
Abstract
Melanoma is the most deadly type of skin cancer, constituting annually ∼75% of all cutaneous cancer-related deaths due to metastatic spread. Currently, because of metastatic spread, there are no effective treatment options for late-stage metastatic melanoma patients. Studies over the past two decades have provided insight into several complex molecular mechanisms as to how these malignancies evade immunological control, indicating the importance of immune escape or suppression for tumor survival. Thus, it is essential to develop innovative cancer strategies and address immune obstacles with the goal of generating more effective immunotherapies. One important area of study is to further elucidate the role and significance of myeloid-derived suppressor cells (MDSCs) in the maintenance of the tumor microenvironment. These cells possess a remarkable ability to suppress immune responses and, as such, facilitate tumor growth. Thus, MDSCs represent an important new target for preventing tumor progression and escape from immune control. In this study, we investigated the role of MDSCs in immune suppression of T cells in an antigen-specific B16 melanoma murine system utilizing a novel synthetic tyrosinase (Tyr) DNA vaccine therapy in both prophylactic and therapeutic models. This Tyr vaccine induced a robust and broad immune response, including directing CD8 T-cell infiltration into tumor sites. The vaccine also reduced the number of MDSCs in the tumor microenvironment through the downregulation of monocyte chemoattractant protein 1, interleukin-10, CXCL5 and arginase II, factors important for MDSC expansion. This novel synthetic DNA vaccine significantly reduced the melanoma tumor burden and increased survival in vivo, due likely, in part, to the facilitation of a change in the tumor microenvironment through MDSC suppression.
Similar content being viewed by others

Introduction
Skin cancer is one of the most common types of cancers diagnosed in the United States, with an estimated 2 million new cases annually. Melanoma is the most deadly type of skin cancer, constituting 5% of diagnoses but >75% of all skin-cancer related deaths because of systemic metastatic spread.1, 2 Of the 76 100 new melanoma diagnoses estimated to occur in 2014, ∼12% will progress to stage III or IV melanoma with a 5-year survival rate of 15%.1 Treatment options for disseminated melanoma remain limited. Currently, ∼17 therapies have been approved for the treatment of metastatic melanoma.3 These include more conventional anti-neoplastic agents such as dacarbazine (DTIC) as well as immunotherapies such as interleukin-2 (IL-2) and the anti-CTLA-4 human monoclonal antibody ipilimumab (Yervoy).2, 3, 4, 5 However, because metastatic melanoma is resistant to native apoptotic and antigrowth signals, is highly metastatic and deploys several local immune evasion mechanisms,1, 6 these therapies, overall, have been demonstrated to be only marginally successful. Consequently, it is essential to develop innovative anticancer strategies that address such obstacles and induce cytotoxicity against tumor cells while minimally affecting normal cells.6
Recent developments in cancer research have focused on using immunotherapies to target specific tumor antigens.7, 8, 9, 10, 11 In the case of melanoma, the tumor-differentiation antigens gp100, Melan-A/MART1 and tyrosinase (Tyr) have been determined to be highly expressed in over 75% of all melanoma tumor cells.12, 13, 14 Past studies using vaccines against MART1 and gp100 have reported the induction of antigen-specific T-cell responses, as measured by interferon-γ (IFN-γ) expression levels.15, 16, 17 Even so, many tumorigenic models demonstrate poor responses to these vaccines. Despite the induction of a cell-mediated immune response against tumor cell antigens, there still remains a need for the development of enhanced and effective immunogenicity of vaccines as well as more efficient delivery systems and a better characterization of vaccine-induced mechanisms for immune activation.18, 19
In this regard, a novel synthetic consensus DNA vaccine against the tumor antigen Tyr was developed and characterized as to its in vivo antitumor efficacy in a prophylactic and therapeutic B16 melanoma challenge model. The B16F10 murine tumor model is particularly useful for these studies as this tumor is highly aggressive and metastatic, and is also poorly immunogenic.11 Furthermore, this tumor appears to exhibit immunosuppressive properties, similar to those observed in human cancer. In this study, a consensus, enhanced Tyr DNA vaccine (designated pTyr) was developed and delivered by optimized in vivo electroporation (EP) in order to enhance efficacy. The vaccine induced highly potent CD8+ T-cell responses as measured by IFN-γ, tumor necrosis factor-α (TNF-α) and IL-2 production, as well as augmented both CD8+ T-cell effector memory and central memory populations. In a prophylactic setting, the vaccine significantly reduced tumor burden in pTyr immunized mice as well as increased survival in a CD8+ T cell-dependent manner. Mice immunized with pTyr had reduced frequencies of myeloid-derived suppressor cells (MDSCs) in tumor tissue, as well as reductions in levels of IL-10, monocyte chemoattractant protein 1 (MCP-1)/CCL2, CXCL-5 and arginase II, suggesting possible mechanisms for the effective reduction in overall tumor burden. Specifically, levels of IL-10, MCP-1/CCL2, CXCL-5 and arginase II were measured in the study reported here as the presence and concentrations of these molecules are indicative of inhibitory activity of MDSCs on T-cell responses that would normally function in the control of tumor growth.20, 21, 22, 23 Similar responses were also observed using a therapeutic vaccination model, with significantly reduced tumor sizes, lower levels of MDSC population and reduced concentrations of MCP-1.
Materials and methods
Tyr DNA vaccine construction
Human tyrosinase gene (Tyr) sequences were collected from GenBank and the resulting enhanced Tyr DNA immunogens were designed using a codon- and RNA-optimized approach, and cloned into a pVax1 expression vector (GenScript, Piscataway, NJ, USA) using BamHI and XhoI under control of the cytomegalovirus immediate-early promoter, as described previously.24 As indicated, this human tyrosinase expressing DNA vaccine is designated pTyr.
Transfection and expression of pTyr DNA vaccine by Western blot
Human Embryonic Kidney (HEK) 293T cells were maintained in Dulbecco’s modified Eagle’s medium (Life Technologies, Grand Island, NY, USA) supplemented with 10% heat-inactivated fetal calf serum, 100 IU of penicillin per ml, 100 μg of streptomycin per ml and 2 mM L-glutamine, and were transfected and harvested as described previously.25 Briefly, cells were transfected using TurboFectin 8.0 (OriGene, Rockville, MD, USA) transfection reagent and 10 μg control vector (pVax1) or the pTyr DNA construct as per the manufacturer’s protocol and then incubated for 48 h. Cells were harvested with ice-cold phosphate-buffered saline (PBS). After transfection with pTyr, cells were washed with PBS, harvested and lysed in RIPA buffer (50 mM Tris-HCl (pH 8.0) with 150 mM NaCl, 1.0% nonidet P-40, 0.5% sodium deoxycholate and 0.1% sodium dodecyl sulfate) containing protease inhibitor and phosphatase inhibitors (Roche Life Sciences, Indianapolis, IN, USA). After centrifugation, the supernatant was separated and stored at −70 °C until use. The protein concentration in samples was quantified by using the Pierce BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Equal amounts of protein were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane. The membrane was blocked and incubated overnight in a 1:1000 dilution of an anti-Tyr primary antibody (Cell Signaling Technology, Danvers, MA, USA) suspended in Tris-buffered saline buffer containing 0.2% Tween-20 and 2.5% nonfat dry milk or 2.5% bovine serum albumin. Following three 10- min washes with Tris-buffered saline containing 0.2% Tween-20, blots were incubated with a horseradish peroxidase-conjugated secondary antibody (Cell Signaling). The blots were washed again for three times in Tris-buffered saline containing 0.2% Tween-20 and visualized with an ECL advance detection system (GE Healthcare Biosciences, Piscataway, NJ, USA).
Animals, DNA plasmid administration and EP delivery
Female C57/B6 mice (8 weeks of age) were purchased from Taconic Biosciences (Hudson, NY ,USA). All animals were housed in a temperature-controlled, light-cycled facility in accordance with the guidelines of the National Institutes of Health and the University of Pennsylvania Institutional Animal Care and Use Committee. C57BL/6 mice were separated into five groups (20 μg of pVax1 and 5, 20, 30 or 60 μg of pTyr) to determine the optimal DNA vaccine dose (n=5 for each group). All groups were immunized intramuscularly three times at 2-week intervals. Based on the dose study, subsequent experiments were carried out with 20 μg of plasmid DNA in 30 μl volume (pVax1 or pTyr) followed by EP (CELLECTRA; Inovio Pharmaceuticals, Plymouth Meeting, PA, USA). Pulsing parameters for delivery were: 2 pulses of 0.1 Amp constant current, 4 s apart and 52 ms in length. All animals received administrations of either experimental or control plasmids by methods described previously.26, 27
Measurement of the anti-Tyr humoral response by enzyme-linked immunosorbent assay (ELISA)
The 96-well microtiter plates (Nunc-Immuno Plate, MaxiSorp, Thermo Fisher Scientific) were coated with purified Tyr protein (Enzo Life Sciences, Farmingdale, NY, USA) by incubating plates overnight at 4 °C with 100 μl of coating buffer (antigen concentration: 2 μg ml−1). The following day, plates were washed three times with 1 × x PBS containing 0.05% Tween 20 (PBS-T, 200 μl per well), blocked for 1 h with 10% fetal bovine serum in PBS (200 μl per well), washed three times again with PBS-T and incubated for 1 h at 37 °C with 1:100 dilutions (in PBS-T with 1% fetal bovine serum FBS (100 μl per well)) of sera from mice immunized with either pTyr or pVax1. Bound IgG was detected using goat anti-mouse IgG horseradish peroxidase (Research Diagnostics, Cleveland, OH, USA) in a dilution of 1:5000. Bound enzyme was detected by the addition of the chromogen substrate solution TMB (R&D Systems, Minneapolis, MN, USA), and optically measured at an absorbance of 450 nm using a Biotek EL312e Bio-Kinetics reader (Biotek US, Winooski,VT, USA). All sera samples were tested in triplicate.28
Measurement of pTyr vaccine induced cellular immune responses quantitated by an IFN-γ enzyme-linked immunospot (ELISpot) assay
Mice were killed 1 week following the last immunization with pTyr or pVax1. Spleens were harvested from each mouse and transferred into R10 media (RPMI-1640) medium, supplemented with 10% fetal bovine serum and 1% antibiotics. Spleens were pulverized using a stomacher (Seward Laboratory Systems, Davie, FL, USA) and transferred through a cell strainer. Erythrocytes were removed by adding ACK lysing buffer (Life Technologies) and splenocytes were isolated and resuspended in R10 media. High-protein IP 96-well Mutiscreen ELISpot plates (EMD Millipore, Billerica, MA, USA) were coated with monoclonal anti-mouse IFN-γ capture antibody (R&D Systems) and incubated overnight at 4 °C. The following day, plates were washed three times with PBS and blocked for 2 h with 1% bovine serum albumin and 5% sucrose in PBS. Isolated splenocytes in R10 medium were counted and added in triplicate wells at 2 × 105 cells per well. Cells were then stimulated with R10 (negative control), Concavalin A (positive control; Sigma-Aldrich, St Louis, MO, USA) or antigenic mouse and human Tyr peptides consisting of 15-mer peptides, overlapping by 11 amino acids (GenScript). Epitope mapping studies were performed to determine dominant epitope within the peptide using the individual peptides pools that were used to stimulate the splenocytes. These individual peptide pools were generated from a peptide matrix according to previously described methodologies.29, 30, 31 The criteria for assigning a dominant epitope designation was based on identification of H2-Db restricted epitopes from the immune epitope database (http://tools.immuneepitope.org). After 24 h of stimulation, the plates were washed with 1 × PBS and incubated overnight with biotinylated anti-mouse IFN-γ antibody (R&D Systems) at 4 °C followed by washing and incubation with streptavidin-alkaline phosphatase (R&D Systems) in a 1:1000 dilution for 2 h in the dark at room temperature. Plates were then washed again three times with PBS followed by addition of BCIP/NBT (5-bromo-4-chloro-3′-indolylphosphate p-toluidine salt and nitro blue tetrazolium chloride) (chromogen color reagent; R&D Systems) for spot development. The plates were incubated for ∼10 min in the dark at room temperatures. The plates were then rinsed with distilled water, dried overnight followed by counting of the spots with an automated ELISpot plate reader system using ImmunoSpot4 software (Cellular Technology Limited, Shaker Heights, OH, USA). Spot-forming counts were then converted to spot-forming cells per 106 splenocytes, as described before.28
Tumor challenge: tumor inoculation and monitoring
B16F10LM3 (B16) is a murine pigmented melanoma derived from the syngeneic C57BL/6 mouse strain that the laboratory has selected from the B16F10 line because of the aggressive in vivo growth and metastatic nature of this tumor line. The B16F line was originally provided by one of the authors, Kenneth E Ugen (University of South Florida, Tampa, FL, USA). For the prophylactic tumor challenge study, C57BL/6 mice were subcutaneously inoculated with 5 × 105 B16 cells (in 200 μl RPMI medium) in the right flank at 7 days after the last immunization and were followed for tumor development and lesion size. In addition, in vivo CD8+ T-cell depletion was performed as described previously using an anti-mouse CD8 (designated 2.43) antibody.32 For the therapeutic model, C57BL/6 mice were subcutaneously inoculated with 5 × 105 B16 cells (in 200 μl RPMI medium) in the right flank at day 0 followed by three vaccinations with pTyr or pVax1 at days 7, 21 and 35.
For all challenge studies, tumor mass (that is, volume) was measured using digital calipers after inoculation and was considered to be significant when visible or palpable, that is, typically a 2-mm diameter lesion size. Specifically, tumor size was calculated by multiplying the vertical length by horizontal length. The survival of the tumor-bearing mice was monitored daily. Mice were killed when the transplanted tumors reached 3 cm in diameter or when severe lesional or perilesional ulceration developed.
Flow cytometric analysis
Tissues were prepared into single-cell suspensions and labeled with appropriate antibodies to assess the level of Gr1+CD11b+-expressing MDSCs in infiltrating B16 tumor cells at 20 days after inoculation with the parental melanoma cell line. MDSCs were isolated as described previously.33 Isolated tumor cells were incubated in minimal essential media containing 2 mg ml−1 collagenase (Roche) and 0.02 mg ml−1 DNase (Roche) and 10% fetal bovine serum for 1 h at room temperature. Cells were pulverized using a stomacher (Seward Laboratory Systems) and transferred through a cell strainer. Isolated cells were counted followed by plating of 5 × 105 cells per well in quadruplicate, for samples from each mouse, in a 96-well U-shaped plate. Plates were centrifuged at 1600 r.p.m. for 5 min and washed with 1 × PBS. After another centrifugation, cells were resuspended in FACS buffer (PBS/1% fetal bovine serum) and three of the four wells were incubated with Live/Dead Fixable Aqua Dead Cell Stain (Life Technologies) for 30 min in the dark at room temperature. Following live/dead cell staining, wells were subsequently stained for surface markers, as previously described.24 The following directly conjugated antibodies were purchased from BD Biosciences (San Jose, CA, USA) or eBioscience and used for the staining and flow cytometry analysis: AF-488 anti-CD11b (clone M1/70, IgG2b), PE-Cy7 anti-Gr1 (clone RB6-8C5, IgG2b) and PerCP-Cy5.5-CD45.2 (clone 104) that detect CD11b, Gr1 and CD45 (on C57BL/6 mouse lymphocytes). Approximately 1 × 105 living tumor cells were counted per sample in a LSRII flow cytometer machine (BD Biosciences). Data were analyzed using Tree Star FlowJo software (TreeStar, Ashland, OR, USA).
For intracellular cytokine staining, cell suspensions (100 ml per well) were transferred into 96-well tissue culture plates and stimulated with Tyr peptide for 6 h. To each well, 0.2 μg of brefeldin A (B7651; Sigma Aldrich) was added for 1 h to allow cytokines to accumulate within the cell by blocking protein transport into post-Golgi compartments. Receptors for antibody Fc were blocked by adding an Fc blocking reagent (01241D; BD Biosciences). Aliquots of cells were washed and then subjected to staining for CD3 and CD8. Cells were then permeabilized by Perm/wash buffer (2091KZ; Pharmingen) and stained for intracellular IFN-γ, IL-2 and TNF-α. Lymphocytes were analyzed on a FACScalibur flow cytometer (BD Biosciences). Data were analyzed using Tree Star FlowJo software (TreeStar).
Measurement of cytokines by ELISA
Samples for cytokine analysis were collected from the tumor microenvironment. Briefly, peritumoral fluid was collected from pTyr or pVax1 vaccinated mice at day 10 after B16 melanoma tumor challenge. Specifically, levels of MCP-1, IL-10, arginase II and CXLC5 in the supernatants from the tumor microenvironments were measured after the samples clarified by centrifugation (1000 g, 5 min). Immunoassays for cytokine quantitation were then performed using ELISA kits according to the manufacturer’s instructions (R&D Systems).
Immunohistochemical analysis
Immunohistological staining for MDSCs was performed by using anti-mouse Gr1 (RB6-8C5: eBioscience), as described previously.33 Images were viewed with Leica DMIRB Inverted Fluorescence Microscope (Buffalo Grove, IL) and processed with Adobe Photoshop software (Adobe Systems, San Jose, CA, USA).
Statistical analysis
Statistical analyses for comparison of tumor growth and mouse survival between pTyr and pVax1 immunization groups were determined using GraphPad Prism Software (GraphPad Software, La Jolla, CA, USA). Statistical comparisons among groups were performed using Student’s t-test. Correlations between the variables in the pVax1 control and pTyr groups were statistically evaluated using Spearman’s rank correlation test. For all the tests, a ‘P’-value of <0.05 was considered to be statistically significant.
Results
Generation of and protein expression from the pTyr DNA vaccine
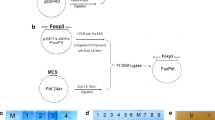
Synthetic full-length human tyrosinase gene (Tyr) sequences were generated as described previously.34 Figure 1a shows a general schematic diagrams for pTyr in a mammalian expression vector. Full-length Tyr genes were successfully synthesized amplified and cloned. To evaluate the expression of pTyr, western blot analysis was performed using pTyr-transfected cell lysates. As indicated in Figure 1b, cell lysates from pTyr-transfected 293T cells revealed the correct molecular size (70 kDa) corresponding to Tyr protein expression. Actin expression in the blotted samples was used as a positive control.

Construction and expression of pTyr (human tyrosinase expressing DNA vaccine) and generation of cellular immune responses. (a) A Tyr-expressing construct (pTyr) was generated using the GenBank and National Center for Biotechnology Information (NCBI) databases. The pTyr construct represents the single sequence that reflects the most conserved amino acids. This synthetic Tyr sequence was then subcloned into the pVax1 expression vector between BamHI and Xho1 restriction sites. (b) Western blot analysis of expression of pTyr. Human 293T cells were transiently transfected with 10 μg of either the control (pVax1) or pTyr vectors, as indicated. Cell lysates were extracted 48 h after transfection and separated by polyacrylamide gel electrophoresis (PAGE), followed by immunoblotting. The immunoblot was incubated with an anti-Tyr antibody that demonstrated staining in the pTyr but not the pVax-1 lane. Staining of electrophoresced and blotted β-actin was used as a control. (c) Schematic representation of immunization and immune analysis time points utilized in this study. (d, e) Interferon-γ (IFN-γ) enzyme-linked immunospot (ELISpot) assay characterization of dominant epitopes analysis: mice were immunized with pVax1 or 20 μg of pTyr using electroporation (EP)-mediated delivery. Splenocytes from individual mice (n=5 per group) were isolated 1 week after third immunization and stimulated with the Tyr peptide matrix mixture pool as indicated. The number of IFN-γ-secreting splenocytes was calculated as the average number of spots in the triplicate wells. Mice vaccinated with pTyr DNA induced higher IFN-γ levels than in the pVax1 control group. The hatched lines in (d, e) represent background levels for spot-forming cells (SFCs) for the assay (that is, ⩽50 IFN-γ SFCs per 106 splenocytes). Dominant epitopes indicated in (d, e) are indicated and described in the Results section.
Magnitude of cellular immune responses elicited after pTyr immunization
The induction of IFN-γ-producing T cells by vaccination was assessed using spleen cells isolated from pVax1- or pTyr-immunized mice using a standard ELISpot assay.24, 35 A schematic diagram of the immunization strategy/protocol for the vaccine dosage study is indicated in Figure 1c. C57BL/6 mice (n=5 each) were grouped and mice in the experimental groups were immunized with 20 μg of pTyr or pVax1 DNA (the dose was selected based on data from a pilot study not shown using the peptide matrix pool). Delivery was enhanced by using EP. Specifically, splenocytes from pTyr- or pVax1-immunized mice were isolated and stimulated overnight with either the human or mouse Tyr matrix peptide pools to characterize the induced cellular immune responses and epitope mapping assays were conducted to determine the immunodominant peptide within the Tyr antigen. Accordingly, based on these results, specific mapping was performed using 24 individual peptide pools (15-mers, with 11 amino acids overlapping) from the matrix, as stimulators for splenocytes from pTyr and pVax1 immunized mice, utilizing the IFN-γ ELISpot assay. The mapping data revealed two dominant Tyr epitopes ‘DWRDAEKCDICTDEY’ and ‘AKHTISSDYVIPIGT’ in both the mouse and human sequences. The dominant peptides listed were confirmed to contain one H2-Db restricted epitope by using Immune Epitope Database analysis resource IDEP:AR http://tools.iedb.org consensus tool, suggesting effective processing of this antigen. These results indicated a breadth and magnitude of cellular immune responses induced by the optimized pTyr vaccine (Figures 1d and e).
Enhanced pTyr vaccine increases secretion of immunomodulatory cytokines
In light of the potent immune responses indicated by the IFN-γ ELISpot assay, CD3+/CD8+ T-cell responses were next evaluated by intracellular cytokine staining assays in order to determine the overall immunomodulatory effects of the enhanced pTyr vaccine. Splenocytes were isolated 1 week following the third immunization and were stimulated with Tyr peptides. Activated IFN-γ, TNF-α and IL-2 secreting Tyr-specific CD8+ T cells were measured by flow cytometric analysis after a 6-h incubation using methods described previously.36 Upon analysis of the polyfunctionality of the CD8+ T cells, it was observed that mice immunized with pTyr exhibited a comparable higher frequency of CD8+ T cells secreting various combinations of TNF-α, IL-2 and IFN-γ. CD8+ T cells from pTyr-vaccinated mice had triple functionality, capable of producing IFN-γ, IL-2 and TNF-α (Figure 2a). Hence, central (TCM; CD8+CD44hiCD62Lhi) and effector (TEM; CD8+CD44hiCD62Llo) memory T-cell responses generated by Tyr peptide-stimulated splenocytes following pTyr vaccination were evaluated. It was observed that pTyr-vaccinated mice elicited significantly (P=0.0001) higher levels of TCM as well as TEM responses compared with the pVax1 control immunized group (Figures 2b and c).

Prophylactic pTyr (human tyrosinase expressing DNA vaccine) vaccination stimulates production of interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), interleukin-2 (IL-2) and anti-Tyr humoral immune responses. (a) Single-cell suspensions of splenocytes (1 × 106 cells per well) were prepared from mice immunized with either pVax1 or pTyr-DNA vaccines as described in the Materials and methods. Lymphocytes were purified from splenocytes by using a T-cell enrichment kit and stained for cell surface marker CD3/CD8-(PerCP) as well as for intracellular measurement of IFN-γ-(PE), TNF-α-(FITC) and IL-2-(APC). The frequency of IFN-γ, TNF-α and IL-2 producing CD8+ T cells was determined within the CD8+ T-cell population. (b, c) Splenocytes from immunized mice were stained with surface markers CD3 (APC), CD8 (PerCP), CD44 (FITC) and CD62L (PE) to measure the percent of (b) central (TCM, CD44hi+CD62L hi) and (c) effector (TEM, CD44hi+CD62L lo) memory CD8+T cells in pTyr- and pVax1-immunized mice. Specifically, cells (5 × 105 ) from mice immunized with pVax1 or pTyr (n=4) were stained, gated and analyzed using FlowJo software. Values from individual mice are expressed as means±s.e. (d, e) Detection of tyrosinase-specific antibodies following prophylactic pTyr vaccination. (d) Production, indicated by OD (450 nm) values, of anti-Tyr total IgG in C57Bl/6 mice (n=4) after three immunizations (at 2–week intervals) with pTyr (closed circle) or pVax1 (closed square). Of each vaccine, 20 μg was delivered with electroporation (EP). The sera samples, diluted as indicated below, from immunized mice were collected and anti-Tyr specific antibody levels were measured by enzyme-linked immunosorbent assay (ELISA) using IgG-specific horseradish peroxidase (HRP)-labeled secondary antibodies. Significant differences in OD (450 nm) values between sera from the pTyr and pVax1 immunized groups were observed. (e) Representative anti-Tyr binding levels in sera from pTyr-immunized mice, from three independent experiments, were measured at day 0 (preimmune) as well as days 35, 90 and 120 after vaccination. Sera were serially diluted at 1:20, 1:40, 1:80, 1:160, 1:320 and 1:640 for the ELISA analysis. Each serum dilution was added in triplicate to individual wells (50 μl per well) coated with Tyr recombinant protein. Peak increases in anti-Tyr-specific antibody levels (indicated by OD (450 nm)), compared with the preimmune serum, were detected for all immunized groups.
Humoral immune responses following immunization with pTyr
Humoral immune responses following vaccination was assessed using an ELISA assay as detailed in the Materials and methods section. Antigen-specific total IgG antibody levels against Tyr were evaluated as previously described.24, 25 Significantly higher anti-Tyr total IgG levels were observed in pTyr-vaccinated mice compared with vector alone for all dilution factors (P=0.0001; Figure 2d). Titers estimated at over 500 were routinely observed. Antibody responses appeared to be long-lived as high levels were continued to be observed 120 days after immunization (Figure 2e), indicating robust humoral immune response elicited by the novel pTyr vaccine.
Enhanced pTyr vaccination protects mice against melanoma tumor growth and is correlated with the generation of protective tumor-specific CD8 + T cells
To investigate the prophylactic effects of pTyr immunization on tumor growth, C57BL/6 mice were immunized three times at 2-week intervals with pVax1 or pTyr followed by challenge with 5 × 105 B16 cells injected subcutaneously into the right flank at 1 week after the third immunization (Figure 3a). C57BL/6 mice were separated into three groups (n=10) as follows: pVax1, pTyr and pTyr immunized that had the CD8+ T-cell population depleted. Tumor calipher measurements and Kaplan–Meier survival curves indicate that mice vaccinated with the pVax1 control construct developed tumors that grew rapidly and reached maximum (that is, 100 mm3) lesion size by day 7. The mean tumor volumes (Figure 3b) and survival rates (Figure 3c) are shown. Immunization with pTyr dramatically slowed tumor growth in mice as compared with pVax1 control immunization (P=0.0049).

In vivo protection against tumor growth following prophylactic pTyr (human tyrosinase expressing DNA vaccine) vaccination. C57Bl/6 mice (n=10) were immunized three times at 2-week intervals with pTyr or pVax1 (20 μg intramuscular injection), delivered with electroporation (EP). At 1 week after third immunization, mice were subcutaneously challenged with B16 melanoma cells. Half of the mice per group (n=5) were killed 20 days after tumor challenge for immune analysis, whereas the remaining mice were monitored up to day 80 for tumor size and survival rates. (a) The schedule for immunization, tumor challenge as well as immune analysis is indicated. (b) Tumor size (that is, volume) was measured using digital calipers every other day following B16 melanoma cell challenge for a period of 20 days typically for the nonvaccinated control groups or until the tumors reached a volume of 1000 mm3, at which point killing was necessitated for humane considerations. Error bars indicate the s.d. values for tumor volumes. Results demonstrate that the pTyr-vaccinated mice (filled circles) had significantly reduced tumor volumes compared with pVax1-vaccinated mice (filled squares). (c) Kaplan–Meier survival curves after B16- melanoma cell challenge. Mice immunized with pTyr DNA had improvement in tumor-free survivals compared with pVax1-vaccinated mice or pTyr-vaccinated and CD8-depleted mice. The data are a representation of three independent experiments on groups of 10 animals each. (d) CD8+ T-cell depletion experiments were performed using the same prophylactic pTyr immunization and B16 melanoma cell challenge schedule as indicated in Figure 4a in order to determine the CD8+ T-cell immune-mediated contribution to pTyr vaccine-associated protection from tumor challenge. Tumor volumes of the pTyr plus CD8+ T cell-depleted vaccinated mice were similar to tumor volumes measured in pVax1-immunized mice, both of which were significantly higher than the pTyr-vaccinated mice without CD8+ T-cell depletion. These results confirm the requirement for CD8+ T cells in generating antitumor immunity mediated by the pTyr vaccine.
Data from this study clearly demonstrated that pTyr vaccination can generate IFN-γ systemically. As IFN-γ is an effector function of CD8+ T cells, it is possible that the efficacy of the pTyr vaccine results mainly from CD8+ T cells. To test this hypothesis, CD8+ T-cell depletion experiments were performed. In vivo monoclonal antibody treatment-mediated ablation of CD8+ (clone 2.43; BD Pharmingen) cells was performed by intraperitoneal injection of 100 μg of purified endotoxin-free monoclonal antibody/mouse/injection on days −3 and −1 before tumor inoculation. Depletion of CD8+ T cells completely abrogated the immune responses elicited by pTyr, indicating that CD8+ T cells are required for vaccine-induced immune responses (data not shown). Tumor volumes in CD8-depleted pTyr-vaccinated mice were similar to those in pVax1 mice, both significantly greater than in the pTyr-vaccinated mice without CD8 depletion (P=0.0008; Figure 3d). These results confirm the requirement for CD8+ T cells in generating antitumor immunity elicited by pTyr vaccination.
pTyr vaccination reduces MDSC accumulation in B16 tumor
Previous research has demonstrated that MDSC accumulation promotes tumor immune evasion and tumor progression. Thus, the accumulation of MDSCs in the tumor tissues and splenocytes 20 days after inoculation of the C57BL/6 mice with B16 cells was measured using flow cytometric analysis. As per procedures outlined in the Materials and methods section, the MDSC population was determined for Gr-1+CD11b+-expressing cells on gated CD45.2-expressing cells (Figure 4a). Mice immunized with pTyr had significantly lower percentages of Gr-1+ and CD11b+-expressing cells than pVax1-immunized mice in both the pre-centrifuged tumor microenvironment sample (P=0.0002; Figure 4b) and circulating cells (P=0.0019; Figure 4c), suggesting that inhibition of CD11b+Gr1+ cell recruitment following pTyr is important for the control of tumor growth.

Reduced myeloid-derived suppressor cell (MDSC) levels in a prophylactic pTyr (human tyrosinase expressing DNA vaccine) vaccination model. (a) Flow cytometry was utilized to analyze the accumulation of MDSCs in the tumoral and peritumoral tissues 20 days after inoculation of B16 melanoma cells into C57BL/6 mice. Gating strategies to separate out MDSCs positive for Gr-1 and CD11b analyses are indicated and described in detail in the Materials and methods section. (b, c) Samples of tumor cells from pVax1- and pTyr-vaccinated mice were stained for Gr-1 and CD11b markers to determine the proportion of MDSCs in the tumor microenvironment (b) and among circulating cells from blood samples (c). This experiment was performed three times. Immunohistochemistry was performed to visualize MDSC localization in the tumor microenvironment. (d) Images indicated that MDSCs had marked tumor infiltration from the pVax1-immunized mice but no obvious infiltration in the pTyr DNA-vaccinated mice. Percentages of MDSCs were calculated. Mice vaccinated with pTyr had significantly lower percentages of CD11b+ cells than did pVax1-vaccinated mice, suggesting that the pTyr vaccine is inhibiting MDSC migration and infiltration to the tumor with a subsequent reduction in the size and progression of the malignancy.
Subsequently, immunohistochemistry was performed to visualize MDSC localization in the tumor microenvironment. Images indicated that MDSCs infiltrated into the tumor most markedly in the pVax1-immunized mice. However, there was no obvious infiltration of MDSCs in the pTyr vaccinated mice (Figure 4d).
pTyr vaccination reduces intratumoral recruitment of MDSCs correlated with IL-10, MCP-1, CXCL5 and arginase II levels
MDSCs indirectly affect T-cell activation through the production of T regulatory cells that typically results in the suppression of cell-mediated immunity.37, 38 Furthermore, within tumors, MDSCs produce several immunosuppressive molecules that potentially alter or dampen antitumor immune responses. MDSCs perturb antitumor immunity by skewing the tumor microenvironment toward a cancer-promoting Th2 phenotype through the production of IL-10. In addition, MDSCs also suppress the production of the Th1 cytokine IL-12 that further increases MDSC production of IL-10.37, 39, 40, 41, 42 Accordingly, we sought to determine whether vaccination with pTyr is able to overcome this MDSC-mediated immunosuppressive environment. MDSCs isolated from pVax1-immunized animals were immunosuppressive, as evidenced by significant secretion of IL-10, whereas MDSCs isolated from the pTyr-immunized group showed decreased levels of IL-10 (P=0.0003; Figure 5a).

Reduced levels of interleukin-10 (IL-10), monocyte chemoattractant protein 1 (MCP-1) and arginase II in pTyr (human tyrosinase expressing DNA vaccine)-immunized mice. An enzyme-linked immunosorbent assay (ELISA) was used to measure the levels of IL-10, MCP-1, CXCL5 and arginase II in the tumor microenvironment from both pVax1- and pTyr-vaccinated mice. Samples from mice immunized with the pTyr DNA vaccine or the control pVax1 plasmid and challenged with B16 tumor were collected at day 10 after challenge and measured for the level of the markers listed above by ELISA. This experiment was performed two times.
Furthermore, with the observed accumulation of MDSCs in B16 tumors, the role of MCP-1 was examined as it is an important chemokine for MDSC migration. MCP-1 is a potent angiogenic factor, promoting vascularization ex vivo and in vivo and affecting migration of endothelial cells.43, 44 An ELISA was used to detect the expression of MCP-1 in sera from pVax1- or pTyr-vaccinated mice. A significant increase in MCP-1 levels was observed in serum samples from pVax1 mice compared with pTyr-vaccinated mice (P=0.0034), suggesting that the pTyr vaccine may modulate MCP-1 levels, resulting in the observed decrease in MDSC migration to the tumor tissue (Figure 5b).
CXCL5 is a small cytokine in the CXC family that is produced after cells are stimulated by inflammatory cytokines such as IL-1 or TNF-α. CXCL5 helps to stimulate chemotaxis of neutrophils and indirectly promotes angiogenesis by interacting with the chemokine receptor CXCR5. For some patients, CXCL5 has been noted to reduce sunburn pain and is involved in chemotactic and activating roles on neutrophils during inflammatory responses.45, 46 Because of these properties, we decided to examine CXCL5 levels in inducing MDSC migration to the tumor microenvironment. The results presented demonstrated that pTyr-vaccinated mouse groups had significantly reduced CXCL5 levels in collected sera compared with pVax-1 groups (P=0.0155; Figure 5c), thus demonstrating a potential role for CXCL5 in MDSC recruitment.
Previous studies have also suggested that tumor-associated myeloid cells expressed high levels of protein and mRNA for arginase I and arginase II and that MDSCs are typically characterized by high levels of arginase expression.47 In this regard, we screened arginase activity in the tumor microenvironment population in pVax1- and pTyr-vaccinated mice. Tumor-associated myeloid cell fractions from naive mice demonstrated an ability to produce arginase II level, whereas a lower level of arginase II expression was detected in mice receiving the pTyr vaccine in the tumor-associated myeloid cell fraction compared with the tumor-associated myeloid cell subpopulation of pVax1-vaccinated mice (P=0.0001; Figure 5d). Thus, the data suggest that tumor-associated myeloid cells (that is, MDSCs) are a unique subset of cells that produce high levels of arginase II in the tumor microenvironment. This supports the thesis that the antitumor effect caused by inhibition of arginase is, in part, dependent on Tyr vaccine-mediated immune function.
Intramuscular administration of the pTyr vaccine elicits antitumor immunity against melanoma in a therapeutic mouse vaccination model
The eradication of existing tumors by active immunization remains a daunting clinical challenge. On that point the therapeutic potential of an enhanced consensus pTyr vaccine was evaluated in terms of the ability to eradicate or inhibit the growth of established melanoma tumors. In this study, C57/BL6 mice were inoculated with 5 × 105 B16F10 melanoma cells (in 200 μl RPMI medium) by subcutaneously inoculated with in the right flank at day 0. A schematic diagram of the immunization and challenge strategies for the therapeutic model is provided in Figure 6a. For this therapeutic vaccine challenge approach, tumor cells were inoculated first. When the tumor was palpable (that is, ∼7 days after inoculation), mice were vaccinated intramuscularly 3 times at 1-week intervals with either 20 μg of pTyr DNA or pVax1 mediated by EP-enhanced delivery.

Therapeutic antitumor effects and induced Tyr-specific CD8+ T-cell responses in mice immunized with the pTyr (human tyrosinase expressing DNA vaccine). (a) Schedule for tumor challenge, pTyr immunization and immune analysis in the therapeutic vaccination model. Following B16 tumor challenge on day 0, mice were immunized with pTyr on days 7, 14 and day 21. (b) Kaplan–Meir survival curves for mice immunized with pVax1 or pTyr in the therapeutic vaccination model. (c) Frequencies of Tyr-specific interferon-γ (IFN-γ)-producing CD8+ T cells in mice challenged with B16 melanoma cells and measured 1 week after third immunization. (d) Inhibition of myeloid-derived suppressor cell (MDSC) expansion measured by levels of the MDSC markers Gr1 and CD11b in the tumor microenvironment following pTyr vaccination. Analysis was performed by flow cytometric measurement of Gr-1+CD11b+-expressing MDSCs. (e) Levels of monocyte chemoattractant protein 1 (MCP-1) were measured, by enzyme-linked immunosorbent assay (ELISA), in mice vaccinated with pTyr or pVax-1. The results indicate that pTyr primed and expanded antigen-specific CD8+ T cells that subsequently inhibit MDSC function and expansion. This experiment was performed at least three times.
pTyr vaccination significantly suppressed tumor growth compared with control pVax1-vaccinated group. Interestingly, the combination of novel optimized pTyr vaccination and EP-mediated delivery resulted in enhanced antitumor activity in melanoma-bearing mice compared with control vaccination (pVax1) groups, as indicated by long-term survival (by 60%) of the pTyr-vaccinated mice (Figure 6b).
To investigate the immunological mechanism underlying the therapeutic effect of pTyr vaccination, CD8+ T-cell responses were measured by IFN-γ ELISpot. Splenocytes from immunized mice were isolated 1 week after third immunization and stimulated with a Tyr peptide, as described in the Materials and methods section. The Tyr-specific spot numbers in the assay generated by splenocytes from mice vaccinated with pTyr DNA groups were significantly increased compared with that of the pVax1 control group (Figure 6c; P=0.0001). The high cytotoxic T-lymphocyte response in the therapeutic model indicates the potential for the pTyr vaccine to attenuate melanoma tumor growth or to ideally result in complete tumor regression. This ability to attenuate tumor growth further reveals the possible therapeutic applications of the pTyr vaccine. Furthermore, to investigate further the role of cellular immunity in pTyr immunotherapy, the MDSC population as well as MCP-1 production in the tumor microenvironment was evaluated. A modest but significant decrease in the number of MDSCs (Figure 6d) as well as a diminution in MCP-1 production in tumor microenvironment of the mice that were vaccinated with pTyr (Figure 6e) was observed.
Discussion
The ultimate goal of a cancer vaccine is to prevent or eliminate tumors effectively while causing minimal collateral damage to normal cells and tissues.1, 2, 48, 49 Thus far, other than prophylactic vaccines targeting oncogenic viruses (that is, for hepatitis B and human papillomavirus), cancer vaccines tested in the clinic to date have not been very successful in reaching this goal. In addition, the mechanisms of immunological tolerance against cancers make the generation of antitumor, biologically effective immune responses difficult to generate. Therefore, novel and safe strategies, which can break immune tolerance against tumor antigens, are required in order to improve the efficacy of current cancer vaccines.50, 51, 52 In order to enhance the capability of a tumor antigen to generate antigen-specific immunity, many laboratories have described strategies whereby immune adjuvants, that is, cytokines or other molecules, are fused to targeted antigens.10, 53, 54 Previously, our group has described a strategy for enhancing the immunogenicity of tumor antigens by administration of naked DNA plasmids expressing either specific antigen or immunomodulatory molecules through EP-mediated delivery.48, 55, 56, 57, 58, 59, 60 Thus far, these vaccines have been demonstrated to elicit antigen-specific immunity in clinical studies.56, 61
DNA vaccines are able to induce potent antitumor cell-mediated immune responses against a number of tumor-associated antigens, including Tyr.12, 16, 49, 62 There are also several tumor-specific antigens that are uniquely expressed on a lineage of specific tumor cells.18 Tyr, an enzyme that catalyzes the synthesis of melanin, is an antigen considered to be unique to melanoma, and represents a prototypical differentiation antigen expressed by cells of the melanocytic origin.63, 64 Importantly, this immune target antigen is recognized by CD8+ T cells that were derived from some melanoma patients.15, 64 As Tyr is expressed homogeneously in most melanoma specimens, it is a logical protein to target for melanoma vaccination strategies. Various methods for vaccination have been utilized to immunize against Tyr and, specifically, measurable CD8+ T-cell responses have been induced against a synthetic Tyr peptide matrix mixture with some anecdotal clinical benefits being reported in some of these immune therapy studies.65, 66, 67
In addition, there has been growing motivation to better characterize the role of MDSCs in the tumor microenvironment and the relationship between DNA vaccines and MDSCs in reducing tumor progression.36, 63 In this study, a novel and highly enhanced synthetic consensus Tyr DNA vaccine was developed and demonstrated to be highly immunogenic in vivo when delivered with EP. This Tyr expression plasmid significantly increased CD8+ T cell-mediated IFN-γ secretion in an antigen-specific manner in the prophylactic vaccination model. Furthermore, the pTyr vaccine increased survival and attenuated B16 melanoma tumor growth following in vivo tumor challenge in both prophylactic and therapeutic models.
There has been a strong association between multifunctional T cells (producing more than one cytokine) elicited by enhanced DNA immunization and strong and effective antitumor immunity in mice.4, 22 Therefore, in the current study, we characterized the ability of CD8+ T cells to produce cytokines such as IFN-γ, TNF-α and IL-2 at a single cell level by flow cytometry. It was demonstrated that a DNA plasmid-expressing tyrosinase (pTyr) was able to induce, in immunized mice, a significantly higher frequency of these cytokines with a prominent production of the combination of IFN-γ and TNF-α compared with mice immunized with pVax1. Studies with other systems indicate the importance of such T cells for protective immunity.68 To be an effective vaccine, immunization with pTyr should establish long-lasting immunological memory and give rise to a strong and robust response upon challenge with tumor. Higher frequencies of both TCM and TEM in the animals vaccinated with pTyr compared with the pVax1 group were noted.
Recent studies report that MDSCs are involved in producing an immune regulatory cytokine environment that can present antigens to drive immature T cells to become noneffective in tumor clearance.69, 70 Thus, this study aimed to better elucidate the role of the pTyr vaccine in obstructing the tumor-promoting functions of MDSCs. The decrease in the MDSC population observed in tumors from pTyr-vaccinated mice, coupled with the observed decrease of IL-10, MCP-1, arginase II and CXCL5 (Figure 5) in the tumor microenvironment, supports the hypothesis that the novel and enhanced pTyr vaccine is able to induce armed anti-Tyr effector CD8+ T cells to eliminate MDSCs. Together, all of these activities are hypothesized to contribute to tumor growth attenuation and regression. Further studies examining the direct effect of antitumor CD8+ T cells on MDSC populations in the context of a novel DNA plasmid vaccine are in progress. Of particular emphasis will be the goal of inducing complete and durable melanoma tumor regression in a therapeutic vaccine model. In sum, the findings reported in this study as well as the results from future investigations will have implications and applications to other types of malignancies.
References
Hanahan D, Weinberg RA . The hallmarks of cancer. Cell 2000; 100: 57–70.
Rosenberg SA . Finding suitable targets is the major obstacle to cancer gene therapy. Cancer Gene Ther 2014; 21: 45–47.
Bajetta E, Del Vecchio M, Bernard-Marty C, Vitali M, Buzzoni R, Rixe O et al. Metastatic melanoma: chemotherapy. Semin Oncol 2002; 29: 427–445.
Rosenberg SA . IL-2: the first effective immunotherapy for human cancer. J Immunol 2014; 192: 5451–5458.
Yang JC, Sherry RM, Rosenberg SA . Melanoma: why is sentinel lymph node biopsy ‘standard of care’ for melanoma? Nat Rev Clin Oncol 2014; 11: 245–246.
Johnstone RW, Ruefli AA, Lowe SW . Apoptosis: a link between cancer genetics and chemotherapy. Cell 2002; 108: 153–164.
Gerber HP, Olazoglu E, Grewal IS . Targeting inflammatory cells to improve anti-VEGF therapies in oncology. Recent Results Cancer Res 2010; 180: 185–200.
Duffy A, Zhao F, Haile L, Gamrekelashvili J, Fioravanti S, Ma C et al. Comparative analysis of monocytic and granulocytic myeloid-derived suppressor cell subsets in patients with gastrointestinal malignancies. Cancer Immunol Immunother 2012; 62: 299–307.
Chou HS, Hsieh CC, Yang HR, Wang L, Arakawa Y, Brown K et al. Hepatic stellate cells regulate immune response by way of induction of myeloid suppressor cells in mice. Hepatology 2011; 53: 1007–1019.
Dang Y, Wagner WM, Gad E, Rastetter L, Berger CM, Holt GE et al. Dendritic cell-activating vaccine adjuvants differ in the ability to elicit antitumor immunity due to an adjuvant-specific induction of immunosuppressive cells. Clin Cancer Res 2012; 18: 3122–3131.
Wolchok JD, Yuan J, Houghton AN, Gallardo HF, Rasalan TS, Wang J et al. Safety and immunogenicity of tyrosinase DNA vaccines in patients with melanoma. Mol Ther 2007; 15: 2044–2050.
Aris M, Zubieta MR, Colombo M, Arriaga JM, Bianchini M, Alperovich M et al. MART-1- and gp100-expressing and -non-expressing melanoma cells are equally proliferative in tumors and clonogenic in vitro. J Invest Dermat 2012; 132: 365–374.
Yuan J, Ginsberg B, Page D, Li Y, Rasalan T, Gallardo HF et al. CTLA-4 blockade increases antigen-specific CD8(+) T cells in prevaccinated patients with melanoma: three cases. Cancer Immunol Immunother 2011; 60: 1137–1146.
Ginsberg BA, Gallardo HF, Rasalan TS, Adamow M, Mu Z, Tandon S et al. Immunologic response to xenogeneic gp100 DNA in melanoma patients: comparison of particle-mediated epidermal delivery with intramuscular injection. Clin Cancer Res 2010; 16: 4057–4065.
Weber J, Boswell W, Smith J, Hersh E, Snively J, Diaz M et al. Phase 1 trial of intranodal injection of a Melan-A/MART-1 DNA plasmid vaccine in patients with stage IV melanoma. J Immunother 2008; 31: 215–223.
Saenger YM, Li Y, Chiou KC, Chan B, Rizzuto G, Terzulli SL et al. Improved tumor immunity using anti-tyrosinase related protein-1 monoclonal antibody combined with DNA vaccines in murine melanoma. Cancer Res 2008; 68: 9884–9891.
Perales MA, Yuan J, Powel S, Gallardo HF, Rasalan TS, Gonzalez C et al. Phase I/II study of GM-CSF DNA as an adjuvant for a multipeptide cancer vaccine in patients with advanced melanoma. Mol Ther 2008; 16: 2022–2029.
Smith KA, Qiu Z, Wong R, Tam VL, Tam BL, Joea DK et al. Multivalent immunity targeting tumor-associated antigens by intra-lymph node DNA-prime, peptide-boost vaccination. Cancer Gene Ther 2011; 18: 63–76.
Steitz J, Tuting T . Biolistic DNA vaccination against melanoma. Methods Mol Biol 2013; 940: 317–337.
Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 2008; 13: 23–35.
Rosborough BR, Castellaneta A, Natarajan S, Thomson AW, Turnquist HR . Histone deacetylase inhibition facilitates GM-CSF-mediated expansion of myeloid-derived suppressor cells in vitro and in vivo. J Leukoc Biol 2012; 91: 701–709.
Rosati SF, Parkhurst MR, Hong Y, Zheng Z, Feldman SA, Rao M et al. A novel murine T-cell receptor targeting NY-ESO-1. J Immunother 2014; 37: 135–146.
Youn JI, Collazo M, Shalova IN, Biswas SK, Gabrilovich DI . Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J Leukoc Biol 2012; 91: 167–181.
Muthumani K, Wise MC, Broderick KE, Hutnick N, Goodman J, Flingai S et al. HIV-1 Env DNA vaccine plus protein boost delivered by EP expands B- and T-cell responses and neutralizing phenotype in vivo. PLoS One 2013; 8: e84234.
Muthumani K, Flingai S, Wise M, Tingey C, Ugen KE, Weiner DB . Optimized and enhanced DNA plasmid vector based in vivo construction of a neutralizing anti-HIV-1 envelope glycoprotein Fab. Hum Vacc Immunother 2013; 9: 2253–2262.
Draghia-Akli R, Khan AS . In vivo electroporation of gene sequences for therapeutic and vaccination applications. Recent Pat DNA Gene Seq 2007; 1: 207–213.
Hirao LA, Draghia-Akli R, Prigge JT, Yang M, Satishchandran A, Wu L et al. Multivalent smallpox DNA vaccine delivered by intradermal electroporation drives protective immunity in nonhuman primates against lethal monkeypox challenge. J Infect Dis 2011; 203: 95–102.
Mallilankaraman K, Shedlock DJ, Bao H, Kawalekar OU, Fagone P, Ramanathan AA et al. A DNA vaccine against chikungunya virus is protective in mice and induces neutralizing antibodies in mice and nonhuman primates. PLoS Negl Trop Dis 2011; 5: e928.
Shin TH, Pankhong P, Yan J, Khan AS, Sardesai NY, Weiner DB . Induction of robust cellular immunity against HPV6 and HPV11 in mice by DNA vaccine encoding for E6/E7 antigen. Hum Vacc Immunother 2012; 8: 470–478.
Muthumani K, Zhang D, Dayes NS, Hwang DS, Calarota SA, Choo AY et al. Novel engineered HIV-1 East African Clade-A gp160 plasmid construct induces strong humoral and cell-mediated immune responses in vivo. Virology 2003; 314: 134–146.
Anthony DD, Lehmann PV . T-cell epitope mapping using the ELISPOT approach. Methods 2003; 29: 260–269.
Kruisbeek AM . In vivo depletion of CD4- and CD8-specific T cells. Curr Protoc Immunol 2001; Chapter 4: Unit 4 1.
Yi H, Guo C, Yu X, Zuo D, Wang XY . Mouse CD11b+Gr-1+ myeloid cells can promote Th17 cell differentiation and experimental autoimmune encephalomyelitis. J Immunol 2012; 189: 4295–4304.
Yan J, Pankhong P, Shin TH, Obeng-Adjei N, Morrow MP, Walters JN et al. Highly optimized DNA vaccine targeting human telomerase reverse transcriptase stimulates potent antitumor immunity. Cancer Immunol Res 2013; 1: 179–189.
Hirao LA, Wu L, Khan AS, Satishchandran A, Draghia-Akli R, Weiner DB . Intradermal/subcutaneous immunization by electroporation improves plasmid vaccine delivery and potency in pigs and rhesus macaques. Vaccine 2008; 26: 440–448.
Nagaraj S, Schrum AG, Cho HI, Celis E, Gabrilovich DI . Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J Immunol 2010; 184: 3106–3116.
Centuori SM, Trad M, Lacasse CJ, Alizadeh D, Larmonier CB, Hanke NT et al. Myeloid-derived suppressor cells from tumor-bearing mice impair TGF-beta-induced differentiation of CD4+CD25+FoxP3+ Tregs from CD4+CD25-FoxP3- T cells. J Leukoc Biol 2012; 92: 987–997.
Hoechst B, Gamrekelashvili J, Manns MP, Greten TF, Korangy F . Plasticity of human Th17 cells and iTregs is orchestrated by different subsets of myeloid cells. Blood 2011; 117: 6532–6541.
Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S . Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol 2007; 179: 977–983.
Schlecker E, Stojanovic A, Eisen C, Quack C, Falk CS, Umansky V et al. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J Immunol 2012; 189: 5602–5611.
Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW . Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother 2011; 60: 1419–1430.
Tanikawa T, Wilke CM, Kryczek I, Chen GY, Kao J, Nunez G et al. Interleukin-10 ablation promotes tumor development, growth, and metastasis. Cancer Res 2012; 72: 420–429.
Pilon-Thomas S, Nelson N, Vohra N, Jerald M, Pendleton L, Szekeres K et al. Murine pancreatic adenocarcinoma dampens SHIP-1 expression and alters MDSC homeostasis and function. PLoS One 2011; 6: e27729.
Chornoguz O, Grmai L, Sinha P, Artemenko KA, Zubarev RA, Ostrand-Rosenberg S . Proteomic pathway analysis reveals inflammation increases myeloid-derived suppressor cell resistance to apoptosis. Mol Cell Proteomics 2011; 10: M110 002980.
Chang MS, McNinch J, Basu R, Simonet S . Cloning and characterization of the human neutrophil-activating peptide (ENA-78) gene. J Biol Chem 1994; 269: 25277–25282.
Mei J, Liu Y, Dai N, Hoffmann C, Hudock KM, Zhang P et al. Cxcr2 and Cxcl5 regulate the IL-17/G-CSF axis and neutrophil homeostasis in mice. J Clin Invest 2012; 122: 974–986.
Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res 2004; 64: 5839–5849.
Weiner DB, Kim JJ . Cancer vaccines: is the future now? Expert Rev Vaccines 2002; 1: 257–260.
Grosenbaugh DA, Leard AT, Bergman PJ, Klein MK, Meleo K, Susaneck S et al. Safety and efficacy of a xenogeneic DNA vaccine encoding for human tyrosinase as adjunctive treatment for oral malignant melanoma in dogs following surgical excision of the primary tumor. Am J Vet Res 2011; 72: 1631–1638.
Boutte AM, McDonald WH, Shyr Y, Yang L, Lin PC . Characterization of the MDSC proteome associated with metastatic murine mammary tumors using label-free mass spectrometry and shotgun proteomics. PLoS One 2011; 6: e22446.
Bruchard M, Mignot G, Derangere V, Chalmin F, Chevriaux A, Vegran F et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med 2012; 19: 57–64.
Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S . Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res 2007; 67: 10019–10026.
Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med 2010; 207: 2439–2453.
Glass GE, Chan JK, Freidin A, Feldmann M, Horwood NJ, Nanchahal J . TNF-alpha promotes fracture repair by augmenting the recruitment and differentiation of muscle-derived stromal cells. Proc Natl Acad Sci USA 2011; 108: 1585–1590.
Fagone P, Shedlock DJ, Bao H, Kawalekar OU, Yan J, Gupta D et al. Molecular adjuvant HMGB1 enhances anti-influenza immunity during DNA vaccination. Gene Ther 2011; 18: 1070–1077.
Bagarazzi ML, Yan J, Morrow MP, Shen X, Parker RL, Lee JC et al. Immunotherapy against HPV16/18 generates potent TH1 and cytotoxic cellular immune responses. Sci Transl Med 2012; 4: 155ra138.
Kim JJ, Tsai A, Nottingham LK, Morrison L, Cunning DM, Oh J et al. Intracellular adhesion molecule-1 modulates beta-chemokines and directly costimulates T cells in vivo. J Clin Invest 1999; 103: 869–877.
Chattergoon MA, Kim JJ, Yang JS, Robinson TM, Lee DJ, Dentchev T et al. Targeted antigen delivery to antigen-presenting cells including dendritic cells by engineered Fas-mediated apoptosis. Nat Biotechnol 2000; 18: 974–979.
Sin J, Kim JJ, Pachuk C, Satishchandran C, Weiner DB . DNA vaccines encoding interleukin-8 and RANTES enhance antigen-specific Th1-type CD4(+) T-cell-mediated protective immunity against herpes simplex virus type 2 in vivo. J Virol 2000; 74: 11173–11180.
Choo AY, Choo DK, Kim JJ, Weiner DB . DNA vaccination in immunotherapy of cancer. Cancer Treatment Res 2005; 123: 137–156.
Kalams SA, Parker SD, Elizaga M, Metch B, Edupuganti S, Hural J et al. Safety and comparative immunogenicity of an HIV-1 DNA vaccine in combination with plasmid interleukin 12 and impact of intramuscular electroporation for delivery. J Infect Dis 2013; 208: 818–829.
Bellone M, Cantarella D, Castiglioni P, Crosti MC, Ronchetti A, Moro M et al. Relevance of the tumor antigen in the validation of three vaccination strategies for melanoma. J Immunol 2000; 165: 2651–2656.
Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res 2009; 69: 2514–2522.
Godet Y, Bonnin A, Guilloux Y, Vignard V, Schadendorf D, Dreno B et al. A new tyrosinase epitope recognized in the HLA-B*4002 context by CTL from melanoma patients. Cancer Immunol Immunother 2009; 58: 271–280.
Kawakami Y, Robbins PF, Wang X, Tupesis JP, Parkhurst MR, Kang X et al. Identification of new melanoma epitopes on melanosomal proteins recognized by tumor infiltrating T lymphocytes restricted by HLA-A1, -A2, and -A3 alleles. J Immunol 1998; 161: 6985–6992.
Tuting T, Wilson CC, Martin DM, Kasamon YL, Rowles J, Ma DI et al. Autologous human monocyte-derived dendritic cells genetically modified to express melanoma antigens elicit primary cytotoxic T cell responses in vitro: enhancement by cotransfection of genes encoding the Th1-biasing cytokines IL-12 and IFN-alpha. J Immunol 1998; 160: 1139–1147.
Wang S, Bartido S, Yang G, Qin J, Moroi Y, Panageas KS et al. A role for a melanosome transport signal in accessing the MHC class II presentation pathway and in eliciting CD4+ T cell responses. J Immunol 1999; 163: 5820–5826.
Olson JA, McDonald-Hyman C, Jameson SC, Hamilton SE . Effector-like CD8(+) T cells in the memory population mediate potent protective immunity. Immunity 2013; 38: 1250–1260.
Ghansah T . A novel strategy for modulation of MDSC to enhance cancer immunotherapy. Oncoimmunology 2012; 1: 984–985.
LaFace D, Talmadge J . Meeting report: regulatory myeloid cells. Int Immunopharmacol 2011; 11: 780–782.
Acknowledgements
We acknowledge members of the Weiner laboratory for significant contributions and/or critical reading and editing of the manuscript. DBW notes funding from the National Institutes of Health, Basser Research Center for BRCA and Inovio Pharmaceuticals. We also thank Penn CFAR and the ACC core facilities for their support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
DBW has grant funding, participates in industry collaborations and has received speaking honoraria and fees for consulting. This service includes serving on scientific review committees and advisory boards. Remuneration includes direct payments or stock or stock options and, in the interest of disclosure, therefore he notes potential conflicts associated with this work with Pfizer, Medimmune, Inovio, Merck, VGXi, OncoSec, Roche, Aldevron and possibly others. Licensing of technology from his laboratory has created over 100 jobs in the biotech/pharma industry. KEU also reports a conflict in terms of co-inventorship and stock ownership in technology licensed to Oncosec Medical Corporation. The other authors declare no conflict of interest. KAK, JJK and NYS are employees of Inovio Pharmaceuticals, and as such receive salary and benefits including ownership of stock and stock option from the company.
Rights and permissions
About this article

Cite this article
Yan, J., Tingey, C., Lyde, R. et al. Novel and enhanced anti-melanoma DNA vaccine targeting the tyrosinase protein inhibits myeloid-derived suppressor cells and tumor growth in a syngeneic prophylactic and therapeutic murine model. Cancer Gene Ther 21, 507–517 (2014). https://doi.org/10.1038/cgt.2014.56
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cgt.2014.56
- Springer Nature America, Inc.
This article is cited by
-
Booster immunizations with DNA plasmids encoding HER-2/neu prevent spontaneous mammary cancer in HER-2/neu transgenic mice over life span
Scientific Reports (2017)
-
Identification of crucial genes in intracranial aneurysm based on weighted gene coexpression network analysis
Cancer Gene Therapy (2015)