 ; 674 : 143 – 152 .
Guo X, Ma N, Wang J, Song J, Bu X, Cheng Y et al . Increased p38-MAPK is responsible for chemotherapy resistance in human gastric cancer cells. BMC Cancer 2008 ; 8 : 375 .
Shen H, Xu W, Luo W, Zhou L, Yong W, Chen F et al . Upregulation of mdr1 gene is related to activation of the MAPK/ERK signal transduction pathway and YB-1 nuclear translocation in B-cell lymphoma. Exp Hematol 2011 ; 39 : 558 – 569 .
Gong X, Schwartz PH, Linskey ME, Bota DA . Neural stem/progenitors and glioma stem-like cells have differential sensitivity to chemotherapy. Neurology 2011 ; 76 : 1126 – 1134 .
Skarpen E, Flinder LI, Rosseland CM, Orstavik S, Wierod L, Oksvold MP et al . MEK1 and MEK2 regulate distinct functions by sorting ERK2 to different intracellular compartments. FASEB J 2008 ; 22 : 466 – 476 .
Lu Y, Wang L, He M, Huang W, Li H, Wang Y et al . Nix protein positively regulates NF-kappaB activation in gliomas. PLoS One 2012 ; 7 : e44559 .
Jameson KL, Mazur PK, Zehnder AM, Zhang J, Zarnegar B, Sage J, Khavari PA . IQGAP1 scaffold-kinase interaction blockade selectively targets RAS-MAP kinase-driven tumors. Nat Med 2013 ; 19 : 626 – 630 .
Sato A, Sunayama J, Matsuda K, Seino S, Suzuki K, Watanabe E et al . MEK-ERK signaling dictates DNA-repair gene MGMT expression and temozolomide resistance of stem-like glioblastoma cells via the MDM2-p53 axis. Stem Cells 2011 ; 29 : 1942 – 1951 .
Stewart S, Sundaram M, Zhang Y, Lee J, Han M, Guan KL . Kinase suppressor of Ras forms a multiprotein signaling complex and modulates MEK localization. Mol Cell Biol 1999 ; 19 : 5523 – 5534 .
Acknowledgements
This work was supported by NSFC81302187 and CWS14C063.
Author information
Authors and Affiliations
Ethics declarations
Competing interests
The authors declare that there is no conflict of interest in this article.
Rights and permissions
About this article

Cite this article
He, H., Yao, M., Zhang, W. et al. MEK2 is a prognostic marker and potential chemo-sensitizing target for glioma patients undergoing temozolomide treatment. Cell Mol Immunol 13, 658–668 (2016). https://doi.org/10.1038/cmi.2015.46
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cmi.2015.46
- Springer Nature Limited