
Abstract
Transplant-associated thrombotic microangiopathy (TA-TMA) is an early complication of hematopoietic cell transplantation (HCT). A high mortality rate is documented in patients who are refractory to calcineurin inhibitor cessation. Estimates of TA-TMA prevalence vary significantly and are higher in allogeneic compared with autologous HCT. Furthermore, our understanding of the pathophysiology that is strongly related to diagnosis and treatment options is limited. Recent evidence has linked TA-TMA with atypical hemolytic uremic syndrome, a disease of excessive activation of the alternative pathway of complement, opening the Pandora’s box in treatment options. As conventional treatment management is highly inefficient, detection of complement activation may allow for early recognition of patients who will benefit from complement inhibition. Preliminary clinical results showing successful eculizumab administration in children and adults with TA-TMA need to be carefully evaluated. Therefore, realizing the unmet needs of better understanding TA-TMA in this complex setting, we aimed to summarize current knowledge focusing on (1) critical evaluation of diagnostic criteria, (2) epidemiology and prognosis, (3) recent evidence of complement activation and endothelial damage and (4) treatment options.
Similar content being viewed by others
Introduction
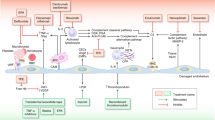
Transplant-associated thrombotic microangiopathy (TA-TMA) represents a life-threatening complication following hematopoietic cell transplantation (HCT).1, 2, 3, 4, 5, 6, 7, 8, 9 The syndrome presents with features of TMA including thrombocytopenia, nonimmune hemolytic anemia, peripheral blood schistocytes and often other end-organ damage to the kidneys and central nervous system. The onset of renal failure and central nervous system dysfunction such as seizure, stroke or encephalopathy, accompanied by hypertension, hemolytic anemia and consumptive thrombocytopenia in the absence of coagulopathy, are the classic hallmarks of the syndrome. Interestingly, this severe clinical manifestation is not evident in all TA-TMA patients, as a number of them manifest a syndrome that resolves after withdrawal of calcineurin inhibitors (CNIs). This fact highlights that our understanding in terms of diagnosis and pathophysiology remains unclear, although it has been recognized for decades.
The lack of reliable diagnostic and prognostic markers hampers prompt clinical management. Recent evidence suggests that the syndrome manifests as a result of endothelial dysfunction because of multiple triggers probably in a genetically predisposed recipient.8, 10 In this context, TA-TMA shares common features with atypical hemolytic uremic syndrome (aHUS), a TMA characterized by excessive activation of the alternative pathway of complement, opening Pandora’s box in available therapeutic options.11 aHUS is treated with a C5 monoclonal antibody that safely and efficiently inhibits terminal complement inhibition, eculizumab.12, 13 Unlike aHUS, conventional therapeutic interventions in TA-TMA have proven inefficient in refractory cases, leading to increased mortality rates. Although successful eculizumab administration has been reported in children and adults with TA-TMA,14, 15 preliminary clinical results need to be carefully evaluated.
Realizing the unmet needs of better understanding TA-TMA in this complex setting, we aimed to summarize current knowledge focusing on (1) critical evaluation of diagnostic criteria, (2) epidemiology and prognosis, (3) recent evidence of complement activation and endothelial damage and (4) treatment options. Accordingly, we performed a systematic MEDLINE search using the terms: thrombotic microangiopathy, hematopoietic cell transplantation, prognosis, GvHD, complement activation and complement inhibition.
Critical evaluation of diagnostic criteria
TA-TMA diagnosis relies on clinical criteria proposed by the Bone Marrow Transplant Clinical Trials Network (BMT-CTN) in 20057 and the International Working Group (IWG) in 2007.16 Several pitfalls have been identified in both diagnostic criteria that limit their diagnostic sensitivity.17, 18 First, schistocytosis that is required by diagnostic criteria may be absent in severe forms of TA-TMA because of the high vascular permeability and extravasation of erythrocytes observed in TMA.11 Second, the criterion of normal coagulation assays that is necessary to exclude disseminated intravascular coagulation from the differential diagnosis is not included in the current diagnostic criteria. Third, the CTN diagnostic criteria require concurrent renal or neurologic dysfunction for diagnosis of TMA. However, several causes of nephropathy not relevant to TMA may be recognized in HCT recipients. In addition, neurologic abnormalities are not as common as in thrombotic thrombocytopenic purpura (TTP).16, 19 Fourth, the IWG criteria require counting of a schistocyte percentage higher than 4% that limits their accuracy because of the lack of a standardized laboratory method for schistocyte counting. Finally, many patients manifest TMA early after bone marrow transplant at a time when their reticulocyte count is low from the conditioning regimen and their transfusion burden is still high, making classic microangiopathic findings on the bloods smear inconspicuous. To overcome these limitations, Cho et al.18 have introduced the entity of probable TMA that requires normal coagulation studies, schistocytes higher than 2 per high-power field but does not require renal or neurologic dysfunction.
Based on their work on TA-TMA pathophysiology in children and young adults, Jodele et al.8 have recently proposed an algorithm for TA-TMA diagnosis. According to this algorithm, clinical criteria for TA-TMA diagnosis are the following: lactate dehydrogenase above normal, presence of schistocytes or histological evidence of TMA, thrombocytopenia, proteinuria and hypertension. Acute elevation of lactate dehydrogenase, proteinuria >30 mg/dL and hypertension more severe than expected with calcineurin or steroid therapy, usually requiring >2 antihypertensive medications should raise clinical suspicion for TA-TMA and be further investigated. Beyond diagnosis, the same group has also proposed risk criteria for TA-TMA that will be further discussed in the prognosis section.8 Table 1 summarizes current diagnostic criteria.
Diversity among diagnostic criteria may lead to difficulty in understanding clinically important syndromes that require immediate intensification of treatment. In addition, in the complicated setting of HCT, cytopenias and organ dysfunction, such as renal, central nervous system and hepatic, are relatively common and multifactorial. Common causes are drugs, infection, or GvHD, making the diagnosis of TA-TMA even more difficult. Furthermore, these criteria are strictly descriptive and do not take into account the pathophysiology of the syndrome, because of the absence of robust diagnostic testing. Therefore, updated consensus criteria that would overcome the existing limitations and take into account recent research findings are warranted.
Epidemiology and prognosis
The lack of solid diagnostic criteria and testing hampers accurate estimation of prevalence that vary significantly among studies. TA-TMA is less common in autologous than in allogeneic HCT (0–27% in autologous compared with 6–76% in allogeneic).1, 2, 3, 4, 5, 6, 7, 8, 9, 19, 20 TA-TMA is considered an early HCT complication that occurs usually within the first 3 months, but late episodes (up to 2 years) have also been described.21, 22
TA-TMA was first recognized in 1980, as a side effect of cyclosporine administration for GvHD prophylaxis in allogeneic HCT.23 Since then, CNIs have been linked to the syndrome and are immediately withdrawn after TA-TMA diagnosis. Other clinical studies have identified a number of additional risk factors for TA-TMA: age, donor type, conditioning regimen, mTOR (mechanistic target of rapamycin) inhibitors, acute GvHD and infections.1, 2, 4, 6, 24, 25, 26, 27 Interestingly, the presence of GvHD is the common denominator in many studies suggesting that successful prevention and treatment strategies for GvHD need to be timely employed. However, the exposure to these factors following allogeneic HCT in all patients is high, making their role in pathogenesis difficult to determine. It is not clear whether one factor alone can trigger the manifestation of TMA in HCT recipients. For example, CNI withdrawal does not reliably reverse TA-TMA. In addition, CNI administration in diseases, such as aplastic anemia or red cell aplasia, does not cause TMA.28
Prognosis is poor with a high mortality rate of roughly 50–75%.1, 2, 3, 4, 5, 6, 7, 8, 9, 29 The prognostic role of schistocytosis percentage remains controversial.17, 30, 31 Nevertheless, accumulating evidence strengthens the role of renal dysfunction as a poor prognostic factor linked to lower survival rates.8, 18, 19 Recently, Jodele et al.8 proposed that patients with proteinuria >30 mg/dL and evidence of terminal complement activation (elevated soluble C5b-9) have poor prognosis and require immediate therapeutic interventions.
Evidence of excessive complement activation and endothelial damage
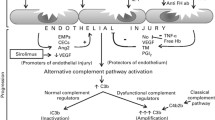
Complement activation
An initial obstacle to our understanding of the syndrome has been its limited association with deficiency of the plasma protease ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 motif, 13). Severe ADAMTS13 deficiency (usually defined as <10%) is observed in TTP and is best treated with plasma exchange.32, 33 In a number of TA-TMA studies, ADAMTS13 has not proven a useful marker or predictor of the disease.34, 35 This evidence is in line with the observation that TA-TMA is generally unresponsive to therapeutic plasma exchange.9, 36
As our understanding of aHUS has evolved, TA-TMA seems to resemble more aHUS than other TMAs. Indeed, Laskin et al.11 have concisely reviewed the analogies between TA-TMA and aHUS in terms of pathophysiological and clinical evidence available until 2011. aHUS is most commonly caused by defects in the regulation of the alternative pathway of complement. These defects are usually inherited, including mutations in complement factor H (CFH) and complement factor I (CFI), complement component C3, membrane cofactor protein or thrombomodulin, but may also be acquired, such as autoantibodies to CFH.37, 38 Interestingly enough, genetic mutations are found in 50–60% of patients diagnosed with aHUS and triggers are considered crucial for the manifestation of the disease (two-hit hypothesis).37, 39
Similarly, accumulating evidence of complement activation has been recently reported in TA-TMA of children and young adults. Jodele et al.40 have identified abnormalities in CFH-related genes (CFHR) and autoantibodies to CFH, a major regulator of the APC, in six children with TA-TMA following HCT. If the two-hit hypothesis of the aHUS is true for TA-TMA, genetic susceptibility may be the first required hit for the development of the syndrome. A recent prospective study of the same cohort has tested variants of genes involved in complement activation before and after transplant. This study provided evidence of pretransplant genetic susceptibility in 65% of patients who developed TA-TMA. In addition, variants in three or more genes were associated with increased mortality.10 Although the functional role of gene variants was evaluated in a subset of patients, connection of genotype to phenotype needs to be further investigated. Complement-related variants implicated in the pathophysiology of TA-TMA by these studies are presented in Table 2. Ethnic differences in complement-related variants may also explain differences in TMA occurrence.
Reliable markers of complement activation have been a long-standing need in the field of TMAs. Recently, products of terminal complement activation, that is, C5a and soluble C5b-9 or membrane attack complex, have been compared in aHUS and TTP. Although C5a and C5b-9 plasma levels were increased in aHUS, these markers were not reliable in distinguishing the two diseases and do not have clear cutoff values.41 However, serum C5b-9 levels have been included in a diagnostic algorithm for the evaluation of TA-TMA.8 In an effort to develop a rapid and simple in vitro diagnostic assay for aHUS, we have recently modified the Ham test, traditionally used for diagnosis of paroxysmal nocturnal hemoglobinuria. The principle of the Ham assay is that paroxysmal nocturnal hemoglobinuria cells are more vulnerable to acidified serum that serves to activate complement.42 The modified Ham test utilizes paroxysmal nocturnal hemoglobinuria-like cell lines that are susceptible to complement-mediated cell death induced by activated serum, such as the aHUS serum. Results in aHUS have been promising, showing that the modified Ham test effectively distinguishes aHUS from TTP.43 Except for aHUS, the modified Ham test has also successfully detected increased complement activation in typical HUS44 and HELLP (hemolysis, elevated liver enzyme levels, and low platelet levels) syndrome.45 Preliminary data utilizing the modified Ham test have also shown increased complement activation in TA-TMA patients compared with other HCT recipients.46 However, these data need to be further validated in larger TMA cohorts.
Other mechanisms of endothelial damage
Beyond complement-induced endothelial damage, HCT recipients are vulnerable to endothelial injury by a number of clinical factors, including CNI and/or mTOR inhibitors, GvHD and infections.
CNI and/or mTOR inhibitors
Cyclosporine, sirolimus and tacrolimus are widely used immunosuppressants in both hematopoietic cell and organ transplantations. Endothelial dysfunction predisposing to TA-TMA is evident post treatment with these agents,47, 48 although mechanistic evidence relies basically in renal transplant studies. It is well known that CNIs decrease prostacyclin, nitric oxide and activated protein C and increase thromboxane A2 and endothelin.49, 50, 51 More recent studies have shown an increase of endothelial cell progenitors, induction of endothelial cell apoptosis and dysregulation of metalloproteinases in endothelial cells.52, 53, 54, 55 Interestingly, thrombomodulin, a protein also involved in complement regulation, has been documented to protect from cyclosporine-induced vascular damage.56 In HCT recipients, tacrolimus and sirolimus had a proinflammatory effect, but only cyclosporine exhibited an additional prothrombotic effect.57
GvHD
The endothelium has been long considered a key mediator of end-organ damage in acute and chronic GvHD.58, 59, 60 Furthermore, endothelial cell vulnerability and dysfunction may also contribute to steroid refractoriness in GvHD.61, 62 More recent studies also support the importance of vascular alterations in GvHD.63, 64 Similar to the above-mentioned study on cyclosporine, the complement regulator thrombomodulin exerts beneficial effects on immune GvHD too.65
Infections
The pathophysiology linking infections to TA-TMA is not well defined. It possibly involves both complement activation and endothelial dysfunction through the cytokine storm. An interesting finding in the field is that neutrophil extracellular traps have been found increased in TA-TMA patients.66
Treatment options
Conventional management
Conventional management of TA-TMA has been unsatisfying, with resultant high mortality rates in patients not responding to CNI cessation. Available treatment strategies require first the withdrawal of CNIs or mTOR inhibitors. Second, treating physicians need to optimally control GvHD or concomitant infections that are common in TA-TMA patients. Among infectious agents, Aspergillus, CMV and adenovirus have been mostly implicated in TA-TMA.1, 6, 9
The next steps in conventional management depend largely on availability of relevant testing (ADAMTS13 activity, complement testing) and the center’s policy. Plasma exchange has been traditionally considered the standard of care in TMA and continues to be used in many centers. Theoretically, plasma exchange would be efficient only in patients with ADAMTS13 deficiency or antibodies to CFH.67, 68 Interestingly, Jodele and colleagues11 in a study of a small pediatric cohort have suggested that earlier the initiation of plasma exchange the better. It has also been hypothesized that plasma exchange might provide additional benefit by removal of excessive complement proteins, inflammatory cytokines or circulating endothelial cells.
However, the role of plasma exchange is largely questioned. A number of studies have shown poor survival in TA-TMA patients managed with plasma exchange, despite initial responses that may also be attributed in part to CNI or sirolimus withdrawal.69, 70, 71, 72, 73, 74 It should also be noted that clinical and laboratory estimation of response to plasma exchange is particularly difficult in HCT recipients. In addition, comorbidities such as severe GvHD or infection might also contribute to morbidity and mortality in these patients. Finally, it is difficult to use plasma exchange simultaneously with novel agents, such as eculizumab, because the dosage of the agents needs to be readministered after each session.
In refractory TA-TMA cases, intensification of immunosuppressive treatment is often recommended. In particular, rituximab, an anti-CD20 antibody, has been successfully used in refractory TTP and other TMA cases, although some patients still remain refractory as shown by a recent phase-II study in nontransplant patients.75 In TA-TMA, successful rituximab administration has also been reported.76, 77 Alternative agents used for refractory cases also include defibrotide and daclizumab. Based on its beneficial effects against endothelial dysfunction, a polydisperse oligonucleotide, defibrotide, has promising results in TA-TMA as reported by retrospective studies.6, 78, 79 Limited reports also exist on the potential benefits of daclizumab, a humanized antibody against interleukin-2 receptor, in patients with TA-TMA and GvHD.80
Complement inhibition
More recently, complement inhibition has been introduced as a novel treatment strategy for complement-mediated diseases. The first-in-class complement inhibitor, eculizumab, is a monoclonal antibody that binds C5 and effectively inhibits the formation of membrane attack complex/C5b-9. Terminal complement inhibition by eculizumab is highly effective and FDA (Food and Drug Administration) approved for treating aHUS.12, 13 However, given the lack of a definitive diagnostic assay and the high cost of the drug therapy is often delayed or not administered.
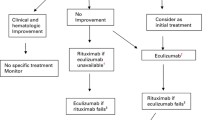
Favorable outcomes of eculizumab treatment have also been described in patients with TA-TMA. Retrospective evaluation of 12 patients who received eculizumab by the French group has shown hematological response and overall survival at 50% and 33%, respectively.81 It should be noted that eculizumab dosage was appropriate in all patients as measured by total hemolytic complement activity. As pointed out by the authors, results are encouraging compared with mortality rates in the pre-eculizumab era, and early initiation of eculizumab treatment may be even more promising. TMA resolution after early administration of eculizumab has also been documented in case reports.82, 83 A more recent case series has reported response to eculizumab in four out of five adult TA-TMA patients achieving transfusion independence and improvement in renal function.15 However, the nonresponder and one responder to eculizumab succumbed to fatal infections. In the pediatric cohort, Jodele et al.14 reported safety of eculizumab administration in 30 pediatric HCT recipients with TA-TMA, even without meningococcal vaccination in the early post transplant period. TMA-related mortality was observed in only 4 out of 30 patients.14 This group adjusted eculizumab dosing using total complement activity (CH50) and terminal complement activation (sC5b-9) monitoring.84 Figure 1 proposes an algorithm for TA-TMA management.

Algorithm for TA-TMA management. Complement studies: complement-related genetic mutations or functional assays (serum C3, C4, C5b-9); CFH, complement factor H; CNI, calcineurin inhibitor; GVHD, graft-versus-host disease; mTOR, mechanistic target of rapamycin; TA-TMA, transplant-associated thrombotic microangiopathy; TPE, therapeutic plasma exchange.
Beyond eculizumab, novel complement inhibitors are in the developmental pipeline for complement-related diseases. Among them, engineered complement receptor 2/factor H fusion protein TT30,85 members of the peptide C3 inhibitor compstatin family,86 C1 esterase inhibitor C1INH (Cinryze)87 and factor D inhibitors88 are promising in terms of overcoming limitations of eculizumab observed in treated patients with paroxysmal nocturnal hemoglobinuria. However, their safety and efficacy remains to be proven in clinical studies. Furthermore, their potential usefulness in patients with TA-TMA will be determined when the role of complement activation in TA-TMA is clarified.
Conclusions and future perspectives
In conclusion, TA-TMA remains an unresolved complication of HCT, leading to increased morbidity and mortality mainly in allogeneic HCT recipients. Its pathophysiology, diagnosis and treatment options have not been fully elucidated. In this complex setting, recent evidence of increased complement activation needs to be confirmed in larger cohorts utilizing both genetic and functional assays. Connecting the genotype to phenotype remains a research challenge in diseases of increased complement activation. In addition, better understanding of the pathophysiology may lead to more accurate diagnostic criteria and targeted treatment. In the era of precision medicine, reliable detection of complement activation may allow for early initiation of complement in selected patients and, thus, to improved clinical outcomes. Patient selection, time of treatment initiation, duration of treatment, response and impact on survival remain to be confirmed in future prospective studies of larger cohorts.
References
Ye Y, Zheng W, Wang J, Hu Y, Luo Y, Tan Y et al. Risk and prognostic factors of transplantation-associated thrombotic microangiopathy in allogeneic haematopoietic stem cell transplantation: a nested case control study. Hematol Oncol (e-pub ahead of print 1 June 2016; doi:10.1002/hon.2310).
Sakellari I, Gavriilaki E, Boussiou Z, Batsis I, Mallouri D, Constantinou V et al. Transplant-associated thrombotic microangiopathy: an unresolved complication of unrelated allogeneic transplant for hematologic diseases. Hematol Oncol (e-pub ahead of print 19 September 2016; doi:10.1002/hon.2346).
Changsirikulchai S, Myerson D, Guthrie KA, McDonald GB, Alpers CE, Hingorani SR . Renal thrombotic microangiopathy after hematopoietic cell transplant: role of GVHD in pathogenesis. Clin J Am Soc Nephrol 2009; 4: 345–353.
Nakamae H, Yamane T, Hasegawa T, Nakamae M, Terada Y, Hagihara K et al. Risk factor analysis for thrombotic microangiopathy after reduced-intensity or myeloablative allogeneic hematopoietic stem cell transplantation. Am J Hematol 2006; 81: 525–531.
Willems E, Baron F, Seidel L, Frere P, Fillet G, Beguin Y . Comparison of thrombotic microangiopathy after allogeneic hematopoietic cell transplantation with high-dose or nonmyeloablative conditioning. Bone Marrow Transplant 2010; 45: 689–693.
Uderzo C, Bonanomi S, Busca A, Renoldi M, Ferrari P, Iacobelli M et al. Risk factors and severe outcome in thrombotic microangiopathy after allogeneic hematopoietic stem cell transplantation. Transplantation 2006; 82: 638–644.
Ho VT, Cutler C, Carter S, Martin P, Adams R, Horowitz M et al. Blood and marrow transplant clinical trials network toxicity committee consensus summary: thrombotic microangiopathy after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2005; 11: 571–575.
Jodele S, Davies SM, Lane A, Khoury J, Dandoy C, Goebel J et al. Diagnostic and risk criteria for HSCT-associated thrombotic microangiopathy: a study in children and young adults. Blood 2014; 124: 645–653.
Fuge R, Bird JM, Fraser A, Hart D, Hunt L, Cornish JM et al. The clinical features, risk factors and outcome of thrombotic thrombocytopenic purpura occurring after bone marrow transplantation. Br J Haematol 2001; 113: 58–64.
Jodele S, Zhang K, Zou F, Laskin B, Dandoy CE, Myers KC et al. The genetic fingerprint of susceptibility for transplant-associated thrombotic microangiopathy. Blood 2016; 127: 989–996.
Laskin BL, Goebel J, Davies SM, Jodele S . Small vessels, big trouble in the kidneys and beyond: hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Blood 2011; 118: 1452–1462.
Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med 2013; 368: 2169–2181.
Rathbone J, Kaltenthaler E, Richards A, Tappenden P, Bessey A, Cantrell A . A systematic review of eculizumab for atypical haemolytic uraemic syndrome (aHUS). BMJ Open 2013; 3: e003573.
Jodele S, Dandoy CE, Danziger-Isakov L, Myers KC, El-Bietar J, Nelson A et al. Terminal complement blockade after hematopoietic stem cell transplantation is safe without meningococcal vaccination. Biol Blood Marrow Transplant 2016; 22: 1337–1340.
Vasu S, Wu H, Satoskar A, Puto M, Roddy J, Blum W et al. Eculizumab therapy in adults with allogeneic hematopoietic cell transplant-associated thrombotic microangiopathy. Bone Marrow Transplant 2016; 51: 1241–1244.
Ruutu T, Barosi G, Benjamin RJ, Clark RE, George JN, Gratwohl A et al. Diagnostic criteria for hematopoietic stem cell transplant-associated microangiopathy: results of a consensus process by an International Working Group. Haematologica 2007; 92: 95–100.
Kennedy GA, Bleakley S, Butler J, Mudie K, Kearey N, Durrant S . Posttransplant thrombotic microangiopathy: sensitivity of proposed new diagnostic criteria. Transfusion 2009; 49: 1884–1889.
Cho BS, Yahng SA, Lee SE, Eom KS, Kim YJ, Kim HJ et al. Validation of recently proposed consensus criteria for thrombotic microangiopathy after allogeneic hematopoietic stem-cell transplantation. Transplantation 2010; 90: 918–926.
Ruutu T, Hermans J, Niederwieser D, Gratwohl A, Kiehl M, Volin L et al. Thrombotic thrombocytopenic purpura after allogeneic stem cell transplantation: a survey of the European Group for Blood and Marrow Transplantation (EBMT). Br J Haematol 2002; 118: 1112–1119.
Laskin BL, Goebel J, Davies SM, Khoury JC, Bleesing JJ, Mehta PA et al. Early clinical indicators of transplant-associated thrombotic microangiopathy in pediatric neuroblastoma patients undergoing auto-SCT. Bone Marrow Transplant 2011; 46: 682–689.
George JN, Li X, McMinn JR, Terrell DR, Vesely SK, Selby GB . Thrombotic thrombocytopenic purpura-hemolytic uremic syndrome following allogeneic HPC transplantation: a diagnostic dilemma. Transfusion 2004; 44: 294–304.
Daly AS, Hasegawa WS, Lipton JH, Messner HA, Kiss TL . Transplantation-associated thrombotic microangiopathy is associated with transplantation from unrelated donors, acute graft-versus-host disease and venoocclusive disease of the liver. Transfus Apher Sci 2002; 27: 3–12.
Powles RL, Clink HM, Spence D, Morgenstern G, Watson JG, Selby PJ et al. Cyclosporin A to prevent graft-versus-host disease in man after allogeneic bone-marrow transplantation. Lancet 1980; 1: 327–329.
Kaloyannidis P, Mallouri D, Hatziioannou K, Batsis I, Yannaki E, Papavasileiou P et al. Low body mass index is an independent risk factor for transplant-associated microangiopathy following total-body irradiation-based conditioning regimens. Biol Blood Marrow Transplant 2008; 14: 1076–1078.
Mii A, Shimizu A, Kaneko T, Fujita E, Fukui M, Fujino T et al. Renal thrombotic microangiopathy associated with chronic graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Pathol Int 2011; 61: 518–527.
Cutler C, Henry NL, Magee C, Li S, Kim HT, Alyea E et al. Sirolimus and thrombotic microangiopathy after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2005; 11: 551–557.
Shayani S, Palmer J, Stiller T, Liu X, Thomas SH, Khuu T et al. Thrombotic microangiopathy associated with sirolimus level after allogeneic hematopoietic cell transplantation with tacrolimus/sirolimus-based graft-versus-host disease prophylaxis. Biol Blood Marrow Transplant 2013; 19: 298–304.
Dezern AE, Brodsky RA . Clinical management of aplastic anemia. Expert Rev Hematol 2011; 4: 221–230.
Jodele S, Fukuda T, Vinks A, Mizuno K, Laskin BL, Goebel J et al. Eculizumab therapy in children with severe hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Biol Blood Marrow Transplant 2014; 20: 518–525.
Kanamori H, Takaishi Y, Takabayashi M, Tanaka M, Yamaji S, Tomita N et al. Clinical significance of fragmented red cells after allogeneic bone marrow transplantation. Int J Hematol 2003; 77: 180–184.
Martinez MT, Bucher C, Stussi G, Heim D, Buser A, Tsakiris DA et al. Transplant-associated microangiopathy (TAM) in recipients of allogeneic hematopoietic stem cell transplants. Bone Marrow Transplant 2005; 36: 993–1000.
Cataland SR, Wu HM . Diagnosis and management of complement mediated thrombotic microangiopathies. Blood Rev 2014; 28: 67–74.
Rock GA, Shumak KH, Buskard NA, Blanchette VS, Kelton JG, Nair RC et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med 1991; 325: 393–397.
Kentouche K, Zintl F, Angerhaus D, Fuchs D, Hermann J, Schneppenheim R et al. von Willebrand factor-cleaving protease (ADAMTS13) in the course of stem cell transplantation. Semin Thromb Hemost 2006; 32: 98–104.
Peyvandi F, Siboni SM, Lambertenghi Deliliers D, Lavoretano S, De Fazio N, Moroni B et al. Prospective study on the behaviour of the metalloprotease ADAMTS13 and of von Willebrand factor after bone marrow transplantation. Br J Haematol 2006; 134: 187–195.
Sarkodee-Adoo C, Sotirescu D, Sensenbrenner L, Rapoport AP, Cottler-Fox M, Tricot G et al. Thrombotic microangiopathy in blood and marrow transplant patients receiving tacrolimus or cyclosporine A. Transfusion 2003; 43: 78–84.
Maga TK, Nishimura CJ, Weaver AE, Frees KL, Smith RJ . Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome. Hum Mutat 2010; 31: E1445–E1460.
Frimat M, Tabarin F, Dimitrov JD, Poitou C, Halbwachs-Mecarelli L, Fremeaux-Bacchi V et al. Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood 2013; 122: 282–292.
Kavanagh D, Goodship T . Genetics and complement in atypical HUS. Pediatr Nephrol 2010; 25: 2431–2442.
Jodele S, Licht C, Goebel J, Dixon BP, Zhang K, Sivakumaran TA et al. Abnormalities in the alternative pathway of complement in children with hematopoietic stem cell transplant-associated thrombotic microangiopathy. Blood 2013; 122: 2003–2007.
Cataland SR, Holers VM, Geyer S, Yang S, Wu HM . Biomarkers of terminal complement activation confirm the diagnosis of aHUS and differentiate aHUS from TTP. Blood 2014; 123: 3733–3738.
Ham TH, Dingle JH . Studies on destruction of red blood cells. II. Chronic hemolytic anemia with paroxysmal nocturnal hemoglobinuria: certain immunological aspects of the hemolytic mechanism with special reference to serum complement. J Clin Invest 1939; 18: 657–672.
Gavriilaki E, Yuan X, Ye Z, Ambinder AJ, Shanbhag SP, Streiff MB et al. Modified Ham test for atypical hemolytic uremic syndrome. Blood 2015; 125: 3637–3646.
Brady TM, Pruette C, Loeffler LF, Weidemann D, Strouse JJ, Gavriilaki E et al. Typical hus: evidence of acute phase complement activation from a daycare outbreak. J Clin Exp Nephrol 2016; 1: 1337–1340.
Vaught AJ, Gavriilaki E, Hueppchen N, Blakemore K, Yuan X, Seifert SM et al. Direct evidence of complement activation in HELLP syndrome: A link to atypical hemolytic uremic syndrome. Exp Hematol 2016; 44: 390–398.
Gavriilaki EI, Imus P, Yuan X, Baines A, Jones R, Brodsky RA . Evidence of complement dysregulation in transplant-associated thrombotic microangiopathy. Haematologica 2016; 101 (suppl 1): 107 (P327).
Sahin G, Akay OM, Bal C, Yalcin AU, Gulbas Z . The effect of calcineurin inhibitors on endothelial and platelet function in renal transplant patients. Clin Nephrol 2011; 76: 218–225.
Rodriguez R, Nakamura R, Palmer JM, Parker P, Shayani S, Nademanee A et al. A phase II pilot study of tacrolimus/sirolimus GVHD prophylaxis for sibling donor hematopoietic stem cell transplantation using 3 conditioning regimens. Blood 2010; 115: 1098–1105.
Brown Z, Neild GH . Cyclosporine inhibits prostacyclin production by cultured human endothelial cells. Transplant Proc 1987; 19 (1 Pt 2): 1178–1180.
Garcia-Maldonado M, Kaufman CE, Comp PC . Decrease in endothelial cell-dependent protein C activation induced by thrombomodulin by treatment with cyclosporine. Transplantation 1991; 51: 701–705.
Burke GW, Ciancio G, Cirocco R, Markou M, Olson L, Contreras N et al. Microangiopathy in kidney and simultaneous pancreas/kidney recipients treated with tacrolimus: evidence of endothelin and cytokine involvement. Transplantation 1999; 68: 1336–1342.
Macunluoglu B, Atakan A, Gokce I, Ari E, Tulunay A, Demiralp E et al. Effects of rapamycin and tacrolimus on mature endothelial cells and endothelial progenitor cells. J Pak Med Assoc 2012; 62: 822–825.
Hwang EA, Kim HS, Ha E, Mun KC . Apoptosis in endothelial cells by cyclosporine. Transplant Proc 2012; 44: 982–984.
Ha E, Mun KC . Effects of cyclosporine on metalloproteinase in endothelial cells. Transplant Proc 2012; 44: 991–992.
Rafiee P, Johnson CP, Li MS, Ogawa H, Heidemann J, Fisher PJ et al. Cyclosporine A enhances leukocyte binding by human intestinal microvascular endothelial cells through inhibition of p38 MAPK and iNOS. Paradoxical proinflammatory effect on the microvascular endothelium. J Biol Chem 2002; 277: 35605–35615.
Ikezoe T, Yang J, Nishioka C, Honda G, Furihata M, Yokoyama A . Thrombomodulin protects endothelial cells from a calcineurin inhibitor-induced cytotoxicity by upregulation of extracellular signal-regulated kinase/myeloid leukemia cell-1 signaling. Arterioscler Thromb Vasc Biol 2012; 32: 2259–2270.
Carmona A, Diaz-Ricart M, Palomo M, Molina P, Pino M, Rovira M et al. Distinct deleterious effects of cyclosporine and tacrolimus and combined tacrolimus-sirolimus on endothelial cells: protective effect of defibrotide. Biol Blood Marrow Transplant 2013; 19: 1439–1445.
Dumler JS, Beschorner WE, Farmer ER, Di Gennaro KA, Saral R, Santos GW . Endothelial-cell injury in cutaneous acute graft-versus-host disease. Am J Pathol 1989; 135: 1097–1103.
Holler E, Kolb HJ, Hiller E, Mraz W, Lehmacher W, Gleixner B et al. Microangiopathy in patients on cyclosporine prophylaxis who developed acute graft-versus-host disease after HLA-identical bone marrow transplantation. Blood 1989; 73: 2018–2024.
Biedermann BC, Sahner S, Gregor M, Tsakiris DA, Jeanneret C, Pober JS et al. Endothelial injury mediated by cytotoxic T lymphocytes and loss of microvessels in chronic graft versus host disease. Lancet 2002; 359: 2078–2083.
Luft T, Dietrich S, Falk C, Conzelmann M, Hess M, Benner A et al. Steroid-refractory GVHD: T-cell attack within a vulnerable endothelial system. Blood 2011; 118: 1685–1692.
Dietrich S, Falk CS, Benner A, Karamustafa S, Hahn E, Andrulis M et al. Endothelial vulnerability and endothelial damage are associated with risk of graft-versus-host disease and response to steroid treatment. Biol Blood Marrow Transplant 2013; 19: 22–27.
Schmid PM, Bouazzaoui A, Doser K, Schmid K, Hoffmann P, Schroeder JA et al. Endothelial dysfunction and altered mechanical and structural properties of resistance arteries in a murine model of graft-versus-host disease. Biol Blood Marrow Transplant 2014; 20: 1493–1500.
Schmid PM, Bouazzaoui A, Schmid K, Birner CM, Schach C, Maier LS et al. Vascular alterations in a murine model of acute graft-versus-host disease are associated with decreased serum levels of adiponectin and an increased activity and vascular expression of indoleamine 2,3-dioxygenase. Cell Transplant 2016; 25: 2051–2062.
Ikezoe T, Yang J, Nishioka C, Yokoyama A . Thrombomodulin alleviates murine GVHD in association with an increase in the proportion of regulatory T cells in the spleen. Bone Marrow Transplant 2015; 50: 113–120.
Arai Y, Yamashita K, Mizugishi K, Watanabe T, Sakamoto S, Kitano T et al. Serum neutrophil extracellular trap levels predict thrombotic microangiopathy after allogeneic stem cell transplantation. Biol Blood Marrow Transplant 2013; 19: 1683–1689.
Bell WR, Braine HG, Ness PM, Kickler TS . Improved survival in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Clinical experience in 108 patients. N Engl J Med 1991; 325: 398–403.
Jodele S, Bleesing JJ, Mehta PA, Filipovich AH, Laskin BL, Goebel J et al. Successful early intervention for hyperacute transplant-associated thrombotic microangiopathy following pediatric hematopoietic stem cell transplantation. Pediatr Transplant 2012; 16: E39–E42.
Daly AS, Xenocostas A, Lipton JH . Transplantation-associated thrombotic microangiopathy: twenty-two years later. Bone Marrow Transplant 2002; 30: 709–715.
George JN, Terrell DR, Vesely SK, Kremer Hovinga JA, Lammle B . Thrombotic microangiopathic syndromes associated with drugs, HIV infection, hematopoietic stem cell transplantation and cancer. Presse Med 2012; 41 (3 Pt 2): e177–e188.
Iacopino P, Pucci G, Arcese W, Bosi A, Falda M, Locatelli F et al. Severe thrombotic microangiopathy: an infrequent complication of bone marrow transplantation. Gruppo Italiano Trapianto Midollo Osseo (GITMO). Bone Marrow Transplant 1999; 24: 47–51.
Jodele S, Laskin BL, Goebel J, Khoury JC, Pinkard SL, Carey PM et al. Does early initiation of therapeutic plasma exchange improve outcome in pediatric stem cell transplant-associated thrombotic microangiopathy? Transfusion 2013; 53: 661–667.
Sarode R, McFarland JG, Flomenberg N, Casper JT, Cohen EP, Drobyski WR et al. Therapeutic plasma exchange does not appear to be effective in the management of thrombotic thrombocytopenic purpura/hemolytic uremic syndrome following bone marrow transplantation. Bone Marrow Transplant 1995; 16: 271–275.
Mulay S, Kreuter JD, Bryant SC, Elliott MA, Hogan WJ, Winters JL et al. Outcomes of plasma exchange in patients with transplant-associated thrombotic microangiopathy based on time of presentation since transplant. J Clin Apher 2015; 30: 147–153.
Clark WF, Rock G, Barth D, Arnold DM, Webert KE, Yenson PR et al. A phase-II sequential case-series study of all patients presenting to four plasma exchange centres with presumed relapsed/refractory thrombotic thrombocytopenic purpura treated with rituximab. Br J Haematol 2015; 170: 208–217.
Au WY, Ma ES, Lee TL, Ha SY, Fung AT, Lie AK et al. Successful treatment of thrombotic microangiopathy after haematopoietic stem cell transplantation with rituximab. Br J Haematol 2007; 137: 475–478.
Vischini G, Cudillo L, Ferrannini M, Di Daniele N, Cerretti R, Arcese W . Rituximab in post allogeneic hematopoietic stem cell transplantation membranous nephropathy: a case report. J Nephrol 2009; 22: 160–163.
Uderzo C, Fumagalli M, De Lorenzo P, Busca A, Vassallo E, Bonanomi S et al. Impact of thrombotic thrombocytopenic purpura on leukemic children undergoing bone marrow transplantation. Bone Marrow Transplant 2000; 26: 1005–1009.
Yeates L, Slatter MA, Bonanomi S, Lim FL, Ong SY, Dalissier A et al. Use of defibrotide to treat transplant-associated thrombotic microangiopathy: a retrospective study of the Paediatric Diseases and Inborn Errors Working Parties of the European Society of Blood and Marrow Transplantation. Bone Marrow Transplant (e-pub ahead of print 16 January 2017; doi:10.1038/bmt.2016.351).
Wolff D, Wilhelm S, Hahn J, Gentilini C, Hilgendorf I, Steiner B et al. Replacement of calcineurin inhibitors with daclizumab in patients with transplantation-associated microangiopathy or renal insufficiency associated with graft-versus-host disease. Bone Marrow Transplant 2006; 38: 445–451.
de Fontbrune FS, Galambrun C, Sirvent A, Huynh A, Faguer S, Nguyen S et al. Use of eculizumab in patients with allogeneic stem cell transplant-associated thrombotic microangiopathy: a study from the SFGM-TC. Transplantation 2015; 99: 1953–1959.
Sevindik OG, Alacacioglu I, Katgi A, Solmaz SM, Acar C, Piskin O et al. Renal and neurological response with eculizumab in a patient with transplant associated thrombotic microangiopathy after allogeneic hematopoietic progenitor cell transplantation. Case Rep Hematol 2015; 2015: 425410.
Fernandez C, Lario A, Fores R, Cabrera R . Eculizumab treatment in a patient with hematopoietic stem cell transplantation-associated thrombotic microangiopathy and steroid-refractory acute graft versus host disease. Hematol Rep 2015; 7: 6107.
Jodele S, Fukuda T, Mizuno K, Vinks AA, Laskin BL, Goebel J et al. Variable eculizumab clearance requires pharmacodynamic monitoring to optimize therapy for thrombotic microangiopathy after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2016; 22: 307–315.
Risitano AM, Notaro R, Pascariello C, Sica M, del Vecchio L, Horvath CJ et al. The complement receptor 2/factor H fusion protein TT30 protects paroxysmal nocturnal hemoglobinuria erythrocytes from complement-mediated hemolysis and C3 fragment. Blood 2012; 119: 6307–6316.
Risitano AM, Ricklin D, Huang Y, Reis ES, Chen H, Ricci P et al. Peptide inhibitors of C3 activation as a novel strategy of complement inhibition for the treatment of paroxysmal nocturnal hemoglobinuria. Blood 2014; 123: 2094–2101.
DeZern AE, Uknis M, Yuan X, Mukhina GL, Varela J, Saye J et al. Complement blockade with a C1 esterase inhibitor in paroxysmal nocturnal hemoglobinuria. Exp Hematol 2014; 42: 857–61.e1.
Yuan X, Gavriilaki E, Thanassi JA, Yang G, Baines AC, Podos SD et al. Small-molecule Factor D inhibitors selectively block the alternative pathway of complement in paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. Haematologica (e-pub ahead of print 3 November 2016; doi:10.3324/haematol.2016.153312).
Acknowledgements
This work was supported by a grant from the Aplastic Anemia and MDS International Foundation and R01HL133113 (to RAB). EG is supported by the European Hematology Association Clinical Research Grant.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
RAB is a member of the Scientific Advisory Board of Achillion Pharmaceuticals, Alexion Pharmaceuticals and Apellis Pharmaceuticals. The remaining authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Gavriilaki, E., Sakellari, I., Anagnostopoulos, A. et al. Transplant-associated thrombotic microangiopathy: opening Pandora’s box. Bone Marrow Transplant 52, 1355–1360 (2017). https://doi.org/10.1038/bmt.2017.39
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bmt.2017.39
- Springer Nature Limited
This article is cited by
-
Development and implementation of evidence-based, nurse-leading early warning model and healthcare quality improvement project for transplant-associated thrombotic microangiopathy: a mixed-methods, before-and-after study
BMC Nursing (2024)
-
Hemostasis and complement in allogeneic hematopoietic stem cell transplantation: clinical significance of two interactive systems
Bone Marrow Transplantation (2024)
-
Eculizumab treatment in paediatric patients diagnosed with aHUS after haematopoietic stem cell transplantation: a HSCT-TMA case series from Japanese aHUS post-marketing surveillance
Bone Marrow Transplantation (2024)
-
Clinical characteristics of membranous nephropathy after allogeneic hematopoietic stem cell transplantation: A real-world multicenter study
Annals of Hematology (2024)
-
Treatment outcome and efficacy of therapeutic plasma exchange for transplant-associated thrombotic microangiopathy in a large real-world cohort study
Bone Marrow Transplantation (2022)