
Abstract
It is well accepted that adaptive immunity plays a key role in the control of hepatitis B virus (HBV) infection. In contrast, the contribution of innate immunity has only received attention in recent years. Toll-like receptors (TLRs) sense pathogen-associated molecule patterns and activate antiviral mechanisms, including intracellular antiviral pathways and the production of antiviral effector interferons (IFNs) and pro-inflammatory cytokines. Experimental results from in vitro and in vivo models have demonstrated that TLRs mediate the activation of cellular signaling pathways and the production of antiviral cytokines, resulting in a suppression of HBV replication. However, HBV infection is associated with downregulation of TLR expression on host cells and blockade of the activation of downstream signaling pathways. In primary HBV infection, TLRs may slow down HBV infection, but contribute only indirectly to viral clearance. Importantly, TLRs may modulate HBV-specific T- and B-cell responses in vivo, which are essential for the termination of HBV infection. Thus, TLR agonists are promising candidates to act as immunomodulators for the treatment of chronic HBV infection. Antiviral treatment may recover TLR expression and function in chronic HBV infection and may increase the efficacy of therapeutic approaches based on TLR activation. A combined therapeutic strategy with antiviral treatment and TLR activation could facilitate the restoration of HBV-specific immune responses and thereby, achieve viral clearance in chronically infected HBV patients.
Similar content being viewed by others


Introduction
Hepatitis B virus (HBV), a hepatotropic non-cytopathic DNA virus, affects millions of people worldwide and is one of the major causes of fatal liver diseases, such as cirrhosis and hepatocellular carcinoma.1 The outcome of acute HBV infection is determined by the strength of the specific host immune response against HBV. It is well accepted that cell-mediated immune responses play a major role in viral clearance during acute HBV infection. Patients with chronic HBV infection usually fail to develop adequate HBV-specific immune responses.2 Although the central role of adaptive immunity in controlling HBV infection is well established, the contribution of innate immunity in this regard is largely unexplored.
Pattern recognition receptors (PPRs), including Toll-like receptors (TLRs), RIG-I-like receptors (RLRs) and NOD-like receptors, are essential for sensing invading pathogens, initiating innate immune responses, limiting the spread of infection and promoting efficient adaptive immune responses.3 Due to the lack of cell cultures and small animal models that efficiently supporting the full HBV infection cycle, the role of PPRs in HBV infection and pathogenesis can only be partially studied.4 While the interaction between HBV and TLRs has been examined in a number of studies, the role of RLRs and NOD-like receptors in HBV infection is largely unknown. RLRs can sense viral RNAs in the cytoplasm and play an important role in recognition of hepatitis C virus (HCV) RNA.5 Unlike HCV, the HBV genome replicates in the nucleocapsid and may evade detection by RLRs. Recent studies have indicated that TLR expression in peripheral immune cells, liver immune cells, and hepatocytes changed during chronic HBV infection accompanied by impaired TLR functions.6,7,8,9 Meanwhile, TLR-mediated innate immune responses have been shown to inhibit HBV replication in hepatocytes and animal models.10,11,12,13,14 Stimulation of innate immune responses with TLR agonists may further improve the immunotherapeutic effect of combination strategies against the hepadnaviral infection. A great number of reviews have already described the basic features of the TLR system and related signaling pathways.15,16 Therefore, we will only focus on the interaction between TLR and HBV in the present review.
Interaction between TLRs and HBV
Despite numerous studies of the interaction between TLRs and HBV, no convincing evidence is available that HBV proteins and viral RNAs and DNA are truly recognized by TLRs. TLR2 was found to recognize viral lipoproteins and glycoproteins and lead to the activation of immune cells. Cooper et al.17 reported that full-length HBcAg triggers the production of proinflammatory cytokines, such as TNF-α, IL-6 and IL-12p40, in macrophages in a TLR2-dependent manner in vitro. This finding was challenged by other scientists, as recombinant HBcAg was derived from a bacterial expression system with risk of contamination.18,19 In contrast, Lee et al.20 indicated that the HBV nucleocapsid does not activate TLR2 but activates TLR7 with packaged single-stranded RNA (ssRNA) as a TLR7 agonists. However, these findings were not confirmed in other systems. It has been shown that HBV interacts with hepatic non-parenchymal cells (NPCs) and induces the production of IL-6, though it is not clear how hepatic NPCs sense HBV.21 However, in a mouse model based on hydrodynamic injection, persistent HBV replication in the mouse liver does not induce measurable TLR activation, such as IL-6 or interferon (IFN) production, after an initial cytokine induction.22
HBV is claimed to be a ‘stealth virus' in the early phase of infection because it does not induce IFNs and IFN-stimulated genes (ISGs) in acute HBV-infected chimpanzees23 or patients.24 This fact could be explained by two possibilities: inability of HBV to activate PPRs or viral inhibition of PPR signaling pathways. Accumulating evidence supports the latter hypothesis that HBV is able to interfere with PPR signaling pathways. It has been consistently shown that TLR expression and function are reduced during HBV infection. Using hepatocytes and Kupffer cells (KCs) isolated from liver biopsies of patients with chronic hepatitis B (CHB), Visvanathan et al.6 first described significantly decreased TLR2 expression on hepatocytes, KCs, and peripheral monocytes in patients with HBeAg-positive CHB compared with HBeAg negative CHB and controls. HBeAg, when expressed by transduction through a recombinant baculoviral vector, was able to inhibit the induction of TNF-α in hepatic cell lines.6 Subsequently, HBeAg was shown to suppress TIR-mediated activation of NF-κB-dependent and IFN-β promoters through disruption of homotypic TIR–TIR interaction, which is critical for TLR-mediated signaling,25 and also through inhibition of IL-1β-mediated NF-κB activation in hepatocytes.26 Consistently, Chen et al.7 also reported that TLR2 mRNA and protein levels were significantly decreased in PBMCs from CHB patients based on real-time PCR and flow cytometric analysis. Impaired cytokine production was observed in PBMCs from CHB patients after challenged with TLR2 ligand, but correlating with the plasma HBsAg levels. HBsAg not only directly blocks the TLR2-mediated JNK-MAPK pathway but also indirectly induces monocytes to release IL-10.27,28 Thus, TLR2 expression and function apparently represent important targets of viral inhibition.
There are experimental findings supporting the view that HBV is able to block the host IFN responses mediated by TLRs. Plasmacytoid dendritic cells (pDCs), the professional producers of type I IFNs, play a pivotal role in innate and adaptive immune responses against viral infections. TLR9 is a PRR for viral and bacterial DNA motifs and stimulates type I IFN secretion by pDCs via IRF7 activation.15 During chronic HBV infection, pDCs display reduced TLR9 expression and an impaired ability to secrete IFN-α after ex vivo stimulation with TLR9 ligands.8,29,30 HBV virions were able to directly inhibit TLR9 transcription through downregulation of TLR9 promoter activity, by blocking the MyD88-IRAK4 axis and Sendai virus targeting IRF7 to suppress IFN-α production.8 Along the same lines, Xu et al.31 demonstrated that HBsAg can inhibit TLR9-mediated IRF7 expression and nuclear translocation through upregulation of SOCS-1 expression. HBsAg, HBeAg and HBV virions may also inhibit the activation of liver NPCs by TLR3 ligands.32 Coculture of hepatic NPC cell supernatants containing HBsAg, HBeAg and HBV virions resulted in abrogation of TLR-induced antiviral activity, correlating with decreased activation of IRF-3, NF-κB and ERK1/2 in NPCs. Our recent data suggested that HBsAg may trigger IL-10 production in hepatic cells and thereby, attenuate the TLR3-mediated activation of NPCs.33
Additionally, HBV proteins, such as polymerase and HBx protein, could interfere with intracellular signaling pathways, preventing IFN responses in hepatocytes. HBV polymerase was reported to inhibit TLR3-mediated IFN-β induction in human hepatocytes through interference with IRF3 activation.34 Another recent study also demonstrated that HBV polymerase suppressed TNF-α, TLR3 or TLR4-induced NF-κB signaling by inhibiting the activity of IKKs via interaction with Hsp90β in hepatoma cells.35 The RLR-mediated signaling pathway senses intracellular viral RNAs to activate IFN promoter stimulator 1 (IPS-1), IRF3 and NF-κB, leading to the production of type I IFN and pro-inflammatory cytokines.36 HBx protein was reported to inhibit RIG-I-mediated IFN-β induction by promoting ubiquitin-dependent degradation of IPS-1 in hepatoma cells.37,38,39 HBV polymerase was shown to suppress RIG-I-induced IFN-β induction by interference with IRF3 phosphorylation and nuclear translocation and inhibiting the interaction between TBK1/IKKe and DDX3 in human hepatocytes.34,40 The mechanisms employed by HBV to counteract innate immune system are summarized in Table 1.
These findings suggest that HBV evolves to escape from the surveillance of the host innate immune system by targeting PRR signal pathways. However, when considering these results in the context of natural HBV infection, we need to pay more attention, because the majority of experiments were performed either under conditions of over-expressing a single viral protein or in cells that are not normally infected by HBV. Other factors besides HBV proteins may regulate TLR expression and function in patients. For example, the reduced expression or function of individual TLRs during chronic HBV infection may not only be due to the replication of HBV, but also be attributed to the different inflammatory environments present in healthy and chronically HBV-infected subjects. Two recent studies showed decreased expression of TLR signaling molecules, such as MyD88, IRAK1 and IRAK4, in PBMCs from CHB patients compared with healthy controls.41,42 One study showed increased IL-12 and decreased IL-6 levels in the serum of CHB patients compared with that of healthy controls. However, it was not clear whether there is a correlation between the inflammatory cytokines and expression of TLR signaling molecules.42 During chronic HCV infection, TLR2 upregulation in peripheral blood monocytes and liver tissue was correlated with increased levels of serum TNF-α and hepatic inflammation.43,44 These results supported the hypothesis that the changed inflammatory environment may lead to reduced expression or function of individual TLRs in CHB patients. Our recent studies suggest that TLR expression and function may significantly change during the different phases of the natural history of chronic HBV infection (our unpublished results). Regardless, due to the lack of small animal models supporting efficient HBV infection, the interaction between HBV components and TLRs has not been confirmed directly in vivo. Thus, the issue of HBV–TLR interaction requires further investigation.
Activation of TLR-mediated innate immune responses inhibits HBV replication
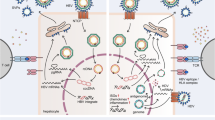
Regardless of the fact that TLR-mediated innate immunity is not or is only weakly activated by HBV infection, experimental activation of the TLR system in hepatic cells and in the liver could suppress HBV replication in vitro and in vivo10,11,12,13,14 (Figure 1). Early experiments in HBV transgenic mice demonstrated the ability of TLR3 ligand poly(I:C) to induce IFN-α-dependent suppression of HBV replication.45 Later, Isogawa et al.10 reported that a single dose intravenous injection of ligands for TLR3, TLR4, TLR5, TLR7 or TLR9 led to a non-cytolytic inhibition of HBV replication in a type I IFN-dependent manner in HBV transgenic mice. Hepatic NPCs, including KCs and liver sinusoidal endothelial cells (LSECs), play a key role in the TLR-mediated inhibition of HBV.11 The supernatants of TLR3- or TLR4-activated KCs and TLR3-activated LSECs efficiently inhibited HBV replication in an immortalized murine hepatocyte cell line derived from HBV transgenic mice (HBV-Met). The antiviral effect of TLR3 was largely mediated by IFN-β, whereas the factor for the TLR4 effect remained undefined.11 Myeloid dendritic cells (DCs) respond to a broad range of TLR ligands, including those for TLR1/2, -3, -4, -7 and -9, to produce antiviral cytokines, which in turn inhibit HBV replication in HBV-Met cells.11 Thus, TLR activation in hepatic NPCs and extra-hepatic DCs could significantly reduce HBV replication through secretion of type I IFNs or other antiviral cytokines.

TLRs and activation of antiviral innate and adaptive immune responses in HBV infection. TLRs are expressed in hepatocytes and hepatic non-parenchymal cells, including LSECs, Kupffer cells, DCs and other cell types. Stimulation of TLRs by their respective ligands leads to the activation of downstream MyD88/TRIF-dependent signaling pathways in hepatic cells and the production of pro-inflammatory cytokines, chemokines and IFNs. Inhibition of HBV replication can be achieved by direct or indirect modes: (1) the intracellular MAPK- and NF-κB-dependent signaling pathways trigger antiviral mechanisms; (2) IFNs and other as yet unknown antiviral factors stimulate the expression of ISGs and other antiviral actions in hepatocytes. However, chemokines and inflammatory cytokines recruit specific T cells into the liver, promote T cell proliferation, and enhance the antiviral functions of HBV-specific CD8+ T cells. Therefore, TLRs inhibit HBV in the liver by activating both innate and adaptive responses. DC, dendritic cell; HBV, hepatitis B virus; IFN, interferon; LSEC, liver sinusoidal endothelial cell; TLR, Toll-like receptor.
In addition to the induction of type I IFNs and pro-inflammatory cytokines, TLR stimulation leads to the activation of intracellular signaling pathways that directly inhibit HBV replication. By over-expression of the TLR adapters MyD88 and TRIF, or the RLR adapter IPS-1, Guo et al.13 clearly demonstrated the ability of intracellular TLR or RLR signal pathways to inhibit HBV replication in hepatoma cells. The over-expression of these adaptors does not lead to the release of antiviral cytokines by transfected hepatoma cells, as HBV replication was not affected by treatment with culture media harvested from cells transfected with each of the three adaptors. In this experimental setting, NF-κB activation was found to play a key role in adaptor-induced antiviral responses.13 Consistently, TLR2 ligands inhibited HBV or woodchuck hepatitis virus (WHV) replication in human hepatoma cells or primary woodchuck hepatocytes through activation of intracellular signaling pathways.12 TLR2-mediated antiviral action is dependent on the presence of adaptor molecules, such as TAK1, IRAK1/4 and TRAF6, and the downstream MAPK and PI-3k/Akt pathways. Silencing the expression of adaptor molecules or blocking the MAPK and PI-3k/Akt pathways with chemical inhibitors abolished the TLR2-mediated antiviral effect and significantly enhanced HBV replication.12 Similarly, TLR4 stimulation with LPS led to a pronounced reduction of WHV replication intermediates without IFN induction in primary woodchuck hepatocytes.46 TLR2 and TLR4 share the cellular MyD88-dependent signaling pathway in mammalian cells and apparently utilize the same intracellular mechanisms to inhibit hepadnaviral replication.
The reports mentioned above demonstrated the ability of TLRs to mediate indirect and direct antiviral activities against HBV. However, TLR-mediated antiviral effects in human hepatoma cells and woodchuck primary hepatocytes were rather low compared with that of potent nucleos/tide analogues. The antiviral effect of TLR ligands in HBV transgenic mice was transient and largely dependent on the induction of IFN production. Therefore, recent results did not conclusively answer the important question about the relevance of TLR-mediated activation of antiviral mechanisms for the control of HBV infection. The innate arm of the immune system usually serves as the first line of defense against microbial invasion and slows down the spread of infection but frequently is overcome by viral evasion strategies. Innate immune responses are generally not long-lasting but rather transient and therefore, essential but not sufficient to control viral infection. It is not yet clear whether a constant or repeated stimulation of TLR-mediated responses is effective over a long period, which may be required for the treatment of chronic HBV infection. Thus, another TLR-mediated function to promote specific adaptive immune responses will need more attention in this context, as discussed in the next section.
Activation of TLR-mediated signaling pathways modulates HBV-specific immune responses
Appropriate HBV-specific T- and B-cell responses are required to control and terminate HBV infection.47,48 Particularly, strong and multispecific CD8+ T-cell responses against HBV proteins are required to clear HBV by both noncytolytic and cytolytic mechanisms.49,50,51 However, HBV-specific CD8+ T-cell responses are characteristically weak, transient and barely detectable in patients with chronic HBV infection.52,53 Therefore, therapeutic restoration of HBV-specific CD8+ T-cell responses in chronically infected HBV patients represents an attractive strategy to terminate HBV persistence.
The liver is recognized as an immunologically privileged organ with a unique composition of innate immune cells. TLRs and RLRs are expressed not only in bone marrow-derived immune cells, such as KCs and hepatic DCs, but also in liver-resident cells, such as hepatocytes, LSECs and hepatic stellate cells.54 Activation of TLRs and RLRs in hepatic cells leads to an induction of type I IFN and a variety of pro-inflammatory cytokines, such as TNF-α, IL-6, IL-1β, IL-12 and IL-18, which play important roles in inhibiting HBV replication,10,11 but also modulate specific immune responses (Figure 1). Priming of naïve CD8+ T cells in the liver usually leads to tolerance instead of immunity, accompanied by poor effector functions and apoptosis of activated CD8+ T cells due to an increased expression of the pro-apoptotic protein Bim.55,56 Consistently, Bim was upregulated on HBV-specific CD8+ T cells during chronic HBV infection and contributed to the deletion of HBV-specific CD8+ T cells.57 LSECs are unique liver-resident antigen-presenting cells that are capable of antigen cross-presentation and induction of CD8+ T-cell tolerance under non-inflammatory conditions58 or promotion of CD8+ T-cell immunity under certain conditions, such as viral infection.59 Recently, we examined functional maturation of LSECs by TLR ligand stimulation, demonstrating that pretreatment of LSECs with P3C (TLR1/2 ligand) but not poly(I:C) (TLR3 ligand) or LPS (TLR4 ligand) reverted their suppressive properties to induce specific T-cell immunity.60 IL-12, which was produced at a low but sustainable level after TLR2 stimulation, was identified as one essential mediator for LSEC-mediated CD8+ T-cell immunity.60 In chronically HBV-infected patients, it was demonstrated that IL-12 but not IFN-α is capable of rescuing the anti-viral function of exhausted HBV-specific CD8+ T cells.61 Therefore, we speculate that activation of the TLR-mediated signaling pathway may lead to an enhancement of HBV-specific T- or B-cell responses in the liver.
Huang et al.62 found that TLR9 activation induced intrahepatic aggregates of myeloid cells that enabled the population expansion of Ag-specific cytotoxic effector CD8+ T cells without causing immunopathology. Importantly, such intrahepatic expansion of DNA vaccine-induced CD8+ T cells controlled chronic HBV infection in a mouse model that was established by the transfer of the HBV genome using an adenovirus vector. In another HBV-persistent mouse model based on hydrodynamic injection of an HBV-genome containing plasmid pAAV/HBV1.2, HBV-specific CD8+ T-cell and anti-HBs antibody responses were impaired. Administration of a dually functioning vector containing both an immunostimulating ssRNA and an HBx-silencing short hairpin RNA induced systemic adaptive immune responses against HBV, including IFN-γ-secreting HBV-specific CD8+ T cells and anti-HBs antibody. The TLR7 signaling pathway and IFN-α/β receptor were found to be important for CD8+ T-cell activation and HBV clearance in this model.63 The same group showed that unmethylated CpG ODNs derived from the HBV genome (HBV-CpG) induced robust secretion of IFN-α by pDCs in a TLR9-dependent manner. Nanoparticles containing HBV-CpG activated DCs, NKs and T cells in vivo, exerting a strong immunostimulatory effect on lymphocytes. Combined with rHBsAg immunization, administration of nanoparticles containing HBV-CpG led to the clearance of HBV and induced an anti-HBsAg response in HBV carrier mice.64 Along the same lines, Wu et al. demonstrated that intrahepatic administration of TLR3 ligand poly(I:C) could recruit CD8+ T cells into the liver and clear HBV in an IFN- and CXCR3-dependent manner.65 These studies indicate that TLR agonists may be used as immunomodulatory agents for the treatment of chronic HBV infection by augmenting the HBV-specific T- or B-cell responses. The mechanisms of TLR-mediated activation of innate immunity or modulation of adaptive immunity to control HBV infection are summarized in Table 2.
Recent evidence has also demonstrated that different cell types of the adaptive immune system, such as effector T cells, also express TLRs.66 Several groups have shown that TLR2 is expressed on activated and memory CD4+ and CD8+ T cells and serves as a costimulatory molecule. TLR2 agonists, such as Pam3CSK4, stimulate activated T cells, thus promoting their proliferation, differentiation and effector function in vitro and in vivo.67,68,69 It has been shown that TLR2 engagement on CD8+ T cells increased T-bet transcription in a MyD88–Akt–mTOR- and protein kinase C-dependent manner.68 Consistently, Pam3CSK4 application in vivo with transferred tumor Ag-specific CD8+ T cells results in enhanced therapeutic efficacy in tumor models.70,71 The expression of TLR3 and TLR9 on human CD8+ T cells was also demonstrated by some studies. TLR stimulation directly promoted IFN-γ production in CD8+ T cells.72,73,74 Thus, the molecular mechanisms underlying TLR-mediated T-cell proliferation and functional differentiation may be further explored for the treatment of chronic HBV infection.
Using TLR agonists for immunotherapy in chronic HBV infection
Based on the findings mentioned above, TLR activation not only induces innate immune responses to limit the replication of HBV in hepatocytes, but could also modulate the adaptive immune response against HBV. Thus, it is rational to discover and develop TLR agonists as antiviral agents as well as adjuvants for preventive and therapeutic vaccination against HBV infection. Before the discovery of TLRs, a TLR3 agonist named polylysine and carboxymethylcellulose-modified polyriboinosinic∶polyribocytidylic acid was recognized as an IFN inducer in chimpanzees.75 Chronically HBV-infected chimpanzees showed transient changes of infection markers when treated with polyriboinosinic∶polyribocytidylic acid. Serum Dane particle-associated DNA polymerase, HBeAg and HBsAg, and intrahepatic hepatitis-B surface and core antigens were transiently diminished during treatment.76 This is the first report demonstrating that TLR activation inhibits HBV replication in vivo. The other TLR3 agonists, such as mismatched double-stranded RNA and Ampligen, induce IFN-like activity and had an antiviral effect in DHBV-infected ducks.77,78 Recently, TLR7 ligand GS-9620 has been examined for its antiviral effect in the woodchuck and chimpanzee models. Interestingly, a 4-week treatment with GS-9620 resulted in a sustained, marked reduction in serum WHV DNA and WHsAg levels and in the induction of anti-WHs antibody responses as well as a markedly decreased incidence of hepatocellular carcinoma in chronic WHV-infected woodchucks.79 Furthermore, GS-9620 induced an increase in serum IFN-α in a dose-dependent manner and triggered ISG expression in PBMCs and the liver in chimpanzees. Short-term oral administration of GS-9620 resulted in a long-term suppression of serum and liver HBV DNA, as well as a reduction in serum levels of HBsAg and HBeAg in 3 chronically HBV-infected chimpanzees.14 In contrast, TLR9 ligand CpG-containing ODNs have been tested in the woodchuck model for treatment of chronic hepatitis B, but failed to show sufficient therapeutic effect when applied alone (Lu M et al., manuscript in preparation). Recently, the TLR8 ligand ssRNA40 was found to selectively activate liver-resident innate immune cells to produce high levels of the antiviral cytokine IFN-γ not only in healthy human livers, but also in chronically HBV- or HCV-infected livers. These results demonstrated the therapeutic implications of TLR8 agonists for the treatment of chronic liver infections.80 Therefore, TLR3, 7 and 8 agonists are promising drug candidates for the treatment of chronic HBV infection if their toxicity can be reduced to a tolerated range.
During chronic HBV infection, the function of innate immunity is impaired due to reduced TLR expression and disturbance of TLR and RLR signaling pathways. However, recent studies have suggested that antiviral treatment in chronic HBV patients can restore the expression of TLRs in PBMCs.9,12,81 Huang et al. demonstrated that chronic HBV patients had lower TLR3 and TLR9 mean fluorescence intensities on PBMCs, liver Kupffer cells and hepatocytes. Hepatic TLR3 and TLR9 mRNA were also significantly reduced in patients. However, pegylated-IFN or entecavir treatment may induce a sustained virological response that is correlated with a restoration of TLR3 and TLR9 expression.9,81 In the WHV-infected woodchuck model, we also found that TLR2 expression in PBMCs was negatively correlated with WHV viral loads during acute WHV infection, and increased if WHV loads were suppressed in woodchucks by antiviral treatment.12 However, these studies did not address the question of whether the functionality of TLRs was also restored by antiviral treatment. Further studies are needed to clarify this question. Regardless, these studies provide a rationale for combination strategies to restore TLR expression and functionality by antiviral treatment followed by TLR stimulation, which could improve the effect of immune modulation in chronic HBV infection.82
A potential use for TLR ligands as adjuvants for prophylactic and therapeutic vaccines has been considered for a long time. The rational design of specific TLR agonists may increase potency and tolerability of new adjuvants and provide the opportunity to meet the stringent safety criteria for a new vaccine formulation.83 Monophosphoryl lipid A, a chemically modified derivative of the lipid A moiety of TLR4 agonist LPS, is considerably less toxic but has similar immunostimulatory activity.84 The AS02 and AS04 adjuvant system, which consists of monophosphoryl lipid A adsorbed on either aluminum or oil, has been evaluated in prophylactic vaccines against HBV.85 The AS04-adjuvant HBV vaccine (Fendrix) boosted the anti-HBs antibody response with an acceptable safety profile not only in liver-transplant candidates,86 who had a suboptimal response to double doses of recombinant hepatitis B vaccine,87 but also in healthy non-responder individuals.88 The AS02-adjuvant HBV vaccine (Supervax) induced more rapid, enhanced, and persistent protection with fewer doses than the currently-licensed AS04-adjuvant HBV vaccine.89 Two TLR9 agonists of class B CpG-ODNs called CPG7909 and 1018 ISS in combination with recombinant HBsAg have been tested in clinical trials. The addition of CPG 7909 to the conventional HBV vaccine (Engerix-B) resulted in higher anti-HBs antibody titers and an enhanced late affinity maturation process to increase the avidity of anti-HBs antibody.90,91 Furthermore, a two-dose regimen of recombinant HBsAg and 1018 ISS (Heplisav) rapidly produced higher anti-HBs titers with generally well-tolerated adverse events, compared with the standard three-dose regimen of Engerix-B.92,93,94 Such a vaccine formulation may be used for patients with an impaired immune system, as it is more effective in the hypo-responsive population, such as HIV-infected patients, than conventional HBV vaccines.95,96,97 The potential therapeutic application of TLR agonists is summarized in Table 3.
Concluding remarks
While the central role of adaptive immunity in controlling HBV infection is well established, the contribution of innate immunity in this regard has been appreciated only in recent years. The development of new in vitro methods of HBV infection will soon shed some light on the recognition of HBV by PRRs.98,99,100 Previous studies indicated that HBV infection apparently does not activate innate immunity in chimpanzees and patients. HBV proteins, such as HBsAg, HBx and HBV polymerase, are associated with the inhibition of TLR or RLR signaling pathways and lead to impaired IFN production. However, it remains to be clarified whether the inhibition of TLR or RLR signaling pathways by HBV is the cause of the lack of early interferon and other innate responses in vivo. Whether the innate immune responses would control HBV infection if initiated in the early phase of HBV infection must be determined. It is well documented that the activation of TLR-mediated signaling pathways not only exhibits direct inhibition of HBV replication, but also enhances HBV-specific T-cell and B-cell responses. Although the expression of TLRs on host cells is downregulated by HBV infection, inhibition of HBV replication by antiviral treatment could partially restore TLR expression in patients. Thus, a treatment strategy with a combination of viral suppression, activation of TLR-mediated innate immunity and restoration of HBV-specific immune responses is needed to achieve effective long-term control of HBV infection and cure chronically HBV-infected patients.
References
Dienstag JL . Hepatitis B virus infection. N Engl J Med 2008; 359: 1486–1500.
Bertoletti A, Ferrari C . Innate and adaptive immune responses in chronic hepatitis B virus infections: towards restoration of immune control of viral infection. Gut 2012; 61: 1754–1764.
Takeuchi O, Akira S . Pattern recognition receptors and inflammation. Cell 2010; 140: 805–820.
Bertoletti A, Maini MK, Ferrari C . The host–pathogen interaction during HBV infection: immunological controversies. Antivir Ther 2010; 15( Suppl 3): 15–24.
Horner SM . Activation and evasion of antiviral innate immunity by hepatitis C virus. J Mol Biol 2014; 426: 1198–1209.
Visvanathan K, Skinner NA, Thompson AJ, Riordan SM, Sozzi V, Edwards R et al. Regulation of Toll-like receptor-2 expression in chronic hepatitis B by the precore protein. Hepatology 2007; 45: 102–110.
Chen Z, Cheng Y, Xu Y, Liao J, Zhang X, Hu Y et al. Expression profiles and function of Toll-like receptors 2 and 4 in peripheral blood mononuclear cells of chronic hepatitis B patients. Clin Immunol 2008; 128( 3): 400–408.
Vincent IE, Zannetti C, Lucifora J, Norder H, Protzer U, Hainaut P et al. Hepatitis B virus impairs TLR9 expression and function in plasmacytoid dendritic cells. PLoS One 2011; 6: e26315.
Huang YW, Lin SC, Wei SC, Hu JT, Chang HY, Huang SH et al. Reduced Toll-like receptor 3 expression in chronic hepatitis B patients and its restoration by interferon therapy. Antivir Ther 2013; 18: 877–884.
Isogawa M, Robek MD, Furuichi Y, Chisari FV . Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J Virol 2005; 79: 7269–7272.
Wu J, Lu M, Meng Z, Trippler M, Broering R, Szczeponek A et al. Toll-like receptor-mediated control of HBV replication by nonparenchymal liver cells in mice. Hepatology 2007; 46: 1769–1778.
Zhang X, Ma Z, Liu H, Liu J, Meng Z, Broering R et al. Role of Toll-like receptor 2 in the immune response against hepadnaviral infection. J Hepatol 2012; 57: 522–528.
Guo H, Jiang D, Ma D, Chang J, Dougherty AM, Cuconati A et al. Activation of pattern recognition receptor-mediated innate immunity inhibits the replication of hepatitis B virus in human hepatocyte-derived cells. J Virol 2009; 83: 847–858.
Lanford RE, Guerra B, Chavez D, Giavedoni L, Hodara VL, Brasky KM et al. GS-9620, an oral agonist of Toll-like receptor-7, induces prolonged suppression of hepatitis B virus in chronically infected chimpanzees. Gastroenterology 2013; 144: 1508–1517, 1517.e1–10.
Akira S, Takeda K . Toll-like receptor signalling. Nat Rev Immunol 2004; 4: 499–511.
Szabo G, Billiar TR, Machida K, Crispe IN, Seki E . Toll-like receptor signaling in liver diseases. Gastroenterol Res Pract 2010; 2010: 971270.
Cooper A, Tal G, Lider O, Shaul Y . Cytokine induction by the hepatitis B virus capsid in macrophages is facilitated by membrane heparan sulfate and involves TLR2. J Immunol 2005; 175: 3165–3176.
Vanlandschoot P, van Houtte F, Serruys B, Leroux-Roels G . Contamination of a recombinant hepatitis B virus nucleocapsid preparation with a human B-cell activator. J Virol 2007; 81: 2535–2536.
Vanlandschoot P, van Houtte F, Ulrichts P, Tavernier J, Leroux-Roels G . Immunostimulatory potential of hepatitis B nucleocapsid preparations: lipopolysaccharide contamination should not be overlooked. J Gen Virol 2005; 86( Pt 2): 323–331.
Lee BO, Tucker A, Frelin L, Sallberg M, Jones J, Peters C et al. Interaction of the hepatitis B core antigen and the innate immune system. J Immunol 2009; 182: 6670–6681.
Hosel M, Quasdorff M, Wiegmann K, Webb D, Zedler U, Broxtermann M et al. Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology 2009; 50: 1773–1782.
Song J, Zhou Y, Li S, Wang B, Zheng X, Wu J et al. Susceptibility of different hepatitis B virus isolates to interferon-alpha in a mouse model based on hydrodynamic injection. PLoS One 2014; 9: e90977.
Wieland S, Thimme R, Purcell RH, Chisari FV . Genomic analysis of the host response to hepatitis B virus infection. Proc Natl Acad Sci USA 2004; 101: 6669–6674.
Dunn C, Peppa D, Khanna P, Nebbia G, Jones M, Brendish N et al. Temporal analysis of early immune responses in patients with acute hepatitis B virus infection. Gastroenterology 2009; 137: 1289–1300.
Lang T, Lo C, Skinner N, Locarnini S, Visvanathan K, Mansell A . The hepatitis B e antigen (HBeAg) targets and suppresses activation of the toll-like receptor signaling pathway. J Hepatol 2011; 55: 762–769.
Wilson R, Warner N, Ryan K, Selleck L, Colledge D, Rodgers S et al. The hepatitis B e antigen suppresses IL-1beta-mediated NF-kappaB activation in hepatocytes. J Viral Hepat 2011; 18: e499–e507.
Shi B, Ren G, Hu Y, Wang S, Zhang Z, Yuan Z . HBsAg inhibits IFN-alpha production in plasmacytoid dendritic cells through TNF-alpha and IL-10 induction in monocytes. PLoS One 2012; 7: e44900.
Wang S, Chen Z, Hu C, Qian F, Cheng Y, Wu M et al. Hepatitis B virus surface antigen selectively inhibits TLR2 ligand-induced IL-12 production in monocytes/macrophages by interfering with JNK activation. J Immunol 2013; 190: 5142–5151.
Xie Q, Shen HC, Jia NN, Wang H, Lin LY, An BY et al. Patients with chronic hepatitis B infection display deficiency of plasmacytoid dendritic cells with reduced expression of TLR9. Microbes Infect 2009; 11: 515–523.
Xu N, Yao HP, Lv GC, Chen Z . Downregulation of TLR7/9 leads to deficient production of IFN-alpha from plasmacytoid dendritic cells in chronic hepatitis B. Inflamm Res 2012; 61: 997–1004.
Xu Y, Hu Y, Shi B, Zhang X, Wang J, Zhang Z et al. HBsAg inhibits TLR9-mediated activation and IFN-alpha production in plasmacytoid dendritic cells. Mol Immunol 2009; 46: 2640–2646.
Wu J, Meng Z, Jiang M, Pei R, Trippler M, Broering R et al. Hepatitis B virus suppresses Toll-like receptor-mediated innate immune responses in murine parenchymal and nonparenchymal liver cells. Hepatology 2009; 49: 1132–1140.
Jiang M, Broering R, Trippler M, Poggenpohl L, Fiedler M, Gerken G et al. Toll-like receptor-mediated immune responses are attenuated in the presence of high levels of hepatitis B virus surface antigen. J Viral Hepat 2014; in press.
Yu S, Chen J, Wu M, Chen H, Kato N, Yuan Z . Hepatitis B virus polymerase inhibits RIG-I- and Toll-like receptor 3-mediated beta interferon induction in human hepatocytes through interference with interferon regulatory factor 3 activation and dampening of the interaction between TBK1/IKKepsilon and DDX3. J Gen Virol 2010; 91( Pt 8): 2080–2090.
Liu D, Wu A, Cui L, Hao R, Wang Y, He J et al. Hepatitis B virus polymerase suppresses NF-kappaB signaling by inhibiting the activity of IKKs via interaction with Hsp90beta. PLoS One 2014; 9: e91658.
Loo YM, Gale M Jr . Immune signaling by RIG-I-like receptors. Immunity 2011; 34: 680–692.
Wei C, Ni C, Song T, Liu Y, Yang X, Zheng Z et al. The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein. J Immunol 2010; 185: 1158–1168.
Kumar M, Jung SY, Hodgson AJ, Madden CR, Qin J, Slagle BL . Hepatitis B virus regulatory HBx protein binds to adaptor protein IPS-1 and inhibits the activation of beta interferon. J Virol 2011; 85: 987–995.
Wang X, Li Y, Mao A, Li C, Tien P . Hepatitis B virus X protein suppresses virus-triggered IRF3 activation and IFN-beta induction by disrupting the VISA-associated complex. Cell Mol Immunol 2010; 7: 341–348.
Wang H, Ryu WS . Hepatitis B virus polymerase blocks pattern recognition receptor signaling via interaction with DDX3: implications for immune evasion. PLoS Pathog 2010; 6: e1000986.
Sajadi SM, Mirzaei V, Hassanshahi G, Khorramdelazad H, Daredor HY, Hosseini SM et al. Decreased expressions of Toll-like receptor 9 and its signaling molecules in chronic hepatitis B virus-infected patients. Arch Pathol Lab Med 2013; 137: 1674–1679.
Momeni M, Zainodini N, Bidaki R, Hassanshahi G, Daneshvar H, Khaleghinia M et al. Decreased expression of Toll like receptor signaling molecules in chronic HBV infected patients. Hum Immunol 2014; 75: 15–19.
Riordan SM, Skinner NA, Kurtovic J, Locarnini S, McIver CJ, Williams R et al. Toll-like receptor expression in chronic hepatitis C: correlation with pro-inflammatory cytokine levels and liver injury. Inflamm Res 2006; 55: 279–285.
Berzsenyi MD, Roberts SK, Preiss S, Woollard DJ, Beard MR, Skinner NA et al. Hepatic TLR2 & TLR4 expression correlates with hepatic inflammation and TNF-alpha in HCV & HCV/HIV infection. J Viral Hepat 2011; 18: 852–860.
Wieland SF, Guidotti LG, Chisari FV . Intrahepatic induction of alpha/beta interferon eliminates viral RNA-containing capsids in hepatitis B virus transgenic mice. J Virol 2000; 74: 4165–4173.
Zhang X, Meng Z, Qiu S, Xu Y, Yang D, Schlaak JF et al. Lipopolysaccharide-induced innate immune responses in primary hepatocytes downregulates woodchuck hepatitis virus replication via interferon-independent pathways. Cell Microbiol 2009; 11: 1624–1637.
Guidotti LG, Chisari FV . Immunobiology and pathogenesis of viral hepatitis. Annu Rev Pathol 2006; 1: 23–61.
Rehermann B, Nascimbeni M . Immunology of hepatitis B virus and hepatitis C virus infection. Nat Rev Immunol 2005; 5: 215–229.
Webster GJ, Reignat S, Maini MK, Whalley SA, Ogg GS, King A et al. Incubation phase of acute hepatitis B in man: dynamic of cellular immune mechanisms. Hepatology 2000; 32: 1117–1124.
Guidotti LG, Rochford R, Chung J, Shapiro M, Purcell R, Chisari FV . Viral clearance without destruction of infected cells during acute HBV infection. Science 1999; 284: 825–829.
Thimme R, Wieland S, Steiger C, Ghrayeb J, Reimann KA, Purcell RH et al. CD8+ T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J Virol 2003; 77: 68–76.
Webster GJ, Reignat S, Brown D, Ogg GS, Jones L, Seneviratne SL et al. Longitudinal analysis of CD8+ T cells specific for structural and nonstructural hepatitis B virus proteins in patients with chronic hepatitis B: implications for immunotherapy. J Virol 2004; 78: 5707–5719.
Boni C, Fisicaro P, Valdatta C, Amadei B, Di Vincenzo P, Giuberti T et al. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J Virol 2007; 81: 4215–4225.
Protzer U, Maini MK, Knolle PA . Living in the liver: hepatic infections. Nat Rev Immunol 2012; 12: 201–213.
Tiegs G, Lohse AW . Immune tolerance: what is unique about the liver. J Autoimmun 2010; 34: 1–6.
Holz LE, Benseler V, Bowen DG, Bouillet P, Strasser A, O'Reilly L et al. Intrahepatic murine CD8 T-cell activation associates with a distinct phenotype leading to Bim-dependent death. Gastroenterology 2008; 135: 989–997.
Lopes AR, Kellam P, Das A, Dunn C, Kwan A, Turner J et al. Bim-mediated deletion of antigen-specific CD8 T cells in patients unable to control HBV infection. J Clin Invest 2008; 118: 1835–1845.
Limmer A, Ohl J, Kurts C, Ljunggren HG, Reiss Y, Groettrup M et al. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat Med 2000; 6: 1348–1354.
Kern M, Popov A, Scholz K, Schumak B, Djandji D, Limmer A et al. Virally infected mouse liver endothelial cells trigger CD8+ T-cell immunity. Gastroenterology 2010; 138: 336–346.
Liu J, Jiang M, Ma Z, Dietze KK, Zelinskyy G, Yang D et al. TLR1/2 ligand-stimulated mouse liver endothelial cells secrete IL-12 and trigger CD8+ T cell immunity in vitro. J Immunol 2013; 191: 6178–6190.
Schurich A, Pallett LJ, Lubowiecki M, Singh HD, Gill US, Kennedy PT et al. The third signal cytokine IL-12 rescues the anti-viral function of exhausted HBV-specific CD8 T cells. PLoS Pathog 2013; 9: e1003208.
Huang LR, Wohlleber D, Reisinger F, Jenne CN, Cheng RL, Abdullah Z et al. Intrahepatic myeloid-cell aggregates enable local proliferation of CD8+ T cells and successful immunotherapy against chronic viral liver infection. Nat Immunol 2013; 14: 574–583.
Lan P, Zhang C, Han Q, Zhang J, Tian Z . Therapeutic recovery of hepatitis B virus (HBV)-induced hepatocyte-intrinsic immune defect reverses systemic adaptive immune tolerance. Hepatology 2013; 58: 73–85.
Lv S, Wang J, Dou S, Yang X, Ni X, Sun R et al. Nanoparticles encapsulating hepatitis B virus cytosine-phosphate-guanosine induce therapeutic immunity against HBV infection. Hepatology 2014; 59: 385–394.
Wu J, Huang S, Zhao X, Chen M, Lin Y, Xia Y et al. Polyinosinic–polycytidylic acid treatment leads to the interferon-dependent clearance of hepatitis B virus in a hydrodynamic injection mouse model. J Virol 2014; in press.
Reynolds JM, Dong C . Toll-like receptor regulation of effector T lymphocyte function. Trends Immunol 2013; 34: 511–519.
Cottalorda A, Verschelde C, Marcais A, Tomkowiak M, Musette P, Uematsu S et al. TLR2 engagement on CD8 T cells lowers the threshold for optimal antigen-induced T cell activation. Eur J Immunol 2006; 36( 7): 1684–1693.
Geng D, Zheng L, Srivastava R, Asprodites N, Velasco-Gonzalez C, Davila E . When Toll-like receptor and T-cell receptor signals collide: a mechanism for enhanced CD8 T-cell effector function. Blood 2010; 116: 3494–3504.
Komai-Koma M, Jones L, Ogg GS, Xu D, Liew FY . TLR2 is expressed on activated T cells as a costimulatory receptor. Proc Natl Acad Sci USA 2004; 101: 3029–3034.
Asprodites N, Zheng L, Geng D, Velasco-Gonzalez C, Sanchez-Perez L, Davila E . Engagement of Toll-like receptor-2 on cytotoxic T-lymphocytes occurs in vivo and augments antitumor activity. FASEB J 2008; 22: 3628–3637.
Geng D, Zheng L, Srivastava R, Velasco-Gonzalez C, Riker A, Markovic SN et al. Amplifying TLR–MyD88 signals within tumor-specific T cells enhances antitumor activity to suboptimal levels of weakly immunogenic tumor antigens. Cancer Res 2010; 70: 7442–7454.
Babu S, Blauvelt CP, Kumaraswami V, Nutman TB . Cutting edge: diminished T cell TLR expression and function modulates the immune response in human filarial infection. J Immunol 2006; 176: 3885–3889.
Bendigs S, Salzer U, Lipford GB, Wagner H, Heeg K . CpG-oligodeoxynucleotides co-stimulate primary T cells in the absence of antigen-presenting cells. Eur J Immunol 1999; 29: 1209–1218.
Tabiasco J, Devevre E, Rufer N, Salaun B, Cerottini JC, Speiser D et al. Human effector CD8+ T lymphocytes express TLR3 as a functional coreceptor. J Immunol 2006; 177: 8708–8713.
Levy HB, Baer G, Baron S, Buckler CE, Gibbs CJ, Iadarola MJ et al. A modified polyriboinosinic–polyribocytidylic acid complex that induces interferon in primates. J Infect Dis 1975; 132: 434–439.
Purcell RH, London WT, McAuliffe VJ, Palmer AE, Kaplan PM, Gerin JL et al. Modification of chronic hepatitis-B virus infection in chimpanzees by administration of an interferon inducer. Lancet 1976; 2: 757–761.
Ijichi K, Mitamura K, Ida S, Machida H, Shimada K . In vivo antiviral effects of mismatched double-stranded RNA on duck hepatitis B virus. J Med Virol 1994; 43: 161–165.
Niu J, Wang Y, Dixon R, Bowden S, Qiao M, Einck L et al. The use of ampligen alone and in combination with ganciclovir and coumermycin A1 for the treatment of ducks congenitally-infected with duck hepatitis B virus. Antiviral Res 1993; 21: 155–171.
Menne S, Tennant BC, Liu KH, Ascenzi MA, Baldwin BH, Bellezza CA et al. Antiviral efficacy and induction of an antibody response against surface antigen with the TLR7 agonist GS-9620 in the woodchuck model of chronic HBV infection. J Hepatol 2011; 54: S441.
Jo J, Tan AT, Ussher JE, Sandalova E, Tang XZ, Tan-Garcia A et al. Toll-like receptor 8 agonist and bacteria trigger potent activation of innate immune cells in human liver. PLoS Pathog 2014; 10: e1004210.
Huang YW, Hsu CK, Lin SC, Wei SC, Hu JT, Chang HY et al. Reduced Toll-like receptor-9 expression on peripheral CD14+ monocytes of chronic hepatitis B patients and its restoration by effective therapy. Antivir Ther 2014; in press.
Durantel D, Zoulim F . Interplay between hepatitis B virus and TLR2-mediated innate immune responses: can restoration of TLR2 functions be a new therapeutic option? J Hepatol 2012; 57: 486–489.
Romagne F . Current and future drugs targeting one class of innate immunity receptors: the Toll-like receptors. Drug Discov Today 2007; 12: 80–87.
Kanzler H, Barrat FJ, Hessel EM, Coffman RL . Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med 2007; 13: 552–559.
Hedayat M, Netea MG, Rezaei N . Targeting of Toll-like receptors: a decade of progress in combating infectious diseases. Lancet Infect Dis 2011; 11: 702–712.
Nevens F, Zuckerman JN, Burroughs AK, Jung MC, Bayas JM, Kallinowski B et al. Immunogenicity and safety of an experimental adjuvanted hepatitis B candidate vaccine in liver transplant patients. Liver Transpl 2006; 12: 1489–1495.
Arslan M, Wiesner RH, Sievers C, Egan K, Zein NN . Double-dose accelerated hepatitis B vaccine in patients with end-stage liver disease. Liver Transpl 2001; 7: 314–320.
Jacques P, Moens G, Desombere I, Dewijngaert J, Leroux-Roels G, Wettendorff M et al. The immunogenicity and reactogenicity profile of a candidate hepatitis B vaccine in an adult vaccine non-responder population. Vaccine 2002; 20: 3644–3649.
Surquin M, Tielemans CL, Kulcsar I, Ryba M, Voros P, Mat O et al. Rapid, enhanced, and persistent protection of patients with renal insufficiency by AS02(V)-adjuvanted hepatitis B vaccine. Kidney Int 2010; 77: 247–255.
Cooper CL, Davis HL, Morris ML, Efler SM, Adhami MA, Krieg AM et al. CPG 7909, an immunostimulatory TLR9 agonist oligodeoxynucleotide, as adjuvant to Engerix-B HBV vaccine in healthy adults: a double-blind phase I/II study. J Clin Immunol 2004; 24: 693–701.
Siegrist CA, Pihlgren M, Tougne C, Efler SM, Morris ML, AlAdhami MJ et al. Co-administration of CpG oligonucleotides enhances the late affinity maturation process of human anti-hepatitis B vaccine response. Vaccine 2004; 23: 615–622.
Halperin SA, Dobson S, McNeil S, Langley JM, Smith B, McCall-Sani R et al. Comparison of the safety and immunogenicity of hepatitis B virus surface antigen co-administered with an immunostimulatory phosphorothioate oligonucleotide and a licensed hepatitis B vaccine in healthy young adults. Vaccine 2006; 24: 20–26.
Halperin SA, Ward B, Cooper C, Predy G, Diaz-Mitoma F, Dionne M et al. Comparison of safety and immunogenicity of two doses of investigational hepatitis B virus surface antigen co-administered with an immunostimulatory phosphorothioate oligodeoxyribonucleotide and three doses of a licensed hepatitis B vaccine in healthy adults 18–55 years of age. Vaccine 2012; 30: 2556–2563.
Barry M, Cooper C . Review of hepatitis B surface antigen-1018 ISS adjuvant-containing vaccine safety and efficacy. Expert Opin Biol Ther 2007; 7: 1731–1737.
Cooper CL, Davis HL, Angel JB, Morris ML, Elfer SM, Seguin I et al. CPG 7909 adjuvant improves hepatitis B virus vaccine seroprotection in antiretroviral-treated HIV-infected adults. AIDS 2005; 19: 1473–1479.
Cooper CL, Angel JB, Seguin I, Davis HL, Cameron DW . CPG 7909 adjuvant plus hepatitis B virus vaccination in HIV-infected adults achieves long-term seroprotection for up to 5 years. Clin Infect Dis 2008; 46: 1310–1314.
Cooper C, Mackie D . Hepatitis B surface antigen-1018 ISS adjuvant-containing vaccine: a review of HEPLISAV safety and efficacy. Expert Rev Vaccines 2011; 10: 417–427.
Shlomai A, Schwartz RE, Ramanan V, Bhatta A, de Jong YP, Bhatia SN et al. Modeling host interactions with hepatitis B virus using primary and induced pluripotent stem cell-derived hepatocellular systems. Proc Natl Acad Sci USA 2014; 111: 12193–1218.
Konig A, Doring B, Mohr C, Geipel A, Geyer J, Glebe D . Kinetics of the bile acid transporter and hepatitis B virus receptor Na+/taurocholate cotransporting polypeptide (NTCP) in hepatocytes. J Hepatol 2014; 61: 867–875.
Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012; 1: e00049.
Acknowledgements
This work was supported by grants from Deutsche Forschungsgemeinschaft TRR60, GK1045 and RTG 1949.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ma, Z., Zhang, E., Yang, D. et al. Contribution of Toll-like receptors to the control of hepatitis B virus infection by initiating antiviral innate responses and promoting specific adaptive immune responses. Cell Mol Immunol 12, 273–282 (2015). https://doi.org/10.1038/cmi.2014.112
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cmi.2014.112
- Springer Nature Limited
Keywords
This article is cited by
-
Reverse Vaccinology and Immunoinformatic Approach for Designing a Bivalent Vaccine Candidate Against Hepatitis A and Hepatitis B Viruses
Molecular Biotechnology (2023)
-
Mesenchymal stem cells-based therapy in liver diseases
Molecular Biomedicine (2022)
-
Contribution of T- and B-cell intrinsic toll-like receptors to the adaptive immune response in viral infectious diseases
Cellular and Molecular Life Sciences (2022)
-
Ag-specific CD4 T cells promote innate immune responses in liver ischemia reperfusion injury
Cellular & Molecular Immunology (2019)
-
Polymorphisms in the Toll-like receptor 3 (TLR3) gene are associated with the natural course of hepatitis B virus infection in Caucasian population
Scientific Reports (2018)