
Abstract
The c-Abl tyrosine kinase participates in a variety of cellular functions, including regulation of the actin cytoskeleton, regulation of the cell cycle, and the apoptotic/cell cycle arrest response to stress, and the Abl family of kinases has been shown to play a crucial role in development of the central nervous system. Recent studies have shown c-Abl activation in human Alzheimer’s and Parkinson’s diseases and c-Abl activation in mouse models and neuronal culture in response to amyloid beta fibrils and oxidative stress. Overexpression of active c-Abl in adult mouse neurons results in neurodegeneration and neuroinflammation. Based on this evidence, a potential role for c-Abl in the pathogenesis of neurodegenerative disease is discussed, and we attempt to place activation of c-Abl in context with other known contributors to neurodegenerative pathology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
c-Abl Function
The Abelson non-receptor tyrosine kinase (c-Abl, Abl1) gene was first identified as the mammalian homolog of the oncogenic gene product of the Abelson murine leukemia virus (Ozanne et al. 1982). Since its discovery, the c-Abl family of tyrosine kinases, including c-Abl (Abl, Abl1) and Abl-related gene (ARG, Abl2) (Kruh et al. 1990), has been shown to be highly conserved across species and has been implicated in a wide variety of cellular processes including regulation of the actin cytoskeleton (Van Etten et al. 1994; Woodring et al. 2003), regulation of the cell cycle (Welch and Wang 1993; Silberman et al. 2008), and apoptotic/cell cycle arrest response to stress (Huang et al. 1996, 1997; Kharbanda et al. 1997; Yuan et al. 1997; Kharbanda et al. 1998; Barila et al. 2003; Cao et al. 2003). The Abl family of kinases has been shown to play an important role in neuronal development and recent studies have shown that c-Abl, specifically, may be an important player in neurodegenerative diseases (Alvarez et al. 2004; Cancino et al. 2009; Jing et al. 2009; Ko et al. 2010; Imam et al. 2011; Schlatterer et al. 2011).
The function of c-Abl is dependent upon its subcellular localization. Cytoplasmic localization appears to be necessary for the transforming and cell survival functions of c-Abl (Van Etten et al. 1989; Wetzler et al. 1993). Nuclear localization of c-Abl typically occurs in response to stress or overexpression and results in growth inhibitory functions, including cell cycle arrest and apoptosis (Van Etten et al. 1989; Wen et al. 1996).
Cytoplasmic c-Abl can be activated during the G1-S phase transition of the cell cycle, when retinoblastoma (Rb) becomes phosphorylated and releases c-Abl from its inhibitory interaction (Welch and Wang 1993). Knockdown of c-Abl in NIH 3T3 cells resulted in a slowed growth rate, and c-Abl knockdown cells entered S phase from G1 earlier than controls, suggesting that c-Abl is important for G1/S checkpoint regulation and that knockdown dysregulates cell growth (Daniel et al. 1995).
Nuclear c-Abl is activated in response to genotoxic stress (Kharbanda et al. 1995). The ataxia–telangectasia mutant protein stimulates activation of c-Abl by genotoxic stress and may partially mediate G1 arrest in response to DNA damage (Shafman et al. 1997). The c-Abl kinase inhibits Rad51, preventing binding to DNA and double-stranded break repair (Yuan et al. 1998). Nuclear c-Abl suppresses growth in fibroblasts in a p53-dependent manner (Goga et al. 1995), and overexpression of wild-type c-Abl and resultant nuclear translocation resulted in slow growth, growth arrest at the G1-S transition, and ultimately cell death in NIH 3T3 cells (Sawyers 1993). c-Abl has been shown to bind p53 and increase p21 in response to DNA damage and decrease cdk2 activity, resulting in G1 arrest (Yuan et al. 1996). Knockout of c-Abl in MCF7 cells impairs apoptotic response to DNA damage, and transfection of these cells with wild-type but not kinase inactive c-Abl induces apoptosis as a result of DNA damage (Yuan et al. 1997). The c-Abl kinase has been shown to activate p73 and participate in apoptosis (Yuan et al. 1999). Interestingly, c-Abl is only stimulated by stress in cells during S phase (Liu et al. 1996).
c-Abl in Neuronal Development
The c-Abl family of kinases plays a role in multiple aspects of nervous system development. In vitro, c-Abl has been shown to localize to synapses in neurons and to regulate clustering of PSD95 postsynaptically, and the inhibition of c-Abl reduced the number of synapses present (de Arce et al. 2010). In mouse embryos, the Abl family of tyrosine kinases, c-Abl and Arg, localize to synaptosomes and growth cone particles (Koleske et al. 1998; Courtney et al. 2000). D-Abl, the Drosophila homolog of mammalian c-Abl, localizes to the CNS in late embryogenesis, and, specifically, to axons growing across the ventral midline (Gertler et al. 1989; Bennett and Hoffmann 1992). The NR2D subunit, expressed mainly during development (Dunah et al. 1996; Wenzel et al. 1996), of the NMDA receptor binds and inhibits the kinase activity of c-Abl (Glover et al. 2000).
Abl−/− Arg−/− mice show a delay in neural tube closure and collapse of the neuroepithelium and exhibit a delay in the appearance of MAP2 positive neurons, indicating that differentiation is inhibited in the absence of these kinases (Koleske et al. 1998). Actin networks in the neuroepitheilum are disrupted in Abl−/− Arg−/− mice, indicating a role for Abl family kinases in neurulation (Koleske et al. 1998).
Transfection with constitutively active c-Abl led to an increase in dendritic complexity in neurons in culture, and inhibition of c-Abl led to decreased dendrite length, decreased branch formation, disrupted dendrite/axon polarity, and an overall decrease in the number of both primary and secondary dendrites compared with controls, indicating a positive role for c-Abl in dendrogenesis (Zukerberg et al. 2000; Jones et al. 2004).
Maternal/zygotic D-Abl mutants have severe CNS defects during development, with a decrease in axons that cross the midline (Grevengoed et al. 2001). Axonal guidance/pathfinding in D-Abl mutant flies is extremely sensitive to mutations of other genes. Drosophila genetic screens indicate that several genes, including disabled (dab), fascilin1, failed axon connections (fax), trio, and prospero enhance the D-Abl mutant phenotype of impaired crossover and axonal outgrowth (Gertler et al. 1989, 1993; Elkins et al. 1990; Hill et al. 1995; Liebl et al. 2000) and overexpression of D-abl leads to increased inappropriate midline crossing (Bashaw et al. 2000).
These numerous studies, taken together, show that c-Abl plays a crucial role in neuronal development. Mutations in c-Abl lead to defects in neurulation, dendrogenesis, and axonal guidance, and aberrant c-Abl activity can cause devastating neurological phenotypes.
c-Abl Is Activated in Alzheimer’s Disease
While the activity of c-Abl is crucial for proper neuronal development, it appears that c-Abl remains relatively quiescent in healthy adult neurons, and there are few known functions of c-Abl in fully differentiated neurons. In recent years, it has been shown that activation of c-Abl in adult brain occurs in the context of human neurodegenerative disease (Jing et al. 2009; Ko et al. 2010; Tremblay et al. 2010; Imam et al. 2011). The role of c-Abl has been most widely studied in Alzheimer’s disease (AD), the most common of the neurodegenerative disorders. The Bowser group has shown that c-Abl phosphorylated at Y412, an indicator of activation, co-localizes granulovacuolar degeneration (GVD) in brains of human AD patients. Additionally, c-Abl phosphorylated at T735, a site necessary for interaction with the 14-3-3 protein and cytoplasmic localization in normal cells, co-localized with amyloid plaques, neurofibrillary tangles (NFTs), and GVD in the entorhinal cortex and hippocampus of AD patients (Jing et al. 2009). c-Abl pT735 staining in AD brain has also been observed in our own laboratory (Fig. 1).

c-Abl is present in AD lesions. c-Abl pT735 and tau pY394/pS396 (YP3) staining of human AD hippocampus. Each row represents a separate case
The c-Abl protein has been shown to phosphorylate tau at tyrosines 18, 197, 310, and 394, and tau pY394 has been shown to be present in NFTs in AD (Derkinderen et al. 2005; Tremblay et al. 2010). Amyloid β and oxidative stress activate c-Abl in neuronal culture (Alvarez et al. 2004), and intrahippocampal injection of Aβ fibrils leads to increased expression of c-Abl and a downstream effector, p73 (Cancino et al. 2008). APP/Swe mouse brains showed higher levels of c-Abl than control mice and, when treated with the c-Abl inhibitor STI571, tau phosphorylation was decreased in the brains of APP/Swe mice (Cancino et al. 2009). A transgenic mouse model expressing constitutively active c-Abl in forebrain neurons under the inducible tet-off system (AblPP/tTA mice) exhibited neuronal loss in the CA1 region of the hippocampus and striatum, severe neuroinflammation, and tyrosine phosphorylation of tau, though no significant tangle pathology was present (Schlatterer et al. 2011). The neurodegenerative/neuroinflammatory phenotype in AblPP/tTA mice was specific to expression of activated c-Abl, as transgenic mice with constitutively active Arg under the same expression system were phenotypically indistinguishable from controls (Schlatterer et al. 2011).
There is emerging evidence that the c-Abl tyrosine kinase may also be activated in other neurodegenerative diseases. Recently, two groups showed that there was an increase in c-Abl in the striatum of patients with Parkinson’s disease (PD) and an increase in the amount of tyrosine phosphorylated parkin in those patients (Ko et al. 2010; Imam et al. 2011). Recent studies in our laboratory have revealed c-Abl pT735 staining in neuronal cell bodies in human frontotemporal dementia (FTDP 17) with both the N278K mutation and P301L mutation and Guam Parkinson-dementia, and Abl pT735 co-localization with Pick bodies in human Picks disease (Fig. 2).

c-Abl is present in tauopathies. Tau pS202 (CP13), c-Abl pT735, and tau pY394/pS396 (YP3) staining (columns) of human frontotemporal dementia with N279K and P301L mutations, Pick’s disease, and Guam Parkinson-dementia (rows)
Multiple studies have shown c-Abl activation in human Alzheimer’s disease and AD models, suggesting that c-Abl may play a role in the pathogenesis of the disease. Exciting new studies suggest a role for c-Abl in a variety of other human neurodegenerative diseases and models of disease, suggesting that aberrant c-Abl activation in fully differentiated neurons may be a unifying factor in the pathogenesis of many neurodegenerative diseases, making it an attractive target for future studies and therapeutics.
Potential Activators of c-Abl in AD
While a multitude of studies have shown a correlation between c-Abl activation in neurons and neurodegenerative disease, the questions of how c-Abl becomes activated in neurodegenerative disease and of precisely how c-Abl contributes to the pathogenesis of these diseases remain.
The mechanism of neuronal loss in AD, the most common of the neurodegenerative diseases, remains unknown. However, there is healthy debate on the topic, and several hypotheses exist. The amyloid cascade hypothesis of AD states that accumulation of amyloid β fibrils leads to neuroinflammation followed by altered neuronal physiology and oxidative stress, resulting in altered kinase activity, tangles, and, ultimately, synaptic dysfunction and neuronal loss (Hardy and Selkoe 2002; Citron 2004). Alternatively, a recent review by Karl Herrup suggested that the pathogenesis of AD may be the result of an inappropriate neuroinflammatory response to an initiating injury followed by alterations in neuronal physiology, with aberrant cell cycle re-entry, synaptic loss and neuronal dysfunction and, ultimately, to neuronal loss (Herrup 2010). While there is debate regarding the initiating event in AD, there is agreement on several common themes. Neuroinflammation and neuronal injury via oxidative stress, DNA damage, or other mechanisms seem to play a role in the disease, resulting in altered neuronal cell state (i.e., cell cycle activation), synaptic dysfunction and, ultimately, neuronal loss.
c-Abl Is Activated by and Contributes to Neuroinflammation
Chronic neuroinflammation has been shown to occur in Alzheimer’s disease (reviewed in: (McGeer and McGeer 1995; Akiyama et al. 2000; Bamberger and Landreth 2002; Wyss-Coray and Mucke 2002; Wyss-Coray 2006; Glass et al. 2010)) and in Parkinson’s disease (reviewed in: (McGeer et al. 2001; Nagatsu and Sawada 2005)). A multitude of cytokines, including TNF-α, are upregulated in human AD brain (Griffin et al. 1998; Akiyama et al. 2000). TNF-α has been shown to stimulate caspase cleavage of c-Abl at the C terminus, leading to nuclear accumulation and contributing to apoptosis (Dan et al. 1999; Barila et al. 2003). Mice overexpressing constitutively active c-Abl in forebrain neurons (AblPP/tTA mice) also display florid neuroinflammatory pathology, despite lack of c-Abl in glia (Schlatterer et al. 2011), indicating that activation of c-Abl in neurons may contribute to induction of neuroinflammatory pathology.
c-Abl Is Activated by Oxidative Stress and DNA Damage
With aging and disease, there is a decrease in the body’s ability to handle oxidative stress and DNA damage incurred during normal cellular processes, leading to accumulation of reactive oxygen species and DNA damage. The c-Abl kinase is upregulated in response to oxidative stress and Aβ fibrils in neuronal culture (Alvarez et al. 2004) and is activated in response to DNA damage (Kharbanda et al. 1995), where it appears to play a role in DNA damage-induced apoptosis and cell cycle arrest at the G1-S transition (Yuan et al. 1996, 1997; Shafman et al. 1997). In primary neuronal culture, oxidative and dopaminergic stress resulted in c-Abl activation with subsequent parkin tyrosine phosphorylation, leading to loss of parkin’s protective E3 ubiquitin ligase activity and accumulation of AIMP2 and FBP (Ko et al. 2010; Imam et al. 2011). These data together suggest that neuronal c-Abl can be activated by a variety of oxidative and genotoxic stressors that might be associated with aging or disease and could contribute to neuronal damage or loss as a result of exposure to such damage.
Potential Effects of c-Abl Activation in Neurons
c-Abl and Aberrant Cell Cycle Re-entry
There have been many reports that aberrant cell cycle re-entry occurs in postmitotic neurons in AD and that these events precede neuronal death (Vincent et al. 1996, 1997, 1998; Ding et al. 2000; Husseman et al. 2000; Yang et al. 2001, 2003, 2006; Park et al. 2007). Cell cycle activation in neurons of a transgenic mouse resulted in Alzheimer-like tau and amyloid pathology (Park et al. 2007), and ectopic cell cycle events were shown to occur in neurons in three different transgenic mouse models of APP-induced amyloid plaque formation prior to development of plaques and microgliosis (Yang et al. 2006). However, cell cycle events in postmitotic neurons appear to be dysregulated, with some neurons cycling partially through S phase, but no neurons completing the cell cycle. There appears to be an “arrest” phenotype that eventually leads to neuronal death in lieu of division.
Constitutive activation of cytoplasmic c-Abl is known to stimulate the cell cycle. In neurons in AD, it appears that c-Abl is mainly cytoplasmic (Jing et al. 2009), which correlates with a cell cycle stimulatory function. Unpublished data from AblPP/tTA mice suggest that constitutive activation of c-Abl can lead to expression of cell cycle markers, indicating that activated c-Abl may play a role in aberrant cell cycle re-entry. c-Abl phosphorylated at T735, a modification associated with cytoplasmic localization, is the main form of the protein associated with tangles in severe cases of AD (Jing et al. 2009) and a variety of tauopathies (Fig. 2), suggesting that, at least initially, c-Abl acts in the cytoplasm in neurons to enhance ectopic cell cycle events. However, genotoxic and oxidative stress, Aβ fibrils, and TNF-α have all been shown to activate the nuclear, apoptotic/cell cycle arrest functions of c-Abl, and TNF-α has been shown to cause c-Abl localization to the nucleus. Interestingly, nuclear c-Abl can only be activated in response to genotoxic stress in cells in S phase (Liu et al. 1996), suggesting that ectopic cell cycle activation may be necessary for the apoptotic function of c-Abl.
c-Abl and Tau Phosphorylation
NFTs consisting of hyperphosphorylated tau protein are the characteristic lesion of AD that have been shown to correlate most closely with neurodegeneration and cognitive impairment (Gomez-Isla et al. 1997; Mitchell et al. 2002; Bennett et al. 2004). Transgenic mice expressing human tau develop tau pathology, aberrant cell cycle re-entry in neurons, late-onset neurodegeneration, spatial memory deficits, and synaptic dysfunction (Andorfer et al. 2003, 2005; Polydoro et al. 2009). Tyrosine phosphorylation of tau was shown to be as important as serine/threonine phosphorylation in stabilizing tau aggregation in JNPL3 mice expressing the P301L tau mutation (Vega et al. 2005). The c-Abl protein has been shown to phosphorylate tau at tyrosines 18, 197, 310, and 394, and tau pY394 and pY197 has been shown to be present in NFTs in AD (Derkinderen et al. 2005; Vega et al. 2005; Tremblay et al. 2010). As a kinase that phosphorylates tau, c-Abl may contribute to neurofibrillary tangle pathology and associated cognitive deficits.
Conclusions
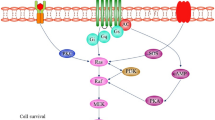
Recent studies show that c-Abl is upregulated in human AD and PD and our findings show that c-Abl is also upregulated in a variety of tauopathies. However, where, precisely, c-Abl fits into the cascade of events leading to neurodegeneration is not yet fully elucidated. A schematic of where c-Abl may fit into the scheme of events leading to neurodegenerative disease is displayed in Fig. 3.

The Potential Role of c-Abl in Neurodegenerative Disease. c-Abl is activated in response to oxidative stress, DNA damage, Aβ fibrils, and the inflammatory cytokine TNF-α. Ectopic cell cycle events induced by aging and other factors listed above contribute to c-Abl activation, and c-Abl activation contributes to aberrant cell cycle events. c-Abl can induce apoptosis, causing direct neuronal loss, through p73 and other mechanisms, and can induce tau phosphorylation, correlating with synaptic dysfunction and dementia
It has been shown that c-Abl can be activated by a variety of known contributors to neurodegenerative pathology, including oxidative stress, genotoxic stress, TNF-α, Aβ fibrils, and NFT, and activation of c-Abl by these events can lead to apoptosis and cell cycle arrest. The implication of these findings is that c-Abl likely acts downstream of known contributors to neurodegenerative pathology to initiate tau phosphorylation and participate in ectopic cell cycle events, eventually leading to neuronal loss, and, possibly, re-activating developmental processes leading to synaptic dysfunction.
Much work is needed in order to elucidate the exact role that c-Abl might play in neurodegenerative disease. Since c-Abl’s effect on the cell cycle can be stimulatory or inhibitory based upon subcellular localization, what role c-Abl might play in ectopic cell cycle events in neurodegeneration is particularly murky. Unpublished data from our laboratory suggest that activation of c-Abl in adult mouse forebrain neurons leads to expression of cell cycle markers, consistent with a positive role for c-Abl in aberrant cell cycle re-entry. Additionally, c-Abl in neurons is localized mainly to the cytoplasm, again consistent with a positive effect on cell cycle re-entry. However, in many cell types, including neurons, oxidative stress and DNA damage stimulate the nuclear, cell cycle inhibitory, and apoptotic functions of c-Abl.
While these data seem opposing, c-Abl cytoplasmic and nuclear effects could ultimately both play a role in ectopic cell cycle events in neurodegeneration. The cell cycle events in neurodegeneration are dysregulated, and it is possible that the nucleocytoplasmic shuttling of c-Abl may allow cytoplasmic c-Abl to play an initial stimulatory role in cell cycle events with subsequent or concurrent activation of c-Abl in the nucleus, contributing to cell cycle arrest and eventual neuronal death. It has been shown that entry into S phase is necessary for the cytotoxic effects of c-Abl to occur, suggesting that the potential detrimental effects of c-Abl would require activation of the cell cycle.
Despite the many questions that still remain regarding the mechanism by which c-Abl acts in neurodegenerative disease, recent studies have made it clear that c-Abl is present in the characteristic lesions of human AD and is increased in human PD, and studies from our laboratory also show that c-Abl is upregulated in a variety of human tauopathies. It is also clear that activation of c-Abl in forebrain neurons in mice can cause neurodegeneration and neuroinflammation, indicating that c-Abl activation alone is sufficient to cause neurodegenerative pathology. These studies taken together suggest that c-Abl is a provocative target for therapeutics for neurodegenerative disease and that further studies of c-Abl mechanism in neurons are warranted.
References
Akiyama H, Barger S, Barnum S et al (2000) Inflammation and Alzheimer’s disease of article. Neurobiol Aging 21:383–421
Alvarez AR, Sandoval PC, Leal NR, Castro PU, Kosik KS (2004) Activation of the neuronal c-Abl tyrosine kinase by amyloid-beta-peptide and reactive oxygen species of article. Neurobiol Dis 17:326–336
Andorfer C, Kress Y, Espinoza M et al (2003) Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms of article. J Neurochem 86:582–590
Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P (2005) Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms of article. J Neurosci 25:5446–5454
Bamberger ME, Landreth GE (2002) Inflammation, apoptosis, and Alzheimer’s disease of article. Neuroscientist 8:276–283
Barila D, Rufini A, Condo I et al (2003) Caspase-dependent cleavage of c-Abl contributes to apoptosis of article. Mol Cell Biol 23:2790–2799
Bashaw GJ, Kidd T, Murray D, Pawson T, Goodman CS (2000) Repulsive axon guidance: Abelson and enabled play opposing roles downstream of the roundabout receptor of article. Cell 101:703–715
Bennett RL, Hoffmann FM (1992) Increased levels of the Drosophila Abelson tyrosine kinase in nerves and muscles: subcellular localization and mutant phenotypes imply a role in cell–cell interactions of article. Development 116:953–966
Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE (2004) Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function of article. Arch Neurol 61:378–384
Cancino GI, Toledo EM, Leal NR et al (2008) STI571 prevents apoptosis, tau phosphorylation and behavioural impairments induced by Alzheimer’s beta-amyloid deposits of article. Brain 131:2425–2442
Cancino GI, Perez de Arce K, Castro PU, Toledo EM, von Bernhardi R, Alvarez AR (2009) c-Abl tyrosine kinase modulates tau pathology and Cdk5 phosphorylation in AD transgenic mice of article. Neurobiol Aging 32(7):1249–1261
Cao C, Leng Y, Kufe D (2003) Catalase activity is regulated by c-Abl and Arg in the oxidative stress response of article. J Biol Chem 278:29667–29675
Citron M (2004) Strategies for disease modification in Alzheimer’s disease of article. Nat Rev Neurosci 5:677–685
Courtney KD, Grove M, Vandongen H, Vandongen A, LaMantia AS, Pendergast AM (2000) Localization and phosphorylation of Abl-interactor proteins, Abi-1 and Abi-2, in the developing nervous system of article. Mol Cell Neurosci 16:244–257
Dan S, Naito M, Seimiya H, Kizaki A, Mashima T, Tsuruo T (1999) Activation of c-Abl tyrosine kinase requires caspase activation and is not involved in JNK/SAPK activation during apoptosis of human monocytic leukemia U937 cells of article. Oncogene 18:1277–1283
Daniel R, Cai Y, Wong PM, Chung SW (1995) Deregulation of c-abl mediated cell growth after retroviral transfer and expression of antisense sequences of article. Oncogene 10:1607–1614
de Arce KP, Varela-Nallar L, Farias O et al (2010) Synaptic clustering of PSD-95 is regulated by c-Abl through tyrosine phosphorylation of article. J Neurosci 30:3728–3738
Derkinderen P, Scales TM, Hanger DP et al (2005) Tyrosine 394 is phosphorylated in Alzheimer’s paired helical filament tau and in fetal tau with c-Abl as the candidate tyrosine kinase of article. J Neurosci 25:6584–6593
Ding XL, Husseman J, Tomashevski A, Nochlin D, Jin LW, Vincent I (2000) The cell cycle Cdc25A tyrosine phosphatase is activated in degenerating postmitotic neurons in Alzheimer’s disease of article. Am J Pathol 157:1983–1990
Dunah AW, Yasuda RP, Wang YH et al (1996) Regional and ontogenic expression of the NMDA receptor subunit NR2D protein in rat brain using a subunit-specific antibody of article. J Neurochem 67:2335–2345
Elkins T, Zinn K, McAllister L, Hoffmann FM, Goodman CS (1990) Genetic analysis of a Drosophila neural cell adhesion molecule: interaction of fasciclin I and Abelson tyrosine kinase mutations of article. Cell 60:565–575
Gertler FB, Bennett RL, Clark MJ, Hoffmann FM (1989) Drosophila abl tyrosine kinase in embryonic CNS axons: a role in axonogenesis is revealed through dosage-sensitive interactions with disabled of article. Cell 58:103–113
Gertler FB, Hill KK, Clark MJ, Hoffmann FM (1993) Dosage-sensitive modifiers of Drosophila abl tyrosine kinase function: prospero, a regulator of axonal outgrowth, and disabled, a novel tyrosine kinase substrate of article. Genes Dev 7:441–453
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH (2010) Mechanisms underlying inflammation in neurodegeneration of article. Cell 140:918–934
Glover RT, Angiolieri M, Kelly S et al (2000) Interaction of the N-methyl-d-aspartic acid receptor NR2D subunit with the c-Abl tyrosine kinase of article. J Biol Chem 275:12725–12729
Goga A, Liu X, Hambuch TM et al (1995) p53 dependent growth suppression by the c-Abl nuclear tyrosine kinase of article. Oncogene 11:791–799
Gomez-Isla T, Hollister R, West H et al (1997) Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease of article. Ann Neurol 41:17–24
Grevengoed EE, Loureiro JJ, Jesse TL, Peifer M (2001) Abelson kinase regulates epithelial morphogenesis in Drosophila of article. J Cell Biol 155:1185–1198
Griffin WS, Sheng JG, Royston MC et al (1998) Glial-neuronal interactions in Alzheimer’s disease: the potential role of a ‘cytokine cycle’ in disease progression of article. Brain Pathol 8:65–72
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics of article. Science 297:353–356
Herrup K (2010) Reimagining Alzheimer’s disease—an age-based hypothesis of article. J Neurosci 30:16755–16762
Hill KK, Bedian V, Juang JL, Hoffmann FM (1995) Genetic interactions between the Drosophila Abelson (Abl) tyrosine kinase and failed axon connections (fax), a novel protein in axon bundles of article. Genetics 141:595–606
Huang TS, Kuo ML, Shew JY, Chou YW, Yang WK (1996) Distinct p53-mediated G1/S checkpoint responses in two NIH3T3 subclone cells following treatment with DNA-damaging agents of article. Oncogene 13:625–632
Huang Y, Yuan ZM, Ishiko T et al (1997) Pro-apoptotic effect of the c-Abl tyrosine kinase in the cellular response to 1-beta-d-arabinofuranosylcytosine of article. Oncogene 15:1947–1952
Husseman JW, Nochlin D, Vincent I (2000) Mitotic activation: a convergent mechanism for a cohort of neurodegenerative diseases of article. Neurobiol Aging 21:815–828
Imam SZ, Zhou Q, Yamamoto A et al (2011) Novel regulation of parkin function through c-Abl-mediated tyrosine phosphorylation: implications for Parkinson's disease of article. J Neurosci 31:157–163
Jing Z, Caltagarone J, Bowser R (2009) Altered subcellular distribution of c-Abl in Alzheimer’s disease of article. J Alzheimers Dis 17:409–422
Jones SB, Lu HY, Lu Q (2004) Abl tyrosine kinase promotes dendrogenesis by inducing actin cytoskeletal rearrangements in cooperation with Rho family small GTPases in hippocampal neurons of article. J Neurosci 24:8510–8521
Kharbanda S, Ren R, Pandey P et al (1995) Activation of the c-Abl tyrosine kinase in the stress response to DNA-damaging agents of article. Nature 376:785–788
Kharbanda S, Yuan ZM, Weichselbaum R, Kufe D (1997) Functional role for the c-Abl protein tyrosine kinase in the cellular response to genotoxic stress of article. Biochim Biophys Acta 1333:O1–O7
Kharbanda S, Yuan ZM, Weichselbaum R, Kufe D (1998) Determination of cell fate by c-Abl activation in the response to DNA damage of article. Oncogene 17:3309–3318
Ko HS, Lee Y, Shin JH et al (2010) Phosphorylation by the c-Abl protein tyrosine kinase inhibits parkin's ubiquitination and protective function of article. Proc Natl Acad Sci USA 107:16691–16696
Koleske AJ, Gifford AM, Scott ML et al (1998) Essential roles for the Abl and Arg tyrosine kinases in neurulation of article. Neuron 21:1259–1272
Kruh GD, Perego R, Miki T, Aaronson SA (1990) The complete coding sequence of arg defines the Abelson subfamily of cytoplasmic tyrosine kinases of article. Proc Natl Acad Sci USA 87:5802–5806
Liebl EC, Forsthoefel DJ, Franco LS et al (2000) Dosage-sensitive, reciprocal genetic interactions between the Abl tyrosine kinase and the putative GEF trio reveal trio's role in axon pathfinding of article. Neuron 26:107–118
Liu ZG, Baskaran R, Lea-Chou ET et al (1996) Three distinct signalling responses by murine fibroblasts to genotoxic stress of article. Nature 384:273–276
McGeer PL, McGeer EG (1995) The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases of article. Brain Res Brain Res Rev 21:195–218
McGeer PL, Yasojima K, McGeer EG (2001) Inflammation in Parkinson's disease of article. Adv Neurol 86:83–89
Mitchell TW, Mufson EJ, Schneider JA et al (2002) Parahippocampal tau pathology in healthy aging, mild cognitive impairment, and early Alzheimer’s disease of article. Ann Neurol 51:182–189
Nagatsu T, Sawada M (2005) Inflammatory process in Parkinson's disease: role for cytokines of article. Curr Pharm Des 11:999–1016
Ozanne B, Wheeler T, Zack J, Smith G, Dale B (1982) Transforming gene of a human leukaemia cell is unrelated to the expressed tumour virus related gene of the cell of article. Nature 299:744–747
Park KH, Hallows JL, Chakrabarty P, Davies P, Vincent I (2007) Conditional neuronal simian virus 40 T antigen expression induces Alzheimer-like tau and amyloid pathology in mice of article. J Neurosci 27:2969–2978
Polydoro M, Acker CM, Duff K, Castillo PE, Davies P (2009) Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology of article. J Neurosci 29:10741–10749
Sawyers CL (1993) Molecular consequences of the BCR-ABL translocation in chronic myelogenous leukemia of article. Leuk Lymphoma 11(Suppl 2):101–103
Schlatterer SD, Tremblay MA, Acker CM, Davies P (2011) Neuronal c-Abl overexpression leads to neuronal loss and neuroinflammation in the mouse forebrain of article. J Alzheimers Dis 25:119–133
Shafman T, Khanna KK, Kedar P et al (1997) Interaction between ATM protein and c-Abl in response to DNA damage of article. Nature 387:520–523
Silberman I, Sionov RV, Zuckerman V et al (2008) T cell survival and function requires the c-Abl tyrosine kinase of article. Cell Cycle 7:3847–3857
Tremblay MA, Acker CM, Davies P (2010) Tau phosphorylated at tyrosine 394 is found in Alzheimer’s disease tangles and can be a product of the Abl-related kinase, Arg of article. J Alzheimers Dis 19:721–733
Van Etten RA, Jackson P, Baltimore D (1989) The mouse type IV c-abl gene product is a nuclear protein, and activation of transforming ability is associated with cytoplasmic localization of article. Cell 58:669–678
Van Etten RA, Jackson PK, Baltimore D, Sanders MC, Matsudaira PT, Janmey PA (1994) The COOH terminus of the c-Abl tyrosine kinase contains distinct F- and G-actin binding domains with bundling activity of article. J Cell Biol 124:325–340
Vega IE, Cui L, Propst JA, Hutton ML, Lee G, Yen SH (2005) Increase in tau tyrosine phosphorylation correlates with the formation of tau aggregates of article. Brain Res Mol Brain Res 138:135–144
Vincent I, Rosado M, Davies P (1996) Mitotic mechanisms in Alzheimer’s disease? of article. J Cell Biol 132:413–425
Vincent I, Jicha G, Rosado M, Dickson DW (1997) Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer’s disease brain of article. J Neurosci 17:3588–3598
Vincent I, Zheng JH, Dickson DW, Kress Y, Davies P (1998) Mitotic phosphoepitopes precede paired helical filaments in Alzheimer’s disease of article. Neurobiol Aging 19:287–296
Welch PJ, Wang JY (1993) A C-terminal protein-binding domain in the retinoblastoma protein regulates nuclear c-Abl tyrosine kinase in the cell cycle of article. Cell 75:779–790
Wen ST, Jackson PK, Van Etten RA (1996) The cytostatic function of c-Abl is controlled by multiple nuclear localization signals and requires the p53 and Rb tumor suppressor gene products of article. EMBO J 15:1583–1595
Wenzel A, Villa M, Mohler H, Benke D (1996) Developmental and regional expression of NMDA receptor subtypes containing the NR2D subunit in rat brain of article. J Neurochem 66:1240–1248
Wetzler M, Talpaz M, Van Etten RA, Hirsh-Ginsberg C, Beran M, Kurzrock R (1993) Subcellular localization of Bcr, Abl, and Bcr-Abl proteins in normal and leukemic cells and correlation of expression with myeloid differentiation of article. J Clin Invest 92:1925–1939
Woodring PJ, Hunter T, Wang JY (2003) Regulation of F-actin-dependent processes by the Abl family of tyrosine kinases of article. J Cell Sci 116:2613–2626
Wyss-Coray T (2006) Inflammation in Alzheimer disease: driving force, bystander or beneficial response? of article. Nat Med 12:1005–1015
Wyss-Coray T, Mucke L (2002) Inflammation in neurodegenerative disease—a double-edged sword of article. Neuron 35:419–432
Yang Y, Geldmacher DS, Herrup K (2001) DNA replication precedes neuronal cell death in Alzheimer’s disease of article. J Neurosci 21:2661–2668
Yang Y, Mufson EJ, Herrup K (2003) Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer’s disease of article. J Neurosci 23:2557–2563
Yang Y, Varvel NH, Lamb BT, Herrup K (2006) Ectopic cell cycle events link human Alzheimer’s disease and amyloid precursor protein transgenic mouse models of article. J Neurosci 26:775–784
Yuan ZM, Huang Y, Whang Y et al (1996) Role for c-Abl tyrosine kinase in growth arrest response to DNA damage of article. Nature 382:272–274
Yuan ZM, Huang Y, Ishiko T, Kharbanda S, Weichselbaum R, Kufe D (1997) Regulation of DNA damage-induced apoptosis by the c-Abl tyrosine kinase of article. Proc Natl Acad Sci USA 94:1437–1440
Yuan ZM, Huang Y, Ishiko T et al (1998) Regulation of Rad51 function by c-Abl in response to DNA damage of article. J Biol Chem 273:3799–3802
Yuan ZM, Shioya H, Ishiko T et al (1999) p73 is regulated by tyrosine kinase c-Abl in the apoptotic response to DNA damage of article. Nature 399:814–817
Zukerberg LR, Patrick GN, Nikolic M et al (2000) Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosine phosphorylation, kinase upregulation, and neurite outgrowth of article. Neuron 26:633–646
Acknowledgments
Supported by Applied Neurosolutions Inc and by NIMH38623 and AG022102. We thank Dr. Dennis Dickson for the human tauopathy cases used for experiments as shown in Fig. 2.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Schlatterer, S.D., Acker, C.M. & Davies, P. c-Abl in Neurodegenerative Disease. J Mol Neurosci 45, 445–452 (2011). https://doi.org/10.1007/s12031-011-9588-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-011-9588-1