
Abstract
Mitochondrial antiviral signaling (MAVS) protein has an important role in antiviral immunity and autoimmunity. However, the pathophysiological role of this signaling pathway, especially in the brain, remains elusive. Here we demonstrated that MAVS signaling existed and mediated poly(I:C)-induced inflammation in the brain. Along with the MAVS signaling activation, there was an induction of autophagic activation. Autophagy negatively regulated the activity of MAVS through direct binding of LC3 to the LIR motif Y(9)xxI(12) of MAVS. We also found that c-Abl kinase phosphorylated MAVS and regulated its interaction with LC3. Interestingly, tyrosine phosphorylation of MAVS was required for downstream signaling activation. Importantly, in vivo data showed that the deficiency of MAVS or c-Abl prevented MPTP-induced microglial activation and dopaminergic neuron loss. Together, our findings reveal the molecular mechanisms underlying the regulation of MAVS-dependent microglial activation in the nervous system, thus providing a potential target for the treatment of microglia-driven inflammatory brain diseases.
Similar content being viewed by others
Main
Mitochondrial antiviral signaling (MAVS) is one of the well-studied signaling pathways, which responses to viral infection and maintains immune homeostasis. Upon infection with an RNA virus, two retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs), RIG-I and melanoma differentiation-associated gene 5 (MDA-5), recognize the viral RNA and interact with MAVS via CARD–CARD domain interaction. The MAVS complex subsequently activates NF-κB and type I interferon by facilitating the nuclear translocation of NF-κB and IRF3, respectively.1, 2, 3, 4 Apart from its function as the essential adaptor of RIG-I and MDA-5, MAVS itself also regulates the translocation and activation of the NLRP3 inflammasome.5 More importantly, MAVS signaling has been found to be critical for immune homeostasis, and dysregualtion of this signaling was associated with type-1 diabetes,6 autoimmune disorders7 and experimental colitis.8
However, it is not known yet the pathophysiological role of this signaling pathway in the brain, especially for the neurodegeneration diseases. Several lines of evidence showed that influenza virus infection contributes to the development of neurodegeneration.9, 10, 11, 12, 13 Moreover, naked poly(I:C), a reagent specifically stimulating TLR3, causes neurodegeneration through TLR3-mediated inflammation.14, 15 Many cytokines also have been found to be involved, including interleukin-1 beta (IL-1β)16 and tumor necrosis factor alpha (TNF-α).17, 18 The above findings linked the inflammatory factors to the development of neurodegeneration. Microglia are the tissue macrophages of the brain, and they have an important role in scavenging pathogens, dying cells and molecules. Activated microglia are observed in many virus infection conditions and neurodegenerative diseases other than Parkinson’s disease (PD), that is, Alzheimer’s disease (AD) and stroke,19, 20, 21, 22 thus representing a potential therapeutic target. Whether MAVS signaling is involved in microglial activation and neurondegeneration diseases still remain elusive.
Taking into account the important role of MAVS signaling, their tight regulation is imperative. Autophagy was reported to regulate the activity of MAVS signaling in MEF cells.23 Also, autophagy defect was found to be involved in the development of neurondegeneration diseases.24, 25, 26 Consistent with this, upregulation of autopahgy by administrating of rapamycin could attenuate oxidative stress-induced neuronal toxicity.27 However, those studies mainly focused on neuronal cells, with few studies on microglial cells. Whether autopahgy regulates MAVS-mediated inflammation in microglia and whether this signaling contributes to the neurondegeneration diseases are still unknown.
In this study, we demonstrate that MAVS signaling is involved in microglial activation in vivo. Along with the inflammation, there is an induction of autophagic activation, and inhibition of autophagy in microglial cells strengthens MAVS-mediated inflammation. Furthermore, we observe that c-Abl kinase phosphorylates MAVS and regulates MAVS signaling activation. Taken together, our findings reveal the molecular mechanisms underlying the regulation of MAVS-dependent microglial activation in the nervous system, with implication of a potential target for the treatment of microglia-driven inflammatory brain diseases.
Results
MAVS signaling is involved in microglial activation in the brain
We first measured protein expression of MAVS in various mouse tissues using immunoblotting. MAVS protein varied widely among the mouse tissues tested, with high levels found in the liver, heart and muscle (Figures 1a and b). There were also considerable levels of MAVS protein in brain samples when compared with organs of the immune system such as the spleen and thymus. To reveal the existence of MAVS signaling pathway in the substantia nigra (SN), we injected poly(I:C)/LyoVec (complexed with transfection reagent, 1 μg for each mice), a specific stimulator of the RLR pathway, into the SN of MAVS−/− or wild-type mice (Figure 1c) and measured IFN-β and TNF-α levels. As shown in Figures 1d and e, poly(I:C)/LyoVec significantly increased IFN-β and TNF-α levels in wild-type mice, but not in MAVS−/− mice, suggesting that MAVSs in the dopaminergic system also mediate poly(I:C)-induced inflammation. Because microglial cells are the main immune cells in the brain,28 we next isolated primary microglial cells from wild-type mice and MAVS−/− mice. Consistent with the role of MAVS in macrophages investigated in this study (Supplementary Figures S1A and B), MAVS knockout significantly decreased the protein levels of IFN-β, TNF-α and IL-6 induced by poly(I:C) (Figures 1f–h), as well as the mRNA levels induced by Sendai virus (SeV) in microglial cells (Figures 1i–k) in the primary microglia. In addition, MAVS knockdown in both BV2 and N9 cells markedly reduced the levels of IFN-β induced by poly(I:C) treatment (Supplementary Figures S1D–G). These results strongly suggested that MAVS is critical for microglial activation induced by either poly(I:C) stimulation or SeV infection in the brain.

MAVS is involved in microglial activation. (a and b) Immunoblotting analysis of MAVS and GAPDH in different mice tissues and quantitative data of MAVS expression. (c) Model of stereotaxic injection of poly(I:C)/LyoVec into the SN (three mice in each group). (d and e) RT-PCR analysis the expression of IFN-β, TNF-α in the SN in MAVS−/− mice and wild-type mice. (n=3 per group). *P<0.05. (f–h) ELISA of IFN-β, TNF-α and IL-6 in the supernatant of primary microglial cells from wild-type mice or MAVS−/− mice treated with transfected poly(I:C) (complexed with Lipofectamine 2000 at a ratio of 1 : 2; 1 μg/ml) for 24 h. *P<0.05. (i, k) Expression of IFN-β, TNF-α and IL-6 in primary microglial cells from wild-type mice or MAVS−/− mice treated with SeV for 12 h. *P<0.05. Error bars are mean±S.E.M. Experiments were carried out in triplicate, at least three independent times
Autophagy inhibits the activation of MAVS signaling in microglial cells
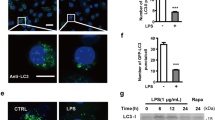
Previous reports implicate that there is functional connection between autophagy and RLR signaling activation.29, 30 Here we observed that treatment of primary microglial cells with poly(I:C) increased the levels of p-IRF3, along with an induction of autophagic activation with the increase of LC3II and the degradation of p62 (Figures 2a–c and Supplementary Figures S2A). A similar observation was made in BV2 cells as seen in Supplementary Figures S2B–D, consistent with the amount of LC3 puncta in cells (Figures 2d and e). Next, we blocked autophagic flux by adding bafilomycin A1 in the culture medium. As shown in Supplementary Figures S2E, poly (I:C) treatment could increase LC3II levels in both normal condition and autophagic flux blockage situation, suggesting that poly (I:C)-induced autophagy activation is due to an enhancement of LC3II levels. Moreover, rapamycin, a well-known autophagy inducer, was used as a positive control (Supplementary Figure S2E). Next, we examined the role of autophagy in the activation of MAVS signaling. As shown in Figures 2f and g, in response to poly(I:C) treatment, LC3 knockdown increased the levels of p-IRF3. IN addition, the levels of both IFN-β and TNF-α increased upon LC3 knockdown (Figures 2h and i), suggesting that autophagy negatively regulates the activation of MAVS signaling in microglial cells. To further confirm this, we generated a mouse that had the deletion of Atg5 gene in microglia, and then isolated primary microglial cells. We observed that Atg5 deletion increased the expression of the IFN-β, TNF-α and IL-6 in poly(I:C)-treated microglial cells (Figures 2j–l). Together, these results confirmed that autophagy inhibits MAVS-mediated inflammation in microglial cells.

Autophagy inhibits the activation of MAVS signaling. (a–c) Immunoblotting analysis of p62, LC3 (rabbit antibody, #2775, Cell Signaling, 1:1000), p-IRF3, IRF3 and GAPDH in primary microglial cells treated with transfected poly(I:C) (1 μg/ml) and quantitative data of p-IRF3 and LC3II expression. *P<0.05. (d and e) Immunofluorescent staining of LC3 and Hoechst in BV2 cells treated with transfected poly(I:C) (1 μg/mL) for 6 h. The images were collected by the confocal laser scanning microscopy (Leica SP5 confocal microscope) using the × 40 objective lens. The quantitative data of LC3 puncta in BV2 cells were analyzed (20 cells were counted in each group). (f and g) Immunoblotting analysis of p-IRF3, IRF3, LC3 and GAPDH in control and LC3 knockdown BV2 cells treated with transfected poly(I:C) (1 μg/ml) and quantitative data of p-IRF3 expression. *P<0.05. (h and i) Expression of IFN-β and TNF-α in control and LC3 knockdown BV2 cells treated with transfected poly(I:C) (1 μg/ml) for 12 h. *P<0.05. (j–l) ELISA of IFN-β, TNF-α and IL-6 in the supernatant of primary microglia from Atg5flox/flox mice and Atg5flox/floxCD11b-Cre mice treated with transfected poly(I:C) (1 μg/ml) for 24 h. *P<0.05. Error bars are mean±S.E.M. Experiments were carried out in triplicate for three independent times
Autophagy regulates MAVS protein level through the direct binding of LC3 to MAVS
We next asked how autophagy inhibits the activity of MAVS signaling in microglial cells. We first measured the protein levels of MAVS under the autophagy deficiency conditions. As shown in Figure 3a, Atg5 knockout increased MAVS protein levels in microglial cells, suggesting that autophagy negatively regulates the protein levels of MAVS in microglial cells. Autophagy receptors, such as NBR, Atg32, p62 and FUNDC1, interact with LC3 through a classical linear motif of W/YxxL/I (LIR motif).31, 32, 33 Interestingly, we found that there was a conserved classical LIR motif Y(9)xxI(12) in the CARD domain of MAVS (Figures 3b and Supplementary Figure S3A), with the motif being exposed to the cytosolic site of the mitochondrial membrane. To confirm this, we transfected the Flag-tagged wild-type MAVS or the CARD deletion mutant of MAVS into HEK293T cells and found that wild-type MAVS interacted with LC3, but not the CARD domain mutant MAVS (Figure 3c), suggesting that MAVS binds to LC3 through its CARD domain. To test whether MAVS binds to LC3 through this LIR motif, we then generated a series of mutants including Y9F, I12A and double mutants Y9F/I12A. We found that mutations in the LIR motif markedly impaired the interactions of MAVS with LC3 (Figure 3d). GST-pull down assay showed that either deletion of the CARD domain or mutation in the LIR motif abolished this interaction (Figure 3e). Furthermore, we found that ectopically expressed wild-type MAVS increased LC3II levels in microglial cells, but not Y9F mutant MAVS (Figure 3f), suggesting that MAVS activation-induced autophagy through direct binding of MAVS and LC3 via its LIR motif. We then asked whether autophagy mediated the degradation of MAVS through its modification in LIR motif. To end this, we determined the expression of MAVS and mutant Y9F MAVS after treating cells with cycloheximide. As shown in Figures 3g and h, we found that mutant Y9F MAVS hold much higher stability compared with wide-type MAVS. Taken together, these results demonstrated that autophagy mediates the degradation of MAVS through this LIR motif, which suggested autophagy negatively regulates microglial inflammation through modulating MAVS protein stability. In addition, reactive oxygen species (ROS) have been regarded as crucial contributors to microglia-driven inflammation.34, 35, 36 Here we found that poly(I:C) treatment increased ROS levels in BV2 cells, and treatment of N-acetyl-l-cysteine, a ROS scavenge reagent, largely inhibited the ROS levels and IFN-β levels (Supplementary Figures S4A and B). Consistently, upregulation of ROS levels in BV2 cells by adding Rotenone increased poly(I:C)-induced IFN-β levels (Supplementary Figure S4C). Next, we found that poly(I:C) treatment increased ROS levels were MAVS-dependent (Supplementary Figure S4D). ROS scavenge largely reduced the level of phosphorylated IRF3 induced by poly(I:C) treatment (Supplementary Figure S4E); however, knockdown of LC3 still increased the phosphorylated IRF3 levels, suggesting that ROS was involved in the regulatory effect of autophagy on MAVS signaling in microglia. Together, these results indicated that autophagy inhibits MAVS signaling activity through regulating the stability of MAVS via the direct binding of LC3 to MAVS in microglial cells.

Autophagy regulates MAVS protein level through the direct binding of LC3 to MAVS. (a) Immunoblotting analysis of MAVS, Atg5 and GAPDH in primary microglia cells from Atg5flox/flox mice and Atg5flox/floxCD11b-Cre mice. (b) Sequence alignment of the LIR motif of MAVS, NBR, Atg32, p62 and FUNDC1. (c) Flag-tagged fully length of MAVS or the CARD domain deleted form was transfected into HEK293T cells. Cell lysates were immunoprecipitated with anti-GFP affinity gel and then blotted for Flag-MAVS. (d) HEK293T cells were transfected with Flag-tagged wild-type MAVS, mutant Y9F, I12A, and Y9FI12A together with GFP-LC3. Cell lysates were immunoprecipitated with anti-Flag M2 affinity gel and then blotted for GFP-LC3. (e) A recombinant GST-LC3 protein was incubated with lysates from wild-type MAVS and mutants in cells then immunoprecipitated with anti-GST antibody-coated protein G gel and then blotted for Flag. (f) BV2 cells were transfected with Flag empty vector, Flag-tagged MAVS or mutant MAVS Y9F, respectively. Forty-eight hours later, cells were lysed and immunobloted of LC3, Flag and GAPDH. (g) HEK293T cells were transfected with Flag-tagged wild-type MAVS or mutant Y9F. Then, cells treated with cycloheximide to inhibit protein synthesis and cell lysates were immunoblotted with Flag or GAPDH. (h) Quantitative expressions of MAVS and mutant Y9F in HEK293T cells. Experiments were carried out for three independent times
c-Abl regulates the interaction of MAVS with LC3 and is required for its signaling activation
Next, we asked how the interaction between MAVS and LC3 is regulated in response to RLR signaling activation. One previous study showed that c-Abl kinase was involved in the activation of MAVS signaling in MCF7 cells;37 however, the precise regulatory mechanism is largely unknown, especially the loss of in vivo evidences. Here as shown in Figure 4a, we found that poly(I:C) treatment increased the binding of MAVS to LC3, suggesting that this interaction was involved the activation of MAVS signaling. Furthermore, we observed that pretreatment of STI571 (a c-Abl-family kinase inhibitor) abolished the interaction between MAVS and LC3. Consistently, induction of c-Abl largely increased the binding of MAVS to LC3 (Figure 4b), suggesting that c-Abl regulates the interaction between MAVS and LC3.

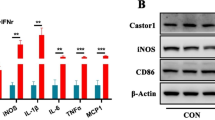
c-Abl regulates the interaction of MAVS with LC3 and is required for its signaling activation. (a) HEK293T cells were transfected with flag-tagged wild-type MAVS together with GFP-LC3 and then treated with transfected poly(I:C) or STI571 as indicated. Cell lysates were immunoprecipitated with anti-Flag M2 affinity gel and then blotted for GFP-LC3. (b) HEK293T cells were transfected with flag-tagged wild-type MAVS and GFP-LC3 together with or without Myc-c-Abl as indicated. Cell lysates were immunoprecipitated with anti-Flag M2 affinity gel and then blotted for GFP-LC3. (c) Immunoblotting analysis of c-Abl, MAVS and GAPDH in primary microglial cells treated with or without STI571 (10 μM for 6 h). (d) Immunoblotting analysis of c-Abl and GAPDH in primary microglial cells from c-Ablflox/flox and c-Abl flox/flox Lyz2-Cre mice. (e) Immunoblot analysis of p-IRF3, IRF3, iNOS and GAPDH in primary microglial cells from c-Ablflox/flox and c-Abl flox/floxLyz2-Cre mice treated with SeV for 24 h. (f) Expression of IFN-β in primary microglial cells from c-Ablflox/flox and c-Abl flox/floxLyz2-Cre mice treated with SeV for 12 h. *P<0.05. (g) ELISA analysis of of IFN-β in the supernatant in primary microglial cells from c-Ablflox/flox and c-Abl flox/floxLyz2-Cre mice treated with SeV for 24 h. *P<0.05. (h–j) Expression of TNF-α, IL-6 and INOS in primary microglial cells from c-Ablflox/flox and c-Abl flox/floxLyz2-Cre mice treated with SeV for 12 h. *P<0.05. Error bars are mean±S.E.M. Experiments were carried out in triplicate with three independent times
As the binding of MAVS to LC3 is important for its stability, we then assayed the protein levels of MAVS under c-Abl deficiency condition. We found pretreatment of STI571 in BV2 cells increased MAVS levels (Figure 4c). To further conform this, we generated a mouse that had the deletion of c-Abl gene in microglia by crossing c-Abl floxed mice with Lyz2-Cre mice.38 The deletion of c-Abl in microglial cells was confirmed in isolated primary microglial cells from c-Ablflox/flox and c-Ablflox/floxLyz2-Cre mice as shown in Figure 4d, and we found that microglial deletion of c-Abl increased MAVS protein levels. To investigate the role of c-Abl on MAVS activity, we treated BV2 cells with STI571 before poly(I:C) stimulation. Surprisingly, as shown in Supplementary Figures S5A and B, we found that c-Abl inhibition significantly reduced the levels of IFN-β and phosphorylated IRF3. Furthermore, knockdown of c-Abl by siRNA also resulted in a decrease of IFN-β mRNA level and p-IRF3 protein level (Supplementary Figures S5C and D), suggesting that c-Abl upregulates the activity of MAVS in microglial cells. Using primary c-Abl deletion microglial cells, we found that c-Abl deletion significantly decreased the expression levels of p-IRF3 and IFN-β upon SeV stimulation (Figures 4e–g). In addition, RT-PCR analysis showed that many downstream cytokines of MAVS were also decreased in c-Abl deleted microglial cells, including TNF-α, IL-6 and iNOS (Figures 4h–j). Taken together, we revealed that c-Abl kinase is required for MAVS signaling activation in microglial cells.
c-Abl interacts with MAVS and phosphorylates MAVS
We next investigated how c-Abl regulates the binding of MAVS with LC3 and how c-Abl regulates the activity of MAVS signaling in microglial cells. First, we found that c-Abl was phosphorylated upon SeV infection and pretreatment of STI571 totally block this increase. Interestingly, MAVS was found to be phosphorylated upon SeV infection, and pretreatment of STI571 also abolished the phosphorylated tyrosine of MAVS, indicating that c-Abl might be involved in the tyrosine phosphorylation of MAVS (Figures 5a and b). Second, we found that c-Abl interacted with MAVS in BV2 cells (Figure 5c). To further map the binding domain of MAVS, we generated two truncated mutants, CARD domain deletion mutant and TM domain deletion mutant (Figure 5d), and examined their interaction with c-Abl. As shown in Figure 5e, deletion of CARD domain of MAVS markedly impaired the binding of MAVS to c-Abl, suggesting that the CARD domain of MAVS is required for the binding. Last, the phosphorylation experiments showed that c-Abl indeed phosphorylated the CARD domain of MAVS (Figure 5f). After analyzing the sequences of MAVS among species (Supplementary Figure S6A), we generated a series of tyrosine mutations of MAVS, including Y9F, Y11F, Y30F and Y71F. When comparing with wild-type MAVS and Y11F mutation, we found the phosphorylation levels in the Y9F, Y30F and Y71F mutant forms were greatly decreased (Figure 5g). Taking into account the requirement of MAVS Y9 for the binding to LC3 (Figures 3d and e), these results indicated that c-Abl-mediated tyrosine phosphorylation for MAVS is critical for the interaction of MAVS with LC3 in microglial cells.

c-Abl interacts with MAVS and phosphorylates MAVS. (a) BV2 cells were infected by SeV with or without pretreatment of STI571 (10 μM for 3 h). Cell lysates were immunoprecipitated with anti-phosphotyrosine antibody-coated gel and then blotted for c-Abl and MAVS. (b) Quantitative data of phosphotyrosine levels of c-Abl and MAVS. *P<0.05. (c) Cell lysates of BV2 cells were immunoprecipitated with anti-c-Abl antibody-coated gel and then blotted for c-Abl and MAVS. (d) Structured region of the MAVS. (e) HEK293T cells were transfected with flag-tagged wild-type MAVS, truncated dCARD and dTM domain of MAVS together with Myc-c-Abl. Cell lysates were immunoprecipitated with anti-Flag M2 affinity gel and then blotted for Myc-c-Abl. (f) HEK293T cells were transfected with flag-tagged wild-type MAVS and truncated dCARD of MAVS together with Myc-c-Abl. Cell lysates were immunoprecipitated with anti-Flag M2 affinity gel and then blotted for phosphotyrosine. (g) HEK293T cells were transfected with flag-tagged wild-type MAVS and mutants as indicated together with Myc-c-Abl or kinase dead form Myc-c-Abl K290R. Cell lysates were immunoprecipitated with anti-Flag M2 affinity gel and then blotted for phosphotyrosine. (h and i) HEK293T cells were transfected with wild-type MAVS and mutants as indicated plus luciferase reporter plasmid for IFN-β activation. Cells were lysed 24 h after transfection for luciferase assays. *P<0.05. (j) Expression of IFN-β in the primary microglial cells of wild-type MAVS, MAVS−/− or MAVS−/− transfected with MAVS and mutant 4YF as indicated. All groups treated with transfected poly(I:C) (1 μg/ml) for 12 h. *P<0.05. (k) HEK293T cells were transfected with wild-type MAVS and mutants as indicated together with or without 3-methyladenine (3-MA) treatment (5 mM for 12 h), the luciferase activity of IFN-β was tested as above. *P<0.05; NS, no significance. Error bars are mean±S.E.M. Experiments were carried out in triplicate with three independent times
We next wanted to know how this tyrosine phosphorylation of MAVS impairs its activity. As shown in Figure 5h, the mutant forms (Y9F, Y30F and Y71F) significantly inhibited the IFN-β promoter activation, with the lowest level in the Y9F mutant. To further study the function of these phosphorylation sites, we generated MAVS 4YF (Y9F/Y11F/Y30F/Y71F). As shown in Figure 5i, MAVS 4YF further decreased the IFN-β promoter activation when compared with MAVS Y9F, suggesting that besides Y9, other tyrosine sites also contributed to the activation of MAVS. Moreover, rescue experiments showed that the ectopically expressed MAVS 4YF failed to activate IFN-β expression (Figure 5j). To investigate whether the status of tyrosine phosphorylation of MAVS affects the connection of MAVS with autophagy, either wild-type MAVS or 4YF mutant MAVS was transfected into HEK293T cells. As shown in Figure 5k, 3-methyladenine (an autophagy inhibitor) treatment only increased wild-type MAVS-induced IFN-β promoter activation, with no effect on mutant 4YF, suggesting that the phosphorylation of the tyrosine sites is required for autophagy inhibition-induced MAVS activation. Taken together, we revealed that autophagy regulated MAVS signaling activation in a c-Abl-mediated phosphorylation manner.
MAVS deficiency attenuates MPTP-induced dopaminergic neuron loss and microglial activation
To investigate the role of MAVS signaling in vivo, a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced mouse model of PD was performed. Wild-type and MAVS−/− mice were treated with MPTP (four intraperitoneal injections of 20 mg/kg at 2 h intervals; Figure 6a). We found that MAVS−/− mice hold more tyrosine hydroxylase (TH)-positive cells in the SN, suggesting that MAVS signaling takes a protective role against MPTP-induced loss of nigral dopaminergic neurons (Figures 6b and c). To examine the role of microglial activation in this process, we stained microglia with ionized calcium-binding adaptor molecule 1 (Iba1), and counted them via stereological analysis. As shown in Figures 6d and e, in the saline-treated group, no differences were observed between wild-type and MAVS−/− mice. However, following MPTP treatment, MAVS−/−mice showed a significant decrease in the number of nigral microglial cells. Furthermore, this difference was associated with a decrease in IFN-β, TNF-α and IL-1β levels in the brain (Figures 6f–h), suggesting that dysregulation of MAVS signaling-driven inflammation contributes to the development of MPTP-induced the activation of microglia. Interestingly, we found that there was a large increase in MAVS expression in αlpha-synuclein transgenic mice and PD patients (Supplementary Figures S7A–D), arguing that this signaling might be involved in the progression of PD.

Deficiency of MAVS attenuates MPTP-induced dopaminergic neuron loss and microglial activation. (a) Model of MPTP-induced dopaminergic neurotoxicity in MAVS−/− and wild-type mice. (b) Immunohistochemical staining for tyrosine hydroxylase (TH) in the SN in wild-type and MAVS−/− mice 7 days after either saline or MPTP treatment. (c) Stereological analysis of TH-positive cells in the SN in wild-type and MAVS−/− mice administrated saline or MPTP (six mice per group). *P<0.05. (d) Immunohistochemical staining for Iba1 in the SN in wild-type and MAVS−/− mice 7 days after either saline or MPTP treatment. (e) Stereological analysis of Iba1-positive cells in the SN in wild-type mice and MAVS−/− mice administrated saline or MPTP (six mice per group). *P<0.05. (f–h) Expression of IFN-β, TNF-α and IL-1β in the striatum in wild-type or MAVS−/− mice administrated saline or MPTP. (i) Schematic model of molecular mechanisms underlying MAVS-dependent microglial activation in the nervous system
As c-Abl is required for the activation of MAVS signaling in microglial cells, we then investigated whether microglial c-Abl knockout also prevent MPTP-induced neuroinflammation and loss of neurons. We found that microglial deletion of c-Abl rendered mice more resistant to MPTP-induced cell loss of nigral dopaminergic neurons (Supplementary Figure S8A and B) and microglial activation (Supplementary Figure S8C and D) within the SN. Importantly, these differences were also associated with a decrease in the levels of IFN-β, TNF-α and IL-6 in the brain tissues (Supplementary Figure S8E), suggesting that the deletion of microglial c-Abl contributes to the development of MPTP-induced microglial activation.
Altogether, these results show that autophagy has a critical role in regulating the activity of MAVS via the binding of LC3 to MAVS, and that c-Abl phosphorylates MAVS and regulates its activity in microglial cells, which contributes to MPTP-induced microglial activation and neurotoxicity in vivo (Figure 6i).
Discussion
The RLR pathway is one of the important innate immunity pathways that is involved in various human diseases; however, its pathophysiological role in the brain remains unclear. Here we found that autophagy regulates MAVS-mediated microglial activation in a phosphorylation-dependent manner: (1) MAVS signaling-driven inflammation contributes to the development of MPTP-induced dopaminergic neurotoxicity; (2) autophagy negatively regulates the activity of MAVS through direct binding of LC3 to the LIR motif of MAVS; and (3) c-Abl regulates the interaction of MAVS with LC3 and is required for its signaling activation.
Evidence is accumulating for the involvement of immune system dysfunction in the development of PD. Here we provide the evidence that MAVS signaling contributed to the microglial activation and MPTP-induced neurotoxicity in vivo. In the absence of MAVS, poly(I:C) and MPTP-induced inflammation were both significantly decreased compared with control mice. Interestingly, the increase in MAVS levels occurred in both αlpha-synuclein transgenic mice and PD patients, indicating that this pathway might be a potential clinical target for PD. Thus, our results suggested that MAVS signaling has important role in the microglia-driven inflammatory brain diseases.
Autophagy is a basic cell biological process, which is upregulated under various stress conditions, including inflammatory responses. The deficiency of autophagy may result in necrotic cell death, a process that can initiate an inflammation reaction. It has been reported that genetic deletion of Atg5, Atg7 or Atg16L1 results in the accumulation of dysfunctional mitochondria and ROS, which have been reported to be essential for the activations of RLR and NLRP3.39, 40, 41 Atg5–Atg12 has also been shown to negatively regulate RLR signaling by direct association with RIG-I and MAVS.30 Tal MC et al.23 reported that autophagy deficiency results in higher levels of MAVS and ROS, leading to amplification of the RLR pathway. However, it remains unclear how this regulation occurs in microglia and whether this signaling contributes to the neurodegenerative disease. Here our results showed that autophagy played critical roles in the activation of MAVS signaling in microglial cells. Autophagy deficiency, either knockdown of LC3 or genetic deletion of Atg5, aggravated the activities of MAVS in microglial cells. Moreover, the finding of the interaction between MAVS and LC3 provides a direct regulatory mechanism between MAVS-induced inflammation and autophagy. It has been reported that autophagy dysfunction increases ROS level, which is involved in the activation of MAVS signaling.23, 42 Here we clearly showed that ROS production is MAVS-dependent and ROS in turn upregulates MAVS-mediated inflammation activation in microglial cells. Taken together, we argue that there might be a feedback regulation loop among MAVS inflammation–autophagy–ROS production in microglial cells upon inflammatory stress.
Phosphorylations of RLR pathway proteins have been reported to be essential for the subsequent innate immune response.43, 44 Here we found that tyrosine kinase c-Abl interacted with and phosphorylated MAVS in microglial cells. Interestingly, one important phosphorylation site, MAVS Y9, is contained in its binding motif with LC3. Inhibition of the activity of c-Abl in microglial cells abolished the interaction between MAVS and LC3, indicating that c-Abl contributes to the inhibitory effect of autophagy on MAVS activation. Recently, MAVS was reported to regulate the localization and inflammasome activation of NLRP3,5 a well-studied inflammasome involved in the development of PD and AD.45, 46 In our study presented here, we found that deletion of MAVS impaired the level of IL-1β in the brain or primary microglial cells (Figure 6h and Supplementary Figure S1C). Whether c-Abl impairs the localization and inflammasome activation of NLRP3, and whether the phosphorylation sites of MAVS are involved in this process are important questions remain to be elucidated. In addition, the role of c-Abl in neurodegenerative disease has been addressed previously.47, 48, 49, 50, 51 We also reported that c-Abl phosphorylates MST1 (mammalian STE20-like kinase 1) and increased MST1-dependent neuronal cell death.52 More recently, we showed that c-Abl/p38 signaling has an important role in the development of PD.53 However, those studies mainly focused on neuronal cells, with few studies on microglial cells. Here we found that c-Abl was required for the activation of MAVS signaling in microglial cells. Furthermore, microglial deletion of c-Abl also protected MPTP-induced neuroinflammation and loss of dopaminergic neurons in vivo, indicating that inhibition of microglial c-Abl also contributes to the neuroprotective effect. It is well-established that MAVS signaling is important for antiviral immunity and autoimmunity.6, 7, 8, 54 It is crucial to further investigate whether MAVS activation via autophagy and phosphorylation is involved in this process.
In summary, our results revealed that the MAVS signaling is involved in the development of microglial activation-induced neurotoxicity. Furthermore, we showed that LC3/autophagy interaction and tyrosine phosphorylation regulate MAVS-mediated microglial activation, suggesting that c-Abl-MAVS signaling is a potential target for the treatment of microglia-driven inflammatory brain diseases.
Materials and Methods
Mice
MAVS−/− mice2 were described previously and kindly provided by Dr Baidong Hou (Institute of Biophysics, Chinese Academy of Science, China). The mice with the deletion of Atg5 in microglia were generated by crossing the Atg5 locus floxed mice (provided by Professor Noboru Mizushima, The Graduate School of Medicine, The University of Tokyo, Tokyo, Japan)25 with CD11b-Cre mice (provided by Dr Lize Xiong, The Fourth Military Medical University, Xian, China). The mice with the deletion of c-Abl in microglia were generated by crossing the c-Abl locus floxed mice (provided by Dr Yong Cang, Zhejiang University, Hangzhou, China) with Lyz2-Cre mice (provided by Model Animal Research Center of Nanjing University, Nanjing, China).
All animals were maintained in the Animal Care Facility at our institute. All animal experiments were approved by the Institutional Animal Care and Use Committee at the Institute of Biophysics, Chinese Academy of Sciences.
MPTP-induced neuroninflammation in vivo
Adult male mice aged 10 weeks were intraperitoneally injected with 20 mg/kg MPTP four times at 2 h intervals. At day 7, animals were killed, and their brains were dissected and processed for immunohistochemistry, RT-PCR or immunoblotting analysis.
Stereotaxic injection of poly(I:C)/LyoVec into SN
All procedures were performed as previously described.55 In brief, adult (10 weeks of age) male MAVS+/+ mice and MAVS−/− mice were anesthetized with pentrobarbital (60 mg/kg). A measure of 1 μl (1 μg/μl) of poly(I:C)/LyoVec (complexed with transfection reagent, InvivoGen, San Diego, CA, USA) was applied stereotactically into the SN at a rate of 0.2 μl/min.
Stereological assessment
All procedures were performed as previously described.55 In brief, 40 μm coronal sections were cut throughout the entire brain, and every fourth section was used for analysis by Stereo Investigator software (MicroBrightfield, Williston, VT, USA).
Cell preparation and stimulation
Microglial cells were prepared from neonatal mice (age 1–3 days) as described previously.56 For inducing immune activation, 5 × 105 cells were plated into 12-well plates and cultured overnight, and medium was exchanged with opti-MEM before cell stimulation.
Immunohistochemistry
All procedures were performed as previously described.55 In brief, mice were perfused with saline and brains were fixed with 4% paraformaldehyde (w/v) for 1 week. Fixed mouse brains were cryoprotected in 30% sucrose. Coronal sections were cut throughout the whole brain and sections were incubated with rabbit polyclonal anti-TH (1:1000; Pel-Freez Biologicals, Rogers, AR) or Iba1 (1:500; WAKO, Richmond, VA, USA) and visualized with biotinylated goat anti-rabbit IgG, followed by streptavidin-conjugated horseradish peroxidase (Vectastain ABC kit, Zhongshanjinqiao, Beijing, China). Positive immunostaining was visualized with 3,30-diaminobenzidine after reaction with hydrogen peroxide (DAB kit, Zhongshanjinqiao). Stained sections were mounted onto slides and analyzed by Stereo Investigator software (MicroBrightfield).
Immunofluorescence
BV2 cells were washed with PBS three times and then blocked with 5% goat serum in PBS containing 0.2% Triton X-100. Cells were then incubated with the primary antibody overnight at 4 °C. After washing with PBS three times, Alexa Fluor 488-conjugated secondary antibody (Invitrogen, Carlsbad, CA, USA) was added and incubated 1 h at room temperature. Finally, nuclear morphology was visualized using Hoechst 33258 (Sigma, St. Louis, MO, USA).
siRNA or shRNA-mediated gene silences in microglial cells
For siRNA transfection, microglial cells were plated onto 12-well plates, and were then transfected with 50 nM siRNA using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions (siRNA sequences in Supplementary Table 1). For selecting stable targeted gene knockdown BV2 cell line, pLKO.1 vector containing shRNA (sequences in Supplementary Table 1) was co-transfected with VSV-G and pCMV-dR8.12 into HEK293T cells and virus was collected at 36 and 72 h after transfection. Stable knockdown cell lines were established by infecting corresponding lenti-virus, before being selected in complete medium containing 5.0 μg/ml puromysin as described previously.57
Immunoprecipitation and immunoblot analysis
Coimmunoprecipitation and immunoblotting were performed as described previously.58 In brief, cells were collected and lysed with IP buffer containing 0.5% NP-40, 150 mM NaCl, 50 mM Tris-HCl, PH 8, 50 mM NaF, 2 mM EDTA, plus a protease inhibitor mixture (Roche Applied Science, Penzberg, Upper Bavaria, Germany). The equivalent amount of cellular extract were subjected to antibody-coated Protein G Sepharose (GE Healthcare Life, Pittsburgh, PA, USA, 1 μg antibody and 25 μl Protein G Sepharose for each sample) and incubated overnight. Immunoprecipitates were washed four times in lysis buffer and eluted by boiling in Laemmli sample buffer (Bio-Rad, Berkeley, CA, USA). Samples were fractionated by SDS-PAGE and transferred to nitrocellulose. Immunoblots were probed with primary antibodies indicated and visualized by ECL (Thermo, Waltham, MA, USA). Rabbit polyclonal anti-p-IRF3 (Ser396, Cell Signaling, Cambridge, MA, USA, 1:1000), anti-IRF3 (FL-425, Santa Cruz, Santa Cruz, CA, USA, 1:1000), GAPDH (CW0100A, Cwbiotech, Beijing, China, 1:1000), mouse MAVS (#4983, Cell Signaling, 1:1000), human MAVS (ab25084, Abcam, Cambridge, MA, USA, 1:1000), rabbit polyclonal LC3 (#2775, Cell Signaling, 1:1000), c-Abl (2862, Cell Signaling, 1:1000), p-tyrosine (4G10, Cell Signaling; 1:1000), p62 (PM045, MBL; Woburn, MA, USA, 1:1000) and Atg5 (M153-3, MBL, 1:1000).
Transfection of BV2 cells by electroporation
Cells were resuspended in 100 μl electroporation buffer (Lonza, Allendale, NJ, USA) at a concentration of 4 × 107 cells per ml. An amount of 5 μg of plasmid DNA was transferred to Gene Pulser cuvettes and transfected into cells (Bio-Rad). Transfected cells were plated into 6 cm plate in DMEM medium containing 10% FBS and utilized 48 h later.
ELISA
Mouse IFN-β, TNF-α and IL-6 were measured in the cell culture supernatants according to the manufacturer’s instructions (Biolegend, San Diego, CA, USA).
ROS detection
Cellular ROS were measured using a commercial kit (ROS-Glo H2O2 assay, Promega, Madison, WI, USA) according to the manufacturer’s instructions.
Quantitative RT-PCR
Total RNA was extracted from cells or brain tissues using Trizol reagent (Invitrogen). cDNA synthesis was performed using a one step first strain cDNA synthesis kit (Transgen, Beijing, China). Quantitative real-time PCR was performed with primers for IFN-β, TNF-α, IL-6, iNOS and GAPDH (Supplementary Table 2). RT-PCR reactions were performed using 2 × SYBR Green PCR master mix (Transgen) and an Agilent Mx3005P RT-PCR system (Santa Clara, CA, USA). The mRNA levels were normalized to GAPDH expression levels.
Luciferase assay
Luciferase activity was measured with the Dual-Luciferase reporter assay system according to the manufacturer’s protocol (Promega).
Statistical analysis
All values are expressed as the mean±S.E.M. Statistical analysis was performed with the t-test for two groups, or one-way ANOVA for multiple groups (Graphpad Software, San Diego, CA, USA). A P-value<0.05 was considered as significant.
Abbreviations
- MAVS:
-
mitochondrial antiviral signaling
- RIG-I:
-
retinoic acid-inducible gene-I
- MDA-5:
-
melanoma differentiation-associated gene 5
- IL-1β:
-
interleukin-1 beta
- TNF-α:
-
tumor necrosis factor alpha
- PD:
-
Parkinson’s disease
- AD:
-
Alzheimer’s disease
- SeV:
-
Sendai virus
- ROS:
-
reactive oxygen species
- MPTP:
-
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- TH:
-
tyrosine hydroxylase
- Iba1:
-
ionized calcium-binding adaptor molecule 1
- MST1:
-
mammalian STE20-like kinase 1
References
Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 2005; 6: 981–988.
Seth RB, Sun L, Ea CK, Chen ZJ . Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005; 122: 669–682.
Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB . VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell 2005; 19: 727–740.
Rehwinkel J, Reis e Sousa C . RIGorous detection: exposing virus through RNA sensing. Science 2010; 327: 284–286.
Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN . The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 2013; 153: 348–361.
Nejentsev S, Walker N, Riches D, Egholm M, Todd JA . Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science 2009; 324: 387–389.
Funabiki M, Kato H, Miyachi Y, Toki H, Motegi H, Inoue M et al. Autoimmune disorders associated with gain of function of the intracellular sensor MDA5. Immunity 2014; 40: 199–212.
Wang Y, Zhang HX, Sun YP, Liu ZX, Liu XS, Wang L et al. Rig-I−/− mice develop colitis associated with downregulation of G alpha i2. Cell Res 2007; 17: 858–868.
Bryson KJ, Ligertwood Y, Quigg-Nicol M, Dutia BM, Manson J, Nash AA . Influenza A virus infection contributes to Parkinson's disease by driving pathological CD4(+) T cell responses in the brain. Immunology 2013; 140: 127–127.
Harris MA, Tsui JK, Marion SA, Shen H, Teschke K . Association of Parkinson's disease with infections and occupational exposure to possible vectors. Mov Disord 2012; 27: 1111–1117.
Jang H, Boltz D, Sturm-Ramirez K, Shepherd KR, Jiao Y, Webster R et al. Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc Natl Acad Sci USA 2009; 106: 14063–14068.
Rohn TT, Catlin LW . Immunolocalization of influenza A virus and markers of inflammation in the human Parkinson's disease brain. Plos One 2011; 6: 5.
Toovey S, Jick SS, Meier CR . Parkinson's disease or Parkinson symptoms following seasonal influenza. Influenza Other Respir Viruses 2011; 5: 328–333.
Deleidi M, Hallett PJ, Koprich JB, Chung CY, Isacson O . The toll-like receptor-3 agonist polyinosinic:polycytidylic acid triggers nigrostriatal dopaminergic degeneration. J Neurosci 2010; 30: 16091–16101.
Field R, Campion S, Warren C, Murray C, Cunningham C . Systemic challenge with the TLR3 agonist poly I:C induces amplified IFN alpha/beta and IL-1 beta responses in the diseased brain and exacerbates chronic neurodegeneration. Brain Behav Immun 2010; 24: 996–1007.
Tanaka S, Ishii A, Ohtaki H, Shioda S, Yoshida T, Numazawa S . Activation of microglia induces symptoms of Parkinson's disease in wild-type, but not in IL-1 knockout mice. J Neuroinflammation 2013; 10: 143.
Borrajo A, Rodriguez-Perez AI, Diaz-Ruiz C, Guerra MJ, Labandeira-Garcia JL . Microglial TNF-alpha mediates enhancement of dopaminergic degeneration by brain angiotensin. Glia 2014; 62: 145–157.
McCoy MK, Ruhn KA, Martinez TN, McAlpine FE, Blesch A, Tansey MG . Intranigral lentiviral delivery of dominant-negative TNF attenuates neurodegeneration and behavioral deficits in hemiparkinsonian rats. Mol Ther 2008; 16: 1572–1579.
Banati RB, Gehrmann J, Czech C, Monning U, Jones LL, Konig G et al. Early and rapid de novo synthesis of Alzheimer beta A4-amyloid precursor protein (APP) in activated microglia. Glia 1993; 9: 199–210.
Noelker C, Morel L, Lescot T, Osterloh A, Alvarez-Fischer D, Breloer M et al. Toll like receptor 4 mediates cell death in a mouse MPTP model of Parkinson disease. Sci Rep 2013; 3: 1393.
Schwab C, Steele JC, McGeer PL . Neurofibrillary tangles of Guam parkinson-dementia are associated with reactive microglia and complement proteins. Brain Res 1996; 707: 196–205.
Wang XJ, Yan ZQ, Lu GQ, Stuart S, Chen SD . Parkinson disease IgG and C5a-induced synergistic dopaminergic neurotoxicity: role of microglia. Neurochem Int 2007; 50: 39–50.
Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A . Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci USA 2009; 106: 2770–2775.
Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441: 880–884.
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441: 885–889.
Friedman LG, Lachenmayer ML, Wang J, He L, Poulose SM, Komatsu M et al. Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of alpha-synuclein and LRRK2 in the brain. J Neurosci 2012; 32: 7585–7593.
Liu K, Shi N, Sun Y, Zhang T, Sun X . Therapeutic effects of rapamycin on MPTP-induced Parkinsonism in mice. Neurochem Res 2013; 38: 201–207.
Tambuyzer BR, Ponsaerts P, Nouwen EJ . Microglia: gatekeepers of central nervous system immunology. J Leukocyte Biol 2009; 85: 352–370.
Lei Y, Wen H, Yu Y, Taxman DJ, Zhang L, Widman DG et al. The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity 2012; 36: 933–946.
Jounai N, Takeshita F, Kobiyama K, Sawano A, Miyawaki A, Xin KQ et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci USA 2007; 104: 14050–14055.
Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282: 24131–24145.
Noda NN, Ohsumi Y, Inagaki F . Atg8-family interacting motif crucial for selective autophagy. FEBS Lett 2010; 584: 1379–1385.
Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 2012; 14: 177–185.
Drechsel DA, Liang LP, Ryan KR, Patel M . Mitochondrial mechanisms of ROS production and oxidative stress by redox cycling herbicides implicated in Parkinson's disease. Free Radical Bio Med 2007; 43: S185–S185.
Guo SH, Yan JQ, Yang TB, Yang XQ, Bezard E, Zhao BL . Protective effects of green tea polyphenols in the 6-OHDA rat model of Parkinson's disease through inhibition of ROS-NO pathway. Biol Psychiat 2007; 62: 1353–1362.
Kim HG, Ju MS, Kim DH, Hong J, Cho SH, Cho KH et al. Protective effects of chunghyuldan against ROS-mediated neuronal cell death in models of Parkinson's disease. Basic Clin Pharmacol Toxicol 2010; 107: 958–964.
Song T, Wei C, Zheng Z, Xu Y, Cheng X, Yuan Y et al. c-Abl tyrosine kinase interacts with MAVS and regulates innate immune response. FEBS Lett 2009; 584: 33–38.
Cho MH, Cho K, Kang HJ, Jeon EY, Kim HS, Kwon HJ et al. Autophagy in microglia degrades extracellular beta-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy 2014; 10: 1761–1775.
Levine B, Mizushima N, Virgin HW . Autophagy in immunity and inflammation. Nature 2011; 469: 323–335.
Virgin HW, Levine B . Autophagy genes in immunity. Nat Immunol 2009; 10: 461–470.
Zhou R, Yazdi AS, Menu P, Tschopp J . A role for mitochondria in NLRP3 inflammasome activation. Nature 2011; 469: 221–225.
Cheng J, Liao Y, Zhou L, Peng S, Chen H, Yuan Z . Amplified RLR signaling activation through an interferon-stimulated gene-endoplasmic reticulum stress-mitochondrial calcium uniporter protein loop. Sci Rep 2016; 6: 20158.
Nistal-Villan E, Gack MU, Martinez-Delgado G, Maharaj NP, Inn KS, Yang H et al. Negative role of RIG-I serine 8 phosphorylation in the regulation of interferon-beta production. J Biol Chem 2010; 285: 20252–20261.
Liu S, Cai X, Wu J, Cong Q, Chen X, Li T et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015.
Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z et al. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 2015; 160: 62–73.
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A et al. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 2012; 493: 674–678.
Schlatterer SD, Acker CM, Davies P . c-Abl in neurodegenerative disease. J Mol Neurosci 2011; 45: 445–452.
Gonzalez-Zuniga M, Contreras PS, Estrada LD, Chamorro D, Villagra A, Zanlungo S et al. c-Abl stabilizes HDAC2 levels by tyrosine phosphorylation repressing neuronal gene expression in Alzheimer's disease. Mol Cell 2014; 56: 163–173.
Imam SZ, Trickler W, Kimura S, Binienda ZK, Paule MG, Slikker W Jr et al. Neuroprotective efficacy of a new brain-penetrating C-Abl inhibitor in a murine Parkinson's disease model. PLoS One 2013; 8: e65129.
Imam SZ, Zhou Q, Yamamoto A, Valente AJ, Ali SF, Bains M et al. Novel regulation of parkin function through c-Abl-mediated tyrosine phosphorylation: implications for Parkinson's disease. J Neurosci 2011; 31: 157–163.
Karuppagounder SS, Brahmachari S, Lee Y, Dawson VL, Dawson TM, Ko HS . The c-Abl inhibitor, nilotinib, protects dopaminergic neurons in a preclinical animal model of Parkinson's disease. Sci Rep 2014; 4: 4874.
Xiao L, Chen D, Hu P, Wu J, Liu W, Zhao Y et al. The c-Abl-MST1 signaling pathway mediates oxidative stress-induced neuronal cell death. J Neurosci 2011; 31: 9611–9619.
Wu R, Chen H, Ma J, He Q, Huang Q, Liu Q et al. c-Abl-p38alpha signaling plays an important role in MPTP-induced neuronal death. Cell Death Differ 2016; 23: 542–552.
Mukherjee P, Woods TA, Moore RA, Peterson KE . Activation of the innate signaling molecule MAVS by bunyavirus infection upregulates the adaptor protein SARM1, leading to neuronal death. Immunity 2013; 38: 705–716.
Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O et al. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson's disease. Cell 2011; 144: 689–702.
Saura J, Tusell JM, Serratosa J . High-yield isolation of murine microglia by mild trypsinization. Glia 2003; 44: 183–189.
Mao B, Hu F, Cheng J, Wang P, Xu M, Yuan F et al. SIRT1 regulates YAP2-mediated cell proliferation and chemoresistance in hepatocellular carcinoma. Oncogene 2014; 33: 1468–1474.
Yuan Z, Kim D, Shu S, Wu J, Guo J, Xiao L et al. Phosphoinositide 3-kinase/Akt inhibits MST1-mediated pro-apoptotic signaling through phosphorylation of threonine 120. J Biol Chem 2010; 285: 3815–3824.
Acknowledgements
We sincerely thank Dr Lize Xiong (The Fourth Military Medical University, Xian, China) for providing us the CD11b-Cre mice, Dr Yong Cang (Zhejiang University, Zhejing, China) for providing the c-Abl flox/flox mice and Dr Zhuohua Zhang (Sanford-Burnham Medical Research Institute, La Jolla, CA, USA) for providing the α-synuclein transgenic mice. We also thank Dr Quan Chen (Institute of Zoology, Chinese Academy of Sciences) for providing PD patients’ samples (Purchased from Netherland Brain Bank) and Dr Shu-Ichi Matsuzawa (Sanford-Burnham Medical Research Institute, La Jolla, CA, USA) for helpful discussions. This work was supported by grants from the National Nature Science Foundation of China (grant nos 81125010, 81030025, 81400987 and 81230026) and the Beijing Nature Science Foundation (7132147).
Author contributions
JC designed and performed the experiments, analyzed the data and wrote the paper. YL, LX and RW performed some experiments and analyzed the data. SZ and HC provided the reagents and analyzed the data. BH, XZ and CL provided mice and advice. JC and ZY conceived the project and wrote the manuscript. Y X and ZY supervised the research.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Piacentini
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article

Cite this article
Cheng, J., Liao, Y., Xiao, L. et al. Autophagy regulates MAVS signaling activation in a phosphorylation-dependent manner in microglia. Cell Death Differ 24, 276–287 (2017). https://doi.org/10.1038/cdd.2016.121
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2016.121
- Springer Nature Limited