
Abstract
Many neurodegenerative diseases are characterized by impairment of protein quality control mechanisms in neuronal cells. Ineffective clearance of misfolded proteins by the proteasome, autophagy pathways and exocytosis leads to accumulation of toxic protein oligomers and aggregates in neurons. Toxic protein species affect various cellular functions resulting in the development of a spectrum of different neurodegenerative proteinopathies, including Huntington's disease (HD). Playing an integral role in proteostasis, dysfunction of the ubiquitylation system in HD is progressive and multi-faceted with numerous biochemical pathways affected, in particular, the ubiquitin-proteasome system and autophagy routes for protein aggregate degradation. Unravelling the molecular mechanisms involved in HD pathogenesis of proteostasis provides new insight in disease progression in HD as well as possible therapeutic avenues. Recent developments of potential therapeutics are discussed in this review.
Similar content being viewed by others


Huntington's disease is an incurable, inherited neuro-degenerative disorder
Huntington's disease (HD) is a devastating inherited neurodegenerative disorder resulting in a diverse range of behavioural, cognitive and physical symptoms. These symptoms are the results of selective neurodegeneration that occurs preferentially in the striatum1. The incidence of HD in different populations around the world varied with epidemiological reports finding between 0.42 to 17.2 cases per 100 0002. Clinical diagnosis of motor onset typically occurs in the prime of adult life, and symptoms progress inexorably, leading to chronic deterioration of patient health. The median timeframe from the manifestation of motor function symptoms to patient death is approximately 18 years3. No cures are currently available.
HD is an autosomal dominant disease caused by a CAG trinucleotide repeat expansion in exon 1 of the huntingtin gene (HTT)4. In healthy individuals, HTT has a CAG tract length of approximately 18 repeats5. Repeat lengths above a critical threshold of 35 CAG triplets in HTT are defined as disease-causing alleles6. At above 40 repeats, the disease is considered highly penetrant and an increased CAG repeat length is correlated with the decreased age of symptom onset as well as the increased rate of pathogenesis7. The CAG repeat in HTT, translates to an expansion of the polyglutamine (polyQ) tract at the N-terminus of huntingtin (HTT). The polyQ expansion mutation is often ascribed as giving a toxic gain-of-function phenotype in HD. PolyQ expanded HTT (hereinafter referred to as mutant HTT, mHTT) is attributed to cytotoxicity and biochemical dysfunction observed in HD models and patients. Significant impairment of the proteostasis network8, dysregulated transcription9, mitochondrial toxicity10,11, cellular energy imbalance12, synaptic dysfunction13 and axonal transport impairment11 are thought to result from aberrant forms of the HTT protein.
Mutant huntingtin protein is polyglutamine expanded
HTT is a 348 kDa protein predicted to be composed almost exclusively of α-helices, organised into namesake HEAT (Huntingtin, elongation factor 3 (EF3), protein phosphatase 2A (PP2A), and the yeast kinase TOR1) repeats14. HEAT repeats are interspersed with unstructured regions, often containing cleavage sites for proteolytic fragmentation as well as post-translational modifications15,16, thought to be responsible for large-scale conformational changes of the protein as well as alterations in protein-protein interaction networks17.
Cells18, model organisms19 and patients20 expressing CAG expanded versions of HTT can generate large protein clusters, fibrils and inclusions, some of which are large enough to be visualised by light microscopy and are composed of 100 000s of HTT molecules20,21. Aggregation has consistently been shown to be dependent on the polyQ expansion of HTT in a range of different experimental systems and environments22,23,24. However, low resolution (> 30 Å) negative stain electron microscopy (EM) data of recombinant full-length HTT protein reveals a globular spherical structure25 with no significant differences between Q23 (general population) and Q46 or Q78 (disease population) HTT EM envelopes resolved at this resolution, suggesting aggregation of mHTT is a complex process requiring specific cellular context. Our unpublished but higher resolution negative stain and cryo-EM data, both at around 15 Å resolution, reveals a curved architecture with a central cavity which varies in size dependent on sample preparation methods, in particular the concentration of cross-linking reagent used in the gradient fixation ultracentrifugation step, but not due to polyQ length26. Beyond this, limited data are available with respect to the structure of the HTT protein. Thus it remains unclear precisely how the polyQ expansion might affect the HTT structure or HTT propensity to form higher order oligomers.
Aggregate species are present in many flavours in various HD models and post-mortem tissue samples although which species of aggregates are damaging or protective to cells remains controversial in the field8,18. The function of HTT, in either the wild type or the disease state, is still poorly understood. It is thought that HTT acts as a scaffolding protein27,28, involved in many protein-protein interactions and the formation of multi-protein complexes. Dysregulation of this interaction network by the polyQ expansion and subsequent aggregation is thought to be responsible for the resultant phenotype29,30, sometimes described as a gain-of-function. Most published work to date on HTT oligomers and their associated neuropathology has focussed on the so-called exon 1 proteins31,32. HTT is cleaved by a variety of caspases, calpains and endopeptidases to yield a variety of N-terminal fragments, including a short sequence encoding exon 1 of the protein, corresponding to the first 90 amino acids of HTT31,32. Exon 1 is comprised of the N-terminal 17 amino acids (N17), the polyQ tract and then a 51-residue proline-rich domain (PRD). The structure of N17 is α-helical and the exon 1 protein adopts a condensed, disordered state at high concentrations33. The exon 1 structure is altered by polyQ expansion although there is no consensus on which morphology of exon 1 represents the toxic aggregate species. Whilst exon 1 HTT protein fragments have been shown to be a degradation product in many HD models and their aggregation underlies disease pathology34,35, larger fragments and full-length HTT are almost certainly involved in this oligomerisation process in human patients36 with post-translational modification, particularly phosphorylation, of HTT playing a crucial role in aggregation and toxicity37. To date, the precise pathogenic mechanism which renders the HTT protein functional with a polyQ tract less than 35 glutamines, but devastatingly damaging above a threshold of 40 glutamines, and which HTT species give rise to this breach in polyQ threshold, remains a fundamental yet unanswered question in the HD field.
Cellular proteostasis is disrupted in a number of neuro-degenerative disorders
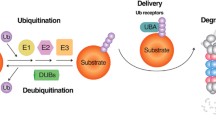
Disruption of protein homeostasis, or proteostasis, is a hallmark of many neurodegenerative diseases, such as Alzheimer's disease and Parkinson's disease as well as HD. Proteostasis is the precise balance of protein expression in cells at the correct concentrations, in the correct localisation and with the appropriate conformation38. Central to proteostasis is the post-translational modification of proteins, by covalent attachment of ubiquitin, ie, ubiquitylation (Figure 1)39. Ubiquitylation is catalysed by a three-enzyme cascade comprised of a ubiquitin activating enzyme (E1), a conjugating enzyme (E2) and a ligase (E3). E3 ligases are believed to be key in substrate recognition. Several E3 ligases have been reported to ubiquitylate HTT, including WWP140, UBE3A41, HACE142, CHIP43,44, Hrd145 and Parkin46 in mammals and Ltn145,47 in yeast. Conjugated ubiquitin can be further modified at one of its seven lysine residues (K6, K11, K27, K29, K33, K48 and K63) as well as the N-terminal methionine (M1) to form polyubiquitin chains of different linkages48. The covalently attached ubiquitin or ubiquitin chain can be removed by a family of enzymes called deubiquitylases (DUBs). Surprisingly, few DUBs have been reported to deubiquitylate HTT. ATXN3, a DUB that itself contains a polyQ tract, and USP19 are the only known DUBs to target HTT49,50,51 besides UPS. Monoubiquitylation or ubiquitin conjugation of proteins by different linkages of chains are recognised by a range of proteins containing ubiquitin binding domains (UBDs) or ubiquitin-interacting motifs (UIMs) and are essential in numerous cell signalling pathways. Ubiquitylated proteins can be targeted for degradation via the ubiquitin proteasome system (UPS) or lysosome/autophagy pathways. Ubiquitin-like modification, such as sumoylation52, is also known to play critical role of HTT degradation.

Overview of the ubiquitin protein system. Target proteins are ubiquitylated at accessible lysine residues by a three-enzyme cascade comprised of a ubiquitin activating enzyme (E1), a conjugating enzyme (E2) and a ubiquitin ligase (E3). Different lysine residues of the tagged ubiquitin can be further polyubiquitylated. The covalently attached ubiquitin modifications can be removed by deubiquitylases (DUBs). Specific covalent linkages of ubiquitin are recognised by a range of proteins containing ubiquitin binding domains (UBDs) or ubiquitin-interacting motifs (UIMs). Examples of known HTT E2 and E3 enzymes as well as DUBs and UBDs are detailed.
A complete understanding of these pathways and how they may be altered in HD patients and disease models can illuminate possible strategies for increasing degradation of mutant HTT (mHTT) through therapeutic intervention to alleviate symptoms. This review aims to describe recent findings in this area, with a focus on ubiquitylation, as well as a discussion of the tractability to develop successful therapeutics for HD modulating these pathways.
Ubiquitin proteasome system function in HD is diminished
The ubiquitin proteasome system (UPS) preferably degrades substrate proteins tagged with K48-linkage polyubiquitin chain although monoubiquitylation was recently also found to target UPS53 besides its function in protein localization and signaling. Pathogenic forms of HTT can be labelled with ubiquitin cross-reacting antibodies20,21 implying that they may be substrates for proteasomal degradation. Inclusion bodies (IBs) containing mHTT can also be labelled with antibodies against proteasome components suggesting direct recruitment of the UPS to some HTT aggregates54. As UPS function is widely reported to be diminished in a variety of HD models55,56, it has been suggested that proteasome sequestration by HTT aggregates is responsible for changes in UPS activity. Disassembly of the proteasome into its substituent components which are then sequestered by HTT aggregates is also postulated as a mechanism to affect UPS activity57. However, studies investigating UPS activity with different proteasome assemblies in the presence of HTT IBs failed to detect significant changes in UPS activity58. The sequestration of proteasomes to ubiquitin conjugated IBs is likely dynamic and reversible59, and some studies show that proteasomes are still able to access substrates in the presence of mHTT species. Recombinantly expressed mHTT aggregates were found not to inhibit UPS function in vitro60 whilst extracted aggregate filaments do58, suggesting that perhaps the ubiquitylation of the aggregates in vivo plays a crucial role. It was postulated polyQ stretches may clog the proteasome as the peptide may not be cleaved by the UPS endopeptidase activity like normal substrates, which may prohibit the diffusion of the polyQ peptide out of the UPS after processing. Contradictory studies show polyQ fragments blocking the proteasome61,62 whilst other show complete degradation of HTT exon 163,64. As such, it is difficult to draw definitive conclusions on UPS activity modulation or a mechanism by which this might occur in HD models, particularly one which is an accurate representation of UPS function in HD patients.
Many of these studies take a simplified view of UPS activity assessing neither the complex E1, E2 and E3 ubiquitin-ligating processes that precede UPS degradation of HTT, nor the complex series of cell signalling pathways which may alter UPS function and processivity. Although there are only 8 subtypes of E1 and about 40 E2s encoded in the human genome, there are more than 600 E3 ligases reflecting the highly-specialised functions of this group of enzymes. In the case of HTT, a unique α-amine E2 enzyme UBE2W has been shown to act on the N-terminus of HTT65. UBE2W targets proteins with disordered N-termini including ATXN3, tau and RPB8 as well as mHTT. UBE2W deficiency correlates with increased mHTT solubility and reduced levels of mHTT insoluble aggregates indicating ubiquitylation by UBE2W plays a role in mHTT proteostasis. UBE3A, a K48-specific E3 ligase, has been shown to specifically target HTT fragments for ubiquitylation and degradation41,66. Overexpression of UBE3A promotes UPS degradation of mHTT and reduce K63-mediated ubiquitylation. Clearance of HTT by UBE3A-mediated ubiquitylation was shown to be age-dependent in HD knock-in mouse models, postulating a mechanism for the age-dependency of HD symptoms and their progression. Yet, the recent finding that both WWP140, an E3 ligase capable of forming K11-, K48- and K63-polyubiquitin chains, and ATXN3, a DUB that prefers K63-chain substrate, function in HTT ubiquitylation, suggests there may be formation of heterotypic polyubiquitin chains67 that will complicates the degradation of HTT by UPS.
HD modulates function of the chaperone and autophagy machinery
HD has been shown to modulate autophagy function to have both toxic and protective effects on cells. Recognition of cargo by autophagosomes68 as well as subsequent axonal transport and substrate degradation69 are all diminished in HD. Rhes GTPase is required for autophagy as it interacts with Beclin-1 to reduce the inhibitory binding of Beclin-1 and Bcl-2 but is sequestered and inactivated by binding to mHTT70. This reduces autophagy function in the striatum where Rhes is expressed, a key tissue affected by HD. Conversely, mHTT also sequesters negative regulator of autophagy mTOR (mammalian target of rapamycin) inducing a higher rate of flux of autophagy and protecting against mHTT cytotoxicity71. mTOR phosphorylates autophagy-initiating kinase ULK1 at Ser757, preventing its interaction and subsequent phosphorylation by AMPK at Ser317 and Ser777, which activate ULK172. This coordinated regulation by phosphorylation is linked to the nutrient status of the cell with mTOR signalling prevailing under conditions of nutrient sufficiency and AMPK signalling in conditions of glucose starvation. Autophagy induction has been shown to relieve cellular toxicity posed by mHTT in the absence of UPS activity73,74. This study achieved increased autophagic flux through the overexpression of HDAC6, a cytosolic deacetylase which intersects both the autophagy and the UPS pathways and promotes aggresomal clearance through tubulin deacetylation (Figure 2). A monomeric or low oligomeric mHTT conformation recognisable by polyQ antibody 3B5H10 was recently shown to be resistant to autophagy, likely due to the lack of K63 polyubiquitylation and its interaction with autophagy receptor SQSTM1, aka. p62, which is essential for cargo recognition and autophagic clearance mediated by the HDAC6 pathway. This might explain why this particular conformation is so cytotoxic75,76.

Degradation pathways for mHTT. mHTT misfolding can lead to subsequent aggregation and ubiquitylation. Smaller ubiquitylated aggregates may be cleared by the proteasome, whereas larger aggregates which form aggresomes or inclusion bodies will be degraded via autophagic routes.
Approximately 90 chaperones and 250 co-chaperones comprise the human chaperome, a class of proteins categorised into families by their dependency on ATP as well as their molecular weight, eg, HSP40, HSP70 and HSP90 representing proteins of 40, 70 and 90 kDa, respectively. Chaperones function to promote proper folding and localisation of proteins and function in response to stresses which might disrupt protein structure or compartmentalisation in cells. Chaperones prevent aberrant protein interactions to hinder protein aggregation and the formation of inclusion bodies or other aggregate species but can also promote degradation of damaged or misfolded proteins. Co-chaperones such as E3 ligases parkin, CHIP and nucleotide exchange factor BAG3 have varied roles in assisting the proteostatic functions of chaperones.
HSP70 and HSP90 play central roles in protein quality control with HSP90 stabilising client proteins and inhibiting their ubiquitylation whilst conversely HSP70 promotes CHIP-dependent ubiquitylation and proteasomal degradation77. HSP90 has been shown to co-immunoprecipitate (co-IP) with both wildtype and polyQ expanded HTT, together with chaperone p2378. Full-length HTT has been shown to be a client of HSP90, and direct binding of HSP90 to exon 1 at N17 was recently demonstrated51. This study also showed that USP19 may be subsequently recruited to HSP90-HTT fragment complexes and can then deubiquitylate HTT, leading to accumulation of insoluble HTT aggregates containing HSP90. Similarly, HSP70 cross-reacting antibodies do bind HTT IBs79,80 suggesting chaperone sequestration by HTT aggregates and HSP70 over-expression can suppress aggregation and toxicity in some HD models80,81,82. Deletion of HSP70 alleles in a R6/2 HD mouse model increases IB size as well as having deleterious effects on the physical, behavioural and neuropathological measures83. CHIP and parkin are implicated as E3 ligases of mHTT degradation. Co-IP of mHTT with CHIP has also been shown to increase mutant exon 1 ubiquitylation and degradation44. HSP70 enhances mHTT's parkin binding and ubiquitylation in vitro84. Overall, it is likely mHTT is a HSP90 client protein and that it is regulated by HSP70/90-based chaperone machinery.
Bcl-2-associated athanogene 3 (BAG3) is a HSP70 co-chaperone which functions in concert with HSP70, HSPB8 and SQSTM1 to target aggregation-prone proteins for autophagic degradation85. BAG3 is crucial for HSPB8 activity in preventing mHTT aggregation, and together, BAG3-HSPB8 promote and facilitate the clearance of mHTT86. Two conserved Ile-Pro-Val motifs of BAG3 bind HSP chaperones, the subsequent interaction promotes chaperone activity towards HTT aggregates as well as their clearance87. BAG3 over-expression has anti-apoptotic effects and promotes the disposal of aggregation-prone proteins, both of which would have positive effects in the case of HD.
Therapeutic strategies for HD targeting ubiquitin and proteostasis pathways
A major focus area for disease-modifying therapeutic development in HD is to lower the levels of mHTT in the cell to alleviate neuropathology88. The monogenic inheritance of HD does favour therapeutic approaches which reduce expression of the mHTT allele by targeting DNA and RNA with gene silencing technologies89,90. Alternative efforts have focussed on lowering mHTT by increasing the degradation of mHTT through increased flux of UPS and autophagic pathways. The therapies discussed below are summarized in Table 1.
Therapeutic approaches in HD targeting UPS
Bortezomib is a well-characterised UPS inhibitor, which binds directly to the proteasome core particle to inhibit protein degradation, and is used in the clinic for treatment of various cancers91,92. Prolonged treatment of patients has been observed to result in peripheral neuropathy indicating the connection between UPS dysfunction and neurotoxicity91. Although direct activation of the UPS can be achieved through the overexpression of proteasome activator PA28 and this has been shown to be beneficial in some models of HD93, most UPS activating molecules act indirectly to increase UPS flux by modulating upstream cellular pathways. For example, sulforaphane increases both proteasome and autophagy activity levels in vivo by activating the Keap1-Nrf2-ARE pathways and inhibiting MAPK and NF-kB pathways, as shown in a number of different HD mouse models94,95. Sulforaphane was also shown to reduce quinolinic acid-induced mitochondrial dysfunction in rat striatum, conferring a neuroprotective effect96. Rolipram inhibits phosphodiesterase 4 (PDE4) thus activating protein kinase A to enhance proteasome activity indirectly and in a HD mouse model was shown to alleviate symptoms of neuronal dysfunction97. Recently, a series of compounds which enhance proteasome activity were identified using a UPS activity probe98,99 with p38 MAPK inhibitor PD169316 proving the most potent. Indirect activation of the UPS with these small molecules led to increased proteasome activity and aggregate clearance in a Parkinson's disease model. PD169316 is untested in an HD model, but p38 inhibition has been shown to be neuroprotective in HD100. Amiloride and its derivative benzamil are also able to rescue acid-sensing ion channel (ASIC)-dependent acidotoxicity which then inhibits UPS function in HD models. Benzamil treatment facilitates proteasomal degradation of mHTT in both cellular and mouse models of HD101. USP14 is a negative regulator of UPS through the deubiquitylation of UPS substrates. USP14 inhibition by IU1 also enhances proteasomal degradation102, but details of the effect of IU1 on HD models have yet to be described despite positive reports for upregulation of UPS activity in models of Alzheimer's disease103,104.
Therapeutic approaches in HD targeting autophagy
mTOR inhibitors with autophagy induction properties can be classified as ATP-competitive, eg, Torin1, or non-ATP-competitive, eg, rapamycin. ATP-competitive inhibitors face issues with toxicity due to their inhibition of mTORC1, mTORC2 and sometimes PI3K, so are usually confined to animal studies only. Rapamycin and its derivatives have been shown to improve HD phenotypes; in particular CCI-779 was shown to reduce mHTT aggregate load in HD mouse models71,105]. Trehalose acts to activate AMPK by inhibiting glucose transporters leading to glucose starvation conditions106,107, which induce autophagy to mitigate toxicity exhibited by mHTT in cell and mouse models108,109. Trehalose was also shown to reverse neurodegenerative phenotypes induced by UPS inhibition, in both normal and HD patient fibroblasts110. Rilmenidine is an mTOR-independent macroautophagy inducer and improves motor function and the clearance of mHTT in HD mouse models111. Lithium induces mTOR-independent autophagy through inhibition of inositol monophophatase therefore reducing inositol and inositol triphosphate (IP3) levels which would inhibit autophagy112. This has been shown to help clear mHTT in drosophila HD models113. Berberine has also shown efficacy in HD models, and is postulated to induce autophagy via AMPK activation114,115. Many other molecules including clonidine116, modulate cAMP or IP3 to induce autophagy and ameliorate phenotypes in HD animal models. Calpain has been described as a rational target for increasing autophagic flux of mHTT. RNAi knock-down of calpain in rodents showed reduced mHTT aggregate burden117 and similar results were observed in transgenic mice which overexpressed calpastatin (CAST), the endogenous inhibitor of calpain. Pramipexole is a dopamine receptor agonist which activates autophagy probably by modulating cAMP signally pathways in R6/1 HD mouse model, resulting in reduced levels of soluble mHTT118. Inhibition of PIP4Kγ by NCT-504 modulates phosphoinositide levels in HD patient fibroblasts and activates basal autophagy, thus reducing the mHTT levels119. An allosteric antagonist of metabotropic glutamate receptor, CTEP, also reduces mHTT aggregate levels in HD mouse models via GSK3β signalling induced autophagic flux120. Metformin is another AMPK activating inducer of autophagy which has been shown to alleviate mHTT associated cytotoxicity121,122. HD patients who are monitored in the Enroll-HD programme (a world-wide observational and longitudinal study of HD patients) who were already taking metformin to treat type II diabetes, were found to have improved cognitive status compared to control patients not on a metformin-regimen123. A great number of small molecules targeting these pathways have been described in the literature but have yet to show efficacy in vivo124. Combination therapies which simultaneously upregulate autophagy through both the mTOR-independent and -dependent pathways have been shown to have synergistic effects on alleviating mHTT associated toxicity. Cooperation has been demonstrated with trehalose-rapamycin108 as well as lithium-rapamycin105 combination treatments in HD models although deleterious side-effects are postulated to worsen in the long-term for these treatment regimens.
Therapeutic approaches in HD targeting chaperone proteins
Elevation of the levels of chaperones including HSP70 and HSP40 has been found to inhibit mHTT aggregation and be neuroprotective in a number of different HD cellular and animal models125,126. However, the tractability of developing small molecules which elevate HSP expression levels is likely limited. HSP90 inhibition, however, is an attractive strategy to treat proteinopathies given its ability to stabilise client proteins, of which mHTT is one78. It was shown that the co-IP of HSP90 and mHTT is abrogated in the presence of HSP90 inhibitors and that mHTT is ubiquitylated and then degraded in a dose-dependent manner with HSP90 inhibitor. HSP90 inhibitors also reduce mutant exon 1 aggregation and toxicity in a variety of HD models80,81,125 due to transcription factor heat-shock factor 1 (HSF1) mediated increases in chaperone expression127,128. HSP90 inhibitor geldanamycin competes with ATP for binding HSP90 and therefore inhibits the binding and stabilising of mHTT81. 17-DMAG and 17-AAG, less toxic geldanamycin derivatives with more favourable pharmacokinetic properties, were also shown to induce expression of molecular chaperones and inhibit mHTT aggregation in cells and Drosophila models of HD respectively125,128 as well as to cross the blood-brain barrier in Alzheimer's models129. HSP90 isoform-specific inhibitors were shown to have improved tolerability compared to pan-HSP90 inhibitors and were also shown to be orally available, cross the blood-brain barrier and reduce HTT levels in rat brains130. Celastrol, a natural product anti-inflammatory agent, binds the C-terminal domain of HSP90 and induces HSP70 expression and protects neurons from mHTT-mediated toxicity131. HSF-1 activating compounds were shown to suppress mHTT associated neurodegeneration in Drosophila HD models through induction of molecular chaperones128.
Proteolysis targeting chimera (PROTAC) can lower specific protein levels with small molecules
PROTAC-based approaches utilise bifunctional molecules to simultaneously bind both the protein of interest and a ubiquitin E3 ligase to promote the ubiquitylation and degradation of the target molecule132,133 by bringing the target protein and E3 ligase into proximity. Recently, a PROTAC-based series of molecules has been described for selective ubiquitylation and then degradation of mHTT via the UPS134. Hybrid molecules that link a ligand for cIAP1 (cellular inhibitor of apoptosis protein 1) E3 ligase to ligands for mHTT were shown to selectively reduce levels of mHTT through E3 ligase recruitment and proteasome degradation pathways in HD patient-derived fibroblasts. Two HD patient-derived fibroblast cell lines were used in this study expressing Q47 and Q68 mHTT, both obtained from biorepository Coriell135, showing that PROTAC-induced degradation could be achieved for adult-onset HD genotypes.
A benefit of the PROTAC approach is that tissue specificity may be exerted by developing molecules that recruit E3 ligases only expressed in a certain tissue. The reported HTT-PROTAC molecules used a bestatin moiety to target cIAP1 E3 ligase, which is expressed in many different tissues throughout the body although not quite as broadly as UBE3A, the HTT-specific E3 ligase66. Modifying these PROTAC molecules to bind a brain-specific E3 ligase, such as TRIM9136 or RNF182137 for example, may reduce systemic effect. It should also be noted that hijacking cIAP1 using bestatin can be problematic due to the fact that bestatin causes the degradation of cIAP1, which can in turn trigger apoptosis. Switching the linker of bestatin in the PROTACs from an ester to an amide reduces cIAP1 degradation in some systems evaluated although the mechanism is currently not understood138. Numerous PROTAC examples illustrate the importance of exploring different linker designs to connect the two functional groups whilst maintaining the desired target protein degradation. Further exploration is warranted for HTT-degrading PROTACs too139,140.
It should also be noted that whilst the described molecules bind and effectively clear mHTT in HD patient fibroblasts, their binding specificity to different HTT aggregate species or aggregates composed of different proteins requires further investigation. The aggregate binding portion of the molecules are composed of previously described ligands phenyldiazenyl benzothiazole (PDB) and benzothiazole-aniline (BTA), which are known to also target amyloid proteins in Alzheimer's disease models141,142. Given the heterogeneous and dynamic nature of HTT aggregate species at different stages of the disease progression, developing small molecules that can bind and target the disease relevant aggregate species for clearance will be challenging. Perhaps finding binders of the expanded polyQ stretch of HTT will instead confer necessary specificity. However, PDB and BTA have both been shown to cross the blood brain barrier in Alzheimer's studies although the pharmacokinetic properties will need to be evaluated as any change to the E3 ligase ligand or linker region of the molecules are made. Overall this study shows the first proof-of-principle HTT degradation by a PROTAC-based approach in HD patient tissues135.
To overcome the need to develop small molecules which potently and selectively bind mHTT aggregates, existing mHTT antibodies could be used via the “TRIM-Away” technology143. Cytosolic antibody-target protein complexes can be recognized by antibody receptor and E3 ligase TRIM21 which then directs the complexes for degradation via the UPS. Whilst a novel approach to targeted protein degradation in cells, this technology is currently only functional in exemplary cases in 2-D cell culture and has yet to be demonstrated on mHTT in known HD disease models. Similarly, when N17 binding HTT intrabody C4 sFv is tagged with the C-terminal PEST region of mouse ornithine decarboxylase, a proteasome directing motif, to target C4 sFv complexes for UPS degradation, HTT exon 1 fragments are significantly reduced in 2D striatal culture144. Whilst C4 sFv and other intrabodies have shown promise in ameliorating HTT aggregation and cytotoxicity145,146,147,148,149, directed and specific mHTT lowering in animal models has not been shown to date with an intrabody-based therapy.
HD genome-wide association studies implicate new, potential therapeutic targets in ubiquitin pathways
HD patient populations with the same CAG repeat length may experience symptom onset in a broad age range, implicating other disease modifying factors. Many recent genome-wide association studies (GWAS) have identified genetic modifiers for the age-of-onset of HD, beyond CAG repeat expansion length, as well as the rate of pathogenic progression. GWAS (GeM-HD consortium, 2015) identified, amongst other modifiers, UBR5 as genetic modifier for HD150. UBR5 is an E3 ubiquitin ligase with key roles in the regulation of the UPS. As discussed, mHTT is degraded by the UPS via E3 ligase UBE3A, an activity which is down-regulated by UBR5151. This finding in the GWAS implies that pharmacological down-regulation of UBR5 function would promote UPS degradation of mHTT. UBR5 is an attractive target in HD as it also regulates PEPCK1 acetylation, which plays a role in gluconeogenesis dysfunction in prodromal HD152. Additionally, ubiquitylation of ATMIN by UBR5 releases ATM kinase allowing activated ATM to recruit the MRN complex153; part of the DNA damage response pathway which is upregulated in HD. Increased p53 levels in HD promotes mHTT aggregation and UBR5 can upregulate p53 ubiquitin-independently by inhibiting ATM-mediated phosphorylation154. As UBR5 plays such a central multi-faceted role in HD phenotype, targeting its activity for inhibition with small molecules seems an obvious therapeutic avenue to be explored (Figure 3).

UBR5 is a genetic modifier of HD and plays a multifaceted role in the disease pathways. UBR5 is an E3 ubiquitin ligase with many roles in HD-related pathways. UBR5 can downregulate UBE3A-mediated degradation of mHTT, regulate PEPCK1 acetylation modulating gluconeogenesis in prodromal HD, ubiquitylate ATMIN to recruit the MRN complex for DNA damage response and upregulate p53 through ATM-phosphorylation inhibition affecting HTT aggregate formation. As UBR5 plays such a central multi-faceted role in HD phenotype, targeting its activity for inhibition with small molecules seems a therapeutic avenue to be explored.
Considerations and limitations of studying and targeting proteostasis in current studies
Elevating protein degradation rates by targeting autophagy and UPS activation has significant therapeutic potential in a range of neurodegenerative proteinopathies, including HD. However, it should be noted that clinical trials in HD have extremely low rates of success155 with many promising therapies failing to translate from the laboratory to the clinic. This is largely attributed to poor understanding of the basic biology of HD, for example, the function of HTT, either wildtype or polyQ expanded, is still largely unknown. Additionally, the disease models used to identify therapeutic targets and evaluate drug efficacy in preclinical research often have critical flaws or caveats in their representation of HD patient neurophysiology and disease progression. Thus, when considering potential avenues for future therapeutic development, it is important to evaluate the published findings carefully.
A key issue with many of the therapeutic reagents described within this review is that these are molecules which have neither defined in vitro and in vivo potency and selectivity for their target proteins nor defined mechanisms of action. Whilst promising phenotypes may be observed in disease models following a particular treatment, target engagement is often not validated, meaning that phenotypic changes observed may be due to polypharmacology: a combination of on- and off-target effects resulting from modulation of both target and peripheral pathways156. Molecules such as rapamycin, which is reported to be both potent and selective, has been observed to have IC50 values for mTORC1 which may range over many orders of magnitude dependent on the cell line or model being used and long-term low dose rapamycin treatment has also been observed to additionally antagonise mTORC2157,158. Therefore, therapeutic dose concentration and regimen should be carefully considered and evaluated to ensure that phenotypes observed from treatments are due to validated, selective target engagement. Ideally, high quality chemical probes for each of the protein targets in question should be used to ensure treatment responses are due to on target effects, are dose-dependent and have a defined mechanism of action159,160,161. The range of commercially available high quality chemical probes is expanding, and information regarding potency and selectively can easily be retrieved through resources such as the chemical probes portal http://www.chemicalprobes.org.
It is increasingly realized that target identification and validation are critical for the success of target-based drug discovery of oncology162 and neurological disorders163. In the case that high quality chemical tools are not available, we and our collaborative labs have developed highly specific and potent proteinaceous biological binder, ubiquitin variants, to interfere with the ubiquitin regulatory enzymes164,165 for target validation purpose. Use of such tool molecules will help identify and validate therapeutic targets in the HTT proteostatis pathway.
Given the progressive and age-dependent nature of HD symptoms, the time-point of targeting HTT with lowering therapies, either via UPS or autophagy upregulation, will likely be critical for the efficacy of any developed treatment. The preference for degradation of smaller, more soluble misfolded ubiquitylated proteins or aggregates by the UPS will mean that later time-points of disease progression will likely show minimal improvement with treatments targeting this pathway166. This is exemplified in the ability of rapamycin to ameliorate cognitive deficits in mouse models of Alzheimer's but only when treatment was given prior to the formation of plaques and tangles of the amyloid-beta protein167. To determine whether autophagy or UPS should be targeted for HTT clearance, it will be critical to analyse the pathway preference for different HTT aggregate species, ideally from studies using tissue relevant patient derived cells, throughout the progression of HD neurodegeneration. Additionally, in considerations of treatment regimen, it is probable that continuously switching on the protein degradation pathways is likely to yield deleterious effects in the cell due to the degradation of off-target substrates. It has been suggested that a pulsatile treatment regimen, likened to periodically removing the rubbish124, would be an effective strategy to allow maximal drug efficacy with reduced toxicity.
The HD models used in each of the studies detailed in this review, report data on HTT clearance from models which can be quite disparate. Some studies are limited to data collected in vitro, using cultured cells, whereas others see effects in vivo, which, in general, use mouse models. Which form of mHTT is overexpressed in these different models also varies. For example, the commonly used R6/2 HD mice express human HTT exon 1 with approximately 150 CAG repeats168, which, whilst sufficient to cause progressive neurodegenerative phenotypes in the mice in question, does not represent the same genetic insult characterised in patients and therefore is unlikely to lead to the same trajectory in disease progression in HD patients. Clearance of HTT aggregates formed solely of HTT exon1 Q150 is not representative of the situation in HD patients which have a smaller polyQ expansion and a heterogeneous mix of aggregate species. Whilst modulation of HTT clearance in the R6/2 mouse model may give indications of possible therapeutic strategies, observations should be made cautiously and ideally verified in alternative disease models and systems. Although other mouse models, such as YAC128 and BACHD do express full-length human HTT, which is 91% sequence identical to the mouse protein, and show progressive late-onset neurodegenerative symptoms, both use polyQ expansions in excess of 100 glutamines which is not representative of the HTT protein expressed in most HD patients169,170. Longer CAG expansions give more robust phenotypes in animal models, and longer polyQ expansions permits easier detection of mHTT compared to wildtype so larger CAG expansions have historically been preferred for study. However, as polyQ stretches might clog the UPS machinery and are regarded as the principal factor in HTT aggregate formation, expression of exaggerated polyQ-length HTT proteins may give rise to different phenotypes when observing the HTT clearance pathways and their modulation by therapeutics compared to the situation in HD patients. Whilst the monogenic nature of HD should simplify the generation of an accurate and disease representative mouse model, it is important to note that the discovery of therapeutics with efficacy in mouse models of HD has yet to translate to success in the clinic. Whilst this is a multi-factorial problem, it does suggest that there are critical differences in neurophysiology, neurodegeneration and ageing between rodents and humans which are deserving of further investigation155.
In recent years, HD patient-derived cell lines became routinely used for in vitro experiments and are available from data and cell repositories such as the National Institute of Neurological Disorders and Stroke (NINDS) which currently hosts 27 HD patient fibroblast cell lines and 23 HD patient induced pluripotent stem cell lines (iPSCs). This collection encompasses samples from both male and female subjects of ages 9–87 representing both control and HD patient samples. HTT gene CAG lengths represented include controls of Q17 to Q29 and HD patient samples spanning Q38 to Q180 although most are Q40 to Q50. The most commonly studied HD iPSC line to date has 60 CAG repeats putting it at the very high end of HD adult onset CAG expansions171. Despite cell lines with more representative repeats for adult onset HD being described, iPSCs with CAG repeats ranging from 43–60 CAGs are less extensively characterised. Perhaps similarly to animal models, this is due to the more robust phenotypes with larger CAG repeat expansions and the greater ease for detecting mHTT.
A number of protocols can be used to differentiate iPSCs into cells exhibiting neuronal features, often referred to as neural precursor cells (NPCs). The variety of methods of differentiation as well as the genetic background of each NPC line gives rise to different disease-associated phenotypes172 so findings derived from study in one NPC line should be viewed cautiously, and a field-wide consensus on a restricted number of phenotypes would improve comparative studies. Ideal experiments would also ensure a selection of NPCs derived from different HD patients as well as controls are differentiated by the same methods and tested in parallel to verify findings although this approach would be resource and time expensive. To date, limited work has been published on mHTT clearance in iPSC derived cells. One study showed that microRNA-196a can decrease levels of HTT aggregate in neuronal cultures differentiated from HD iPSCs, but the mechanism of action for this observed effect is unknown173. Replicating the findings of therapeutics detailed in this review in iPSCs would build confidence that such strategies are deserving of further development as HTT lowering therapies. One key caveat of HD iPSC derived cells for studying HD is that induced pluripotency is reported to reverse age-related phenotypes of the patient sample which could prove problematic given the progressive and age-dependent nature of HD. None-the-less, a mHTT aggregation and clearance study in HD iPSCs with adult-onset polyQ expansions would be helpful to verify and characterise the pathways to target for therapeutic development which are relevant to HD patients.
More recently, cerebral organoids colloquially referred to as “minibrains”, derived from patient iPSCs have been used in the studies of a number neurological disease areas, including Alzheimer's disease174 and Zika infection175. Many diseases of the brain are too complex to be accurately recapitulated in 2-D culture experiments which do not represent the circuitry and intercellular signalling of a 3-D human brain. Whilst minibrains do not replicate human brains, they do succeed in representing a brain-like model of some complexity which may be manipulated and probed by researchers to study their disease of interest. Whilst there are no published studies of the use of minibrains in HD, this would be an interesting future avenue for evaluation of HD phenotype recapitulation in these systems as well as therapeutic study.
Whilst there are obvious commonalities between different neurodegenerative proteinopathies, transfer of knowledge between different diseases should be done cautiously. For example, in the case of rapamycin treatment, HD transgenic mice show improved motor behaviours176 and similarly encouraging results are seen in Alzheimer's159 and Parkinson's177 models of disease. However, rapamycin treatment in an ALS SOD1 transgenic mouse model178 worsens autophagic functions and exacerbates the neurodegenerative phenotype. As such, extrapolation of findings from different diseases, or even different models of the same disease, should be done prudently.
Whilst some therapeutics described in this review are still under active investigation as exemplified by frequent and continuing publication of new findings, some approaches show large gaps or halts in the published literature. This suggests that perhaps findings could not be replicated by other groups or in other HD models studied with negative results not reaching traditional published literature outlets due to publication bias179. Therefore, the validity of certain therapeutics suggested in limited numbers of studies should be analysed and evaluated cautiously.
Conclusion
Alleviating symptoms of Huntington's disease through HTT lowering therapies remains an attractive avenue for therapeutic development and a major focus area for many HD researchers. In the absence of a complete understanding for the biological function of wildtype HTT, nor consensus on the gain-of-function phenotype described for expressed mHTT, eradicating mHTT from the affected tissues in HD patients remains a rational drug discovery strategy.
The monogenic nature of HD makes it an attractive candidate for therapies which might target transcription or translation of the mutant allele90. Current strategies in development include antisense oligonucleotides and RNA interference targeting of mRNA, zinc-finger transcriptional repressors of gene expression as well as more recently CRISPR-Cas9. Whilst these efforts are very promising, the resultant therapies from these approaches will likely be prohibitively expensive for many HD patients, and many currently suffer from issues of effective blood brain barrier penetration via oral or subcutaneous delivery. Recently described outcomes for the Phase 1/2a study for the IONIS-HTTRx antisense oligonucleotide therapy delivered by lumbar puncture indicate that such agents may reduce mutant HTT levels but whether this results in meaningful phenotypic changes in patients remains to be seen. Although small molecule based approaches such as those detailed in this review, may also suffer from ineffective blood brain barrier penetration, the possibility to develop brain penetrant drugs which may be more easily scaled for production is probably more achievable, making these approaches attractive still.
Recent advances permitting researchers greater access to HD patient derived cell lines should allow better scrutiny of therapeutics in disease relevant tissues prior to advancing in preclinical development. GWAS of HD patients have given insight into possible new targets for drug discovery and the recently highlighted UBR5 is one such example of an attractive candidate for inhibition given its multi-faceted role in HD. Developments in PROTAC-based approaches for drug design are making headway in other fields139,140 and represent a new avenue for researchers to explore for HTT lowering therapies. Despite its monogenic inheritance, intensive research into HD in the previous few decades has shown that HD is far from a simple pathology and it is probable that effective disease-modifying therapies are unlikely to be developed in the near future given the complexity of this neurodegenerative disease and our current limited understanding of the biology. However, with respect to HTT lowering therapies targeting the HTT protein itself, the recent advances described in this review should be cause for optimism in their ability to assist researchers in effective, productive and rational drug discovery.
References
Vonsattel JP, DiFiglia M . Huntington disease. J Neuropathol Exp Neurol 1998; 57: 369–84.
Kay C, Hayden MR, Leavitt BR . Chapter 3–Epidemiology of Huntington disease. In: Feigin AS, Anderson KE, editors. Handbook of Clinical Neurology. Elsevier; 2017. p31–46. Available from: http://www.sciencedirect.com/science/article/pii/B9780128018934000031
Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol 2014; 10: 204–16.
A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell 1993; 72: 971–83.
Kay C, Fisher ER, Hayden MR . Huntington's Disease Chapter 7 (eds Bates, G P, Tabrizi, S J & Jones, L). 4th ed. Oxford University Press; 2014.
Semaka A, Kay C, Doty C, Collins JA, Bijlsma EK, Richards F, et al. CAG size-specific risk estimates for intermediate allele repeat instability in Huntington disease. J Med Genet 2013; 50: 696–703.
Lee JM, Ramos EM, Lee JH, Gillis T, Mysore JS, Hayden MR, et al. CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology 2012; 78: 690–5.
Kim M, Lee HS, LaForet G, McIntyre C, Martin EJ, Chang P, et al. Mutant huntingtin expression in clonal striatal cells: dissociation of inclusion formation and neuronal survival by caspase inhibition. J Neurosci Off J Soc Neurosci 1999; 19: 964–73.
Seredenina T, Luthi-Carter R . What have we learned from gene expression profiles in Huntington's disease? Neurobiol Dis 2012; 45: 83–98.
Johri A, Chandra A, Beal MF . PGC-1α, mitochondrial dysfunction, and Huntington's disease. Free Radic Biol Med 2013; 62: 37–46.
Reddy PH, Shirendeb UP . Mutant huntingtin, abnormal mitochondrial dynamics, defective axonal transport of mitochondria, and selective synaptic degeneration in Huntington's disease. Biochim Biophys Acta 2012; 1822: 101–10.
Ju TC, Lin YS, Chern Y . Energy dysfunction in Huntington's disease: insights from PGC-1α, AMPK, and CKB. Cell Mol Life Sci CMLS 2012; 69: 4107–20.
Nithianantharajah J, Hannan AJ . Dysregulation of synaptic proteins, dendritic spine abnormalities and pathological plasticity of synapses as experience-dependent mediators of cognitive and psychiatric symptoms in Huntington's disease. Neuroscience 2013; 251: 66–74.
Andrade MA, Petosa C, O'Donoghue SI, Müller CW, Bork P . Comparison of ARM and HEAT protein repeats. J Mol Biol 2001; 309: 1–18.
Saudou F, Humbert S . The Biology of Huntingtin. Neuron 2016; 89: 910–26.
Ratovitski T, O'Meally RN, Jiang M, Chaerkady R, Chighladze E, Stewart JC, et al. Post-translational modifications (ptms), identified on endogenous Huntingtin, cluster within proteolytic domains between HEAT repeats. J Proteome Res 2017; 16: 2692–708.
DiGiovanni LF, Mocle AJ, Xia J, Truant R . Huntingtin N17 domain is a reactive oxygen species sensor regulating huntingtin phosphorylation and localization. Hum Mol Genet 2016; 25: 3937–45.
Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S . Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004; 431: 805–10.
Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 1997; 90: 537–48.
DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997; 277: 1990–3.
Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, et al. Nuclear and neuropil aggregates in huntington's disease: relationship to neuropathology. J Neurosci 1999; 19: 2522–34.
Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, et al. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo . Cell 1997; 90: 549–58.
Chen S, Berthelier V, Yang W, Wetzel R . Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J Mol Biol 2001; 311: 173–82.
Morley JF, Brignull HR, Weyers JJ, Morimoto RI . The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans . Proc Natl Acad Sci U S A 2002; 99: 10417–22.
Vijayvargia R, Epand R, Leitner A, Jung T-Y, Shin B, Jung R, et al. Huntingtin's spherical solenoid structure enables polyglutamine tract-dependent modulation of its structure and function. eLife 2016; 5: e11184.
Harding R, Deme J, Loppnau P, Ackloo S, Hutchinson A, Hunt B, et al. Pursuit of a high resolution structure of full-length huntingtin by cryo-electron microscopy: CHDI HD Therapeutics Conference 2017. 2017.
Zheng Z, Diamond MI . Huntington disease and the huntingtin protein. Prog Mol Biol Transl Sci 2012; 107: 189–214.
Rui YN, Xu Z, Patel B, Chen Z, Chen D, Tito A, et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat Cell Biol 2015; 17: 262–75.
Kalathur RKR, Pinto JP, Sahoo B, Chaurasia G, Futschik ME . HDNetDB: a molecular interaction database for network-oriented investigations into huntington's disease. Sci Rep 2017; 7: 5216.
Giorgini F . A flexible polyglutamine hinge opens new doors for understanding huntingtin function. Proc Natl Acad Sci U S A 2013; 110: 14516–7.
Trepte P, Strempel N, Wanker EE . Spontaneous self-assembly of pathogenic huntingtin exon 1 protein into amyloid structures. Essays Biochem 2014; 56: 167–80.
Thakur AK, Jayaraman M, Mishra R, Thakur M, Chellgren VM, Byeon IJ, et al. Polyglutamine disruption of the huntingtin exon1 N-terminus triggers a complex aggregation mechanism. Nat Struct Mol Biol 2009; 16: 380–9.
Chen M, Wolynes PG . Aggregation landscapes of Huntingtin exon 1 protein fragments and the critical repeat length for the onset of Huntington's disease. Proc Natl Acad Sci U S A 2017; 114: 4406–11.
Pieri L, Madiona K, Bousset L, Melki R . Fibrillar α-synuclein and Huntingtin exon 1 assemblies are toxic to the cells. Biophys J 2012; 102: 2894–905.
Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996; 87: 493–506.
Daldin M, Fodale V, Cariulo C, Azzollini L, Verani M, Martufi P, et al. Polyglutamine expansion affects huntingtin conformation in multiple Huntington's disease models. Sci Rep 2017; 7: 5070.
Atwal RS, Desmond CR, Caron N, Maiuri T, Xia J, Sipione S, et al. Kinase inhibitors modulate huntingtin cell localization and toxicity. Nat Chem Biol 2011; 7: 453–60.
Yerbury JJ, Ooi L, Dillin A, Saunders DN, Hatters DM, Beart PM, et al. Walking the tightrope: proteostasis and neurodegenerative disease. J Neurochem 2016; 137: 489–505.
Li W, Ye Y . Polyubiquitin chains: functions, structures, and mechanisms. Cell Mol Life Sci CMLS 2008; 65: 2397–406.
Lin L, Jin Z, Tan H, Xu Q, Peng T, Li H . Atypical ubiquitination by E3 ligase WWP1 inhibits the proteasome-mediated degradation of mutant huntingtin. Brain Res 2016; 1643: 103–12.
Bhat KP, Yan S, Wang CE, Li S, Li XJ . Differential ubiquitination and degradation of huntingtin fragments modulated by ubiquitin-protein ligase E3A. Proc Natl Acad Sci U S A 2014; 111: 5706–11.
Rotblat B, Southwell AL, Ehrnhoefer DE, Skotte NH, Metzler M, Franciosi S, et al. HACE1 reduces oxidative stress and mutant Huntingtin toxicity by promoting the NRF2 response. Proc Natl Acad Sci U S A 2014; 111: 3032–7.
Al-Ramahi I, Lam YC, Chen HK, de Gouyon B, Zhang M, Pérez AM, et al. CHIP protects from the neurotoxicity of expanded and wild-type ataxin-1 and promotes their ubiquitination and degradation. J Biol Chem 2006; 281: 26714–24.
Jana NR, Dikshit P, Goswami A, Kotliarova S, Murata S, Tanaka K, et al. Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J Biol Chem 2005; 280: 11635–40.
Yang H, Zhong X, Ballar P, Luo S, Shen Y, Rubinsztein DC, et al. Ubiquitin ligase Hrd1 enhances the degradation and suppresses the toxicity of polyglutamine-expanded huntingtin. Exp Cell Res 2007; 313: 538–50.
Rubio I, Rodríguez-Navarro JA, Tomás-Zapico C, Ruíz C, Casarejos MJ, Perucho J, et al. Effects of partial suppression of parkin on huntingtin mutant R6/1 mice. Brain Res 2009; 1281: 91–100.
Yang J, Hao X, Cao X, Liu B, Nyström T . Spatial sequestration and detoxification of Huntingtin by the ribosome quality control complex. eLife 2016; 5.
Komander D, Rape M . The ubiquitin code. Annu Rev Biochem 2012; 81: 203–29.
Tsou W-L, Ouyang M, Hosking RR, Sutton JR, Blount JR, Burr AA, et al. The deubiquitinase ataxin-3 requires Rad23 and DnaJ-1 for its neuroprotective role in Drosophila melanogaster. Neurobiol Dis 2015; 82: 12–21.
Eckland DJ, Lightman SL . The TSH, T4, T3 and prolactin responses to consecutive infusions of a potent and stabilized thyrotrophin releasing hormone analogue, RX77368, in man. Eur J Clin Invest 1988; 18: 405–9.
He WT, Xue W, Gao YG, Hong JY, Yue HW, Jiang LL, et al. HSP90 recognizes the N-terminus of huntingtin involved in regulation of huntingtin aggregation by USP19. Sci Rep 2017; 7: 14797.
Ochaba J, Monteys AM, O'Rourke JG, Reidling JC, Steffan JS, Davidson BL, et al. PIAS1 regulates mutant Huntingtin accumulation and Huntington's disease-associated phenotypes in vivo . Neuron 2016; 90: 507–20.
Livneh I, Kravtsova-Ivantsiv Y, Braten O, Kwon YT, Ciechanover A . Monoubiquitination joins polyubiquitination as an esteemed proteasomal targeting signal. Bioessays 2017; 39: doi: 10.1002/bies.201700027.
Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, et al. Accumulation of mutant Huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell 2001; 12: 1393–407.
Wang J, Wang CE, Orr A, Tydlacka S, Li SH, Li XJ . Impaired ubiquitin–proteasome system activity in the synapses of Huntington's disease mice. J Cell Biol 2008; 180: 1177–89.
Li XJ, Li S . Proteasomal dysfunction in aging and Huntington disease. Neurobiol Dis 2011; 43: 4–8.
Konstantinova IM, Tsimokha AS, Mittenberg AG . Role of proteasomes in cellular regulation [Internet]. In: International Review of Cell and Molecular Biology. Academic Press; 2008 . p59–124.
Díaz-Hernández M, Valera AG, Morán MA, Gómez-Ramos P, Alvarez-Castelao B, Castaño JG, et al. Inhibition of 26S proteasome activity by huntingtin filaments but not inclusion bodies isolated from mouse and human brain. J Neurochem 2006; 98: 1585–96.
Schipper-Krom S, Juenemann K, Jansen AH, Wiemhoefer A, van den Nieuwendijk R, Smith DL, et al. Dynamic recruitment of active proteasomes into polyglutamine initiated inclusion bodies. FEBS Lett 2014; 588: 151–9.
Bennett EJ, Bence NF, Jayakumar R, Kopito RR . Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol Cell 2005; 17: 351–65.
Holmberg CI, Staniszewski KE, Mensah KN, Matouschek A, Morimoto RI . Inefficient degradation of truncated polyglutamine proteins by the proteasome. EMBO J 2004; 23: 4307–18.
Venkatraman P, Wetzel R, Tanaka M, Nukina N, Goldberg AL . Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol Cell 2004; 14: 95–104.
Michalik A, Van Broeckhoven C . Proteasome degrades soluble expanded polyglutamine completely and efficiently. Neurobiol Dis 2004; 16: 202–11.
Juenemann K, Schipper-Krom S, Wiemhoefer A, Kloss A, Sanz Sanz A, Reits EAJ . Expanded polyglutamine-containing N-terminal Huntingtin fragments are entirely degraded by mammalian proteasomes. J Biol Chem 2013; 288: 27068–84.
Wang B, Zeng L, Merillat SA, Fischer S, Ochaba J, Thompson LM, et al. The ubiquitin conjugating enzyme Ube2W regulates solubility of the Huntington's disease protein, huntingtin. Neurobiol Dis 2018; 109: 127–36.
Maheshwari M, Samanta A, Godavarthi SK, Mukherjee R, Jana NR . Dysfunction of the ubiquitin ligase Ube3a may be associated with synaptic pathophysiology in a mouse model of Huntington disease. J Biol Chem 2012; 287: 29949–57.
Grice GL, Nathan JA . The recognition of ubiquitinated proteins by the proteasome. Cell Mol Life Sci CMLS 2016; 73: 3497–506.
Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat Neurosci 2010; 13: 567–76.
Wong YC, Holzbaur ELF . The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J Neurosci Off J Soc Neurosci 2014; 34: 1293–305.
Mealer RG, Murray AJ, Shahani N, Subramaniam S, Snyder SH . Rhes, a striatal-selective protein implicated in Huntington disease, binds beclin-1 and activates autophagy. J Biol Chem 2014; 289: 3547–54.
Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004; 36: 585–95.
Kim J, Kundu M, Viollet B, Guan KL . AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13: 132–41.
Rubinsztein DC . Autophagy induction rescues toxicity mediated by proteasome inhibition. Neuron 2007; 54: 854–6.
Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007; 447: 859–63.
Fu Y, Wu P, Pan Y, Sun X, Yang H, Difiglia M, et al. A toxic mutant huntingtin species is resistant to selective autophagy. Nat Chem Biol 2017; 13: 1152.
Sun X, Fu Y, Pan Y, Lu B . Conformation-dependent recognition of mutant HTT (huntingtin) proteins by selective autophagy. Autophagy 2017; : 1–2.
Lackie RE, Maciejewski A, Ostapchenko VG, Marques-Lopes J, Choy WY, Duennwald ML, et al. The Hsp70/Hsp90 chaperone machinery in neurodegenerative diseases. Front Neurosci 2017; 11: 254.
Baldo B, Weiss A, Parker CN, Bibel M, Paganetti P, Kaupmann K . A screen for enhancers of clearance identifies huntingtin as a heat shock protein 90 (Hsp90) client protein. J Biol Chem 2012; 287: 1406–14.
Jana NR, Tanaka M, Wang G h, Nukina N . Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Hum Mol Genet 2000; 9: 2009–18.
Hay DG, Sathasivam K, Tobaben S, Stahl B, Marber M, Mestril R, et al. Progressive decrease in chaperone protein levels in a mouse model of Huntington's disease and induction of stress proteins as a therapeutic approach. Hum Mol Genet 2004; 13: 1389–405.
Sittler A, Lurz R, Lueder G, Priller J, Lehrach H, Hayer-Hartl MK, et al. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington's disease. Hum Mol Genet 2001; 10: 1307–15.
Muchowski PJ, Schaffar G, Sittler A, Wanker EE, Hayer-Hartl MK, Hartl FU . Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc Natl Acad Sci U S A 2000; 97: 7841–6.
Wacker JL, Huang SY, Steele AD, Aron R, Lotz GP, Nguyen Q, et al. Loss of Hsp70 exacerbates pathogenesis but not levels of fibrillar aggregates in a mouse model of Huntington's disease. J Neurosci Off J Soc Neurosci 2009; 29: 9104–14.
Tsai YC, Fishman PS, Thakor NV, Oyler GA . Parkin facilitates the elimination of expanded polyglutamine proteins and leads to preservation of proteasome function. J Biol Chem 2003; 278: 22044–55.
Stürner E, Behl C . The role of the multifunctional BAG3 protein in cellular protein quality control and in disease. Front Mol Neurosci 2017; 10: 177.
Carra S, Seguin SJ, Landry J . HspB8 and Bag3: a new chaperone complex targeting misfolded proteins to macroautophagy. Autophagy 2008; 4: 237–9.
Fuchs M, Poirier DJ, Seguin SJ, Lambert H, Carra S, Charette SJ, et al. Identification of the key structural motifs involved in HspB8/HspB6-Bag3 interaction. Biochem J 2009; 425: 245–55.
Ciechanover A, Kwon YT . Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med 2015; 47: e147.
Wyant KJ, Ridder AJ, Dayalu P . Huntington's disease—update on treatments. Curr Neurol Neurosci Rep 2017; 17: 33.
Wild EJ, Tabrizi SJ . Therapies targeting DNA and RNA in Huntington's disease. Lancet Neurol 2017; 16: 837–47.
Richardson PG, Mitsiades C, Hideshima T, Anderson KC . Bortezomib: proteasome inhibition as an effective anticancer therapy. Annu Rev Med 2006; 57: 33–47.
Hideshima T, Bradner JE, Wong J, Chauhan D, Richardson P, Schreiber SL, et al. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci U S A 2005; 102: 8567–72.
Seo H, Sonntag KC, Kim W, Cattaneo E, Isacson O . Proteasome activator enhances survival of Huntington's disease neuronal model cells. PloS One 2007; 2: e238.
Liu Y, Hettinger CL, Zhang D, Rezvani K, Wang X, Wang H . Sulforaphane enhances proteasomal and autophagic activities in mice and is a potential therapeutic reagent for Huntington's disease. J Neurochem 2014; 129: 539–47.
Jang M, Cho IH . Sulforaphane ameliorates 3-nitropropionic acid-induced striatal toxicity by activating the Keap1-Nrf2-ARE pathway and inhibiting the MAPKs and NF-κB pathways. Mol Neurobiol 2016; 53: 2619–35.
Luis-García ER, Limón-Pacheco JH, Serrano-García N, Hernández-Pérez AD, Pedraza-Chaverri J, Orozco-Ibarra M . Sulforaphane prevents quinolinic acid-induced mitochondrial dysfunction in rat striatum. J Biochem Mol Toxicol 2017; 31.
DeMarch Z, Giampà C, Patassini S, Bernardi G, Fusco FR . Beneficial effects of rolipram in the R6/2 mouse model of Huntington's disease. Neurobiol Dis 2008; 30: 375–87.
Berkers CR, van Leeuwen FWB, Groothuis TA, Peperzak V, van Tilburg EW, Borst J, et al. Profiling proteasome activity in tissue with fluorescent probes. Mol Pharm 2007; 4: 739–48.
Leestemaker Y, de Jong A, Witting KF, Penning R, Schuurman K, Rodenko B, et al. Proteasome activation by small molecules. Cell Chem Biol 2017; 24: 725–736.e7.
Taylor DM, Moser R, Régulier E, Breuillaud L, Dixon M, Beesen AA, et al. MAP kinase phosphatase 1 (MKP-1/DUSP1) is neuroprotective in Huntington's disease via additive effects of JNK and p38 inhibition. J Neurosci Off J Soc Neurosci 2013; 33: 2313–25.
Wong HK, Bauer PO, Kurosawa M, Goswami A, Washizu C, Machida Y, et al. Blocking acid-sensing ion channel 1 alleviates Huntington's disease pathology via an ubiquitin-proteasome system-dependent mechanism. Hum Mol Genet 2008; 17: 3223–35.
Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, Chen PC, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 2010; 467: 179–84.
Boselli M, Lee BH, Robert J, Prado MA, Min SW, Cheng C, et al. An inhibitor of the proteasomal deubiquitinating enzyme USP14 induces tau elimination in cultured neurons. J Biol Chem 2017; 292: 179–84.
Kiprowska MJ, Stepanova A, Todaro DR, Galkin A, Haas A, Wilson SM, et al. Neurotoxic mechanisms by which the USP14 inhibitor IU1 depletes ubiquitinated proteins and Tau in rat cerebral cortical neurons: Relevance to Alzheimer's disease. Biochim Biophys Acta 2017; 1863: 1157–70.
Sarkar S, Krishna G, Imarisio S, Saiki S, O'Kane CJ, Rubinsztein DC . A rational mechanism for combination treatment of Huntington's disease using lithium and rapamycin. Hum Mol Genet 2008; 17: 170–8.
DeBosch BJ, Heitmeier MR, Mayer AL, Higgins CB, Crowley JR, Kraft TE, et al. Trehalose inhibits solute carrier 2A (SLC2A) proteins to induce autophagy and prevent hepatic steatosis. Sci Signal 2016; 9: ra21.
Mardones P, Rubinsztein DC, Hetz C . Mystery solved: Trehalose kickstarts autophagy by blocking glucose transport. Sci Signal 2016; 9: fs2.
Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC . Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem 2007; 282: 5641–52.
Tanaka M, Machida Y, Niu S, Ikeda T, Jana NR, Doi H, et al. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med 2004; 10: 148–54.
Fernandez-Estevez MA, Casarejos MJ, López Sendon J, Garcia Caldentey J, Ruiz C, Gomez A, et al. Trehalose reverses cell malfunction in fibroblasts from normal and Huntington's disease patients caused by proteosome inhibition. PloS One 2014; 9: e90202.
Rose C, Menzies FM, Renna M, Acevedo-Arozena A, Corrochano S, Sadiq O, et al. Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington's disease. Hum Mol Genet 2010; 19: 2144–53.
Sarkar S, Rubinsztein DC . Inositol and IP3 levels regulate autophagy: biology and therapeutic speculations. Autophagy 2006; 2: 132–4.
Sarkar S, Krishna G, Imarisio S, Saiki S, O'Kane CJ, Rubinsztein DC . A rational mechanism for combination treatment of Huntington's disease using lithium and rapamycin. Hum Mol Genet 2008; 17: 170–8.
Jiang W, Wei W, Gaertig MA, Li S, Li XJ . Therapeutic effect of berberine on huntington's disease transgenic mouse model. PLoS One 2015; 10: e0134142.
Ahmed T, Gilani AUH, Abdollahi M, Daglia M, Nabavi SF, Nabavi SM . Berberine and neurodegeneration: a review of literature. Pharmacol Rep PR 2015; 67: 970–9.
Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, et al. Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol 2008; 4: 295–305.
Menzies FM, Garcia-Arencibia M, Imarisio S, O'Sullivan NC, Ricketts T, Kent BA, et al. Calpain inhibition mediates autophagy-dependent protection against polyglutamine toxicity. Cell Death Differ 2015; 22: 433–44.
Luis-Ravelo D, Estévez-Silva H, Barroso-Chinea P, Afonso-Oramas D, Salas-Hernández J, Rodríguez-Núñez J, et al. Pramipexole reduces soluble mutant huntingtin and protects striatal neurons through dopamine D3 receptors in a genetic model of Huntington's disease. Exp Neurol 2018; 299: 137–47.
Al-Ramahi I, Panapakkam Giridharan SS, Chen YC, Patnaik S, Safren N, Hasegawa J, et al. Inhibition of PIP4Kγ ameliorates the pathological effects of mutant huntingtin protein. eLife 2017; 6: pii: e29123.
Abd-Elrahman KS, Hamilton A, Hutchinson SR, Liu F, Russell RC, Ferguson SSG . mGluR5 antagonism increases autophagy and prevents disease progression in the zQ175 mouse model of Huntington's disease. Sci Signal 2017; 10.
Jin J, Gu H, Anders NM, Ren T, Jiang M, Tao M, et al. Metformin protects cells from mutant huntingtin toxicity through activation of ampk and modulation of mitochondrial dynamics. Neuromol Med 2016; 18: 581–92.
Vázquez-Manrique RP, Farina F, Cambon K, Dolores Sequedo M, Parker AJ, Millán JM, et al. AMPK activation protects from neuronal dysfunction and vulnerability across nematode, cellular and mouse models of Huntington's disease. Hum Mol Genet 2016; 25: 1043–58.
Hervás D, Fornés-Ferrer V, Gómez-Escribano AP, Sequedo MD, Peiró C, Millán JM, et al. Metformin intake associates with better cognitive function in patients with Huntington's disease. PloS One 2017; 12: e0179283.
Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 2017; 93: 1015–34.
Herbst M, Wanker EE . Small molecule inducers of heat-shock response reduce polyQ-mediated huntingtin aggregation a possible therapeutic strategy. Neurodegener Dis 2007; 4: 254–60.
Lotz GP, Legleiter J, Aron R, Mitchell EJ, Huang S-Y, Ng C, et al. Hsp70 and Hsp40 functionally interact with soluble mutant huntingtin oligomers in a classic ATP-dependent reaction cycle. J Biol Chem 2010; 285: 38183–93.
Labbadia J, Cunliffe H, Weiss A, Katsyuba E, Sathasivam K, Seredenina T, et al. Altered chromatin architecture underlies progressive impairment of the heat shock response in mouse models of Huntington disease. J Clin Invest 2011; 121: 3306–19.
Fujikake N, Nagai Y, Popiel HA, Okamoto Y, Yamaguchi M, Toda T . Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J Biol Chem 2008; 283: 26188–97.
Luo W, Dou F, Rodina A, Chip S, Kim J, Zhao Q, et al. Roles of heat-shock protein 90 in maintaining and facilitating the neurodegenerative phenotype in tauopathies. Proc Natl Acad Sci U S A 2007; 104: 9511–6.
Ernst JT, Neubert T, Liu M, Sperry S, Zuccola H, Turnbull A, et al. Identification of novel HSP90α/β isoform selective inhibitors using structure-based drug design demonstration of potential utility in treating CNS disorders such as Huntington's disease. J Med Chem 2014; 57: 3382–400.
Zhang YQ, Sarge KD . Celastrol inhibits polyglutamine aggregation and toxicity though induction of the heat shock response. J Mol Med Berl Ger 2007; 85: 1421–8.
Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ . Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A 2001; 98: 8554–9.
Deshaies RJ . Protein degradation: Prime time for PROTACs. Nat Chem Biol 2015; 11: 634–5.
Tomoshige S, Nomura S, Ohgane K, Hashimoto Y, Ishikawa M . Discovery of small molecules that induce the degradation of Huntingtin. Angew Chem Int Ed Engl 2017; 56: 11530–3.
Wray S, Self M . NINDS Parkinson's Disease iPSC Consortium, NINDS Huntington's Disease iPSC Consortium, NINDS ALS iPSC Consortium, Lewis PA, et al. Creation of an open-access, mutation-defined fibroblast resource for neurological disease research. PloS One 2012; 7: e43099.
Tanji K, Kamitani T, Mori F, Kakita A, Takahashi H, Wakabayashi K . TRIM9, a novel brain-specific E3 ubiquitin ligase, is repressed in the brain of Parkinson's disease and dementia with Lewy bodies. Neurobiol Dis 2010; 38: 210–8.
Liu QY, Lei JX, Sikorska M, Liu R . A novel brain-enriched E3 ubiquitin ligase RNF182 is up regulated in the brains of Alzheimer's patients and targets ATP6V0C for degradation. Mol Neurodegener 2008; 3: 4.
Itoh Y, Ishikawa M, Kitaguchi R, Sato S, Naito M, Hashimoto Y . Development of target protein-selective degradation inducer for protein knockdown. Bioorg Med Chem 2011; 19: 3229–41.
Ottis P, Crews CM . Proteolysis-targeting chimeras: induced protein degradation as a therapeutic strategy. ACS Chem Biol 2017; 12: 892–8.
Burslem GM, Crews CM . Small-molecule modulation of protein homeostasis. Chem Rev 2017; 117: 11269–301.
Matsumura K, Ono M, Hayashi S, Kimura H, Okamoto Y, Ihara M, et al. Phenyldiazenyl benzothiazole derivatives as probes for in vivo imaging of neurofibrillary tangles in Alzheimer's disease brains. MedChemComm 2011; 2: 596–600.
Klunk WE, Wang Y, Huang G, Debnath ML, Holt DP, Mathis CA . Uncharged thioflavin-T derivatives bind to amyloid-beta protein with high affinity and readily enter the brain. Life Sci 2001; 69: 1471–84.
Clift D, McEwan WA, Labzin LI, Konieczny V, Mogessie B, James LC, et al. A method for the acute and rapid degradation of endogenous proteins. Cell 2017; 171: 1692–706.e18.
Butler DC, Messer A . Bifunctional anti-huntingtin proteasome-directed intrabodies mediate efficient degradation of mutant huntingtin exon 1 protein fragments. PLoS One 2011; 6: e29199.
Butler DC, Snyder-Keller A, De Genst E, Messer A . Differential nuclear localization of complexes may underlie in vivo intrabody efficacy in Huntington's disease. Protein Eng Des Sel PEDS 2014; 27: 359–63.
Southwell AL, Ko J, Patterson PH . Intrabody gene therapy ameliorates motor, cognitive, and neuropathological symptoms in multiple mouse models of Huntington's disease. J Neurosci Off J Soc Neurosci 2009; 29: 13589–602.
Southwell AL, Khoshnan A, Dunn D, Bugg CW, Lo DC, Patterson PH . Intrabodies binding the proline-rich domains of mutant huntingtin increase its turnover and reduce neurotoxicity. J Neurosci Off J Soc Neurosci 2008; 28: 9013–20.
Amaro IA, Henderson LA . An intrabody drug (rAAV6-INT41) reduces the binding of N-terminal Huntingtin fragment(s) to DNA to basal levels in PC12 cells and delays cognitive loss in the R6/2 animal model. J Neurodegener Dis 2016; 2016: 7120753.
Wang CE, Zhou H, McGuire JR, Cerullo V, Lee B, Li SH, et al. Suppression of neuropil aggregates and neurological symptoms by an intracellular antibody implicates the cytoplasmic toxicity of mutant huntingtin. J Cell Biol 2008; 181: 803–16.
Genetic Modifiers of Huntington's Disease (GeM-HD) Consortium. Identification of genetic factors that modify clinical onset of Huntington's disease. Cell 2015; 162: 516–26.
Tomaic V, Pim D, Thomas M, Massimi P, Myers MP, Banks L . Regulation of the human papillomavirus type 18 E6/E6AP ubiquitin ligase complex by the HECT domain-containing protein EDD. J Virol 2011; 85: 3120–7.
Jiang W, Wang S, Xiao M, Lin Y, Zhou L, Lei Q, et al. Acetylation regulates gluconeogenesis by promoting PEPCK1 degradation via recruiting the UBR5 ubiquitin ligase. Mol Cell 2011; 43: 33–44.
Zhang T, Cronshaw J, Kanu N, Snijders AP, Behrens A . UBR5-mediated ubiquitination of ATMIN is required for ionizing radiation-induced ATM signaling and function. Proc Natl Acad Sci U S A 2014; 111: 12091–6.
Ling S, Lin WC . EDD inhibits ATM-mediated phosphorylation of p53. J Biol Chem 2011; 286: 14972–82.
Travessa AM, Rodrigues FB, Mestre TA, Ferreira JJ . Fifteen years of clinical trials in Huntington's disease: a very low clinical drug development success rate. J Huntingt Dis 2017; 6: 157–63.
Garbaccio RM, Parmee ER . The impact of chemical probes in drug discovery: a pharmaceutical industry perspective. Cell Chem Biol 2016; 23: 10–7.
Mukhopadhyay S, Frias MA, Chatterjee A, Yellen P, Foster DA . The enigma of rapamycin dosage. Mol Cancer Ther 2016; 15: 347–53.
Bové J, Martínez-Vicente M, Vila M . Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci 2011; 12: 437–52.
Arrowsmith CH, Audia JE, Austin C, Baell J, Bennett J, Blagg J, et al. The promise and peril of chemical probes. Nat Chem Biol 2015; 11: 536–41.
Workman P, Collins I . Probing the probes: fitness factors for small molecule tools. Chem Biol 2010; 17: 561–77.
Bunnage ME, Chekler ELP, Jones LH . Target validation using chemical probes. Nat Chem Biol 2013; 9: 195–9.
Frigault MM, Barrett JC . Is target validation all we need? Curr Opin Pharmacol 2014; 17: 81–6.
Alguacil LF, González-Martín C . Target identification and validation in brain reward dysfunction. Drug Discov Today 2015; 20: 347–52.
Zhang W, Wu KP, Sartori MA, Kamadurai HB, Ordureau A, Jiang C, et al. System-Wide modulation of HECT E3 ligases with selective ubiquitin variant probes. Mol Cell 2016; 62: 121–36.
Ernst A, Avvakumov G, Tong J, Fan Y, Zhao Y, Alberts P, et al. A strategy for modulation of enzymes in the ubiquitin system. Science 2013; 339: 590–5.
Lu K, Brave F den, Jentsch S . Pathway choice between proteasomal and autophagic degradation. Autophagy 2017; 13: 1799–800.
Majumder S, Richardson A, Strong R, Oddo S . Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One 2011; 6: e25416.
Li JY, Popovic N, Brundin P . The use of the R6 transgenic mouse models of Huntington's disease in attempts to develop novel therapeutic strategies. NeuroRx 2005; 2: 447–64.
Gray M, Shirasaki DI, Cepeda C, André VM, Wilburn B, Lu XH, et al. Full-length human mutant Huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci 2008; 28: 6182–95.
Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, et al. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet 2003; 12: 1555–67.
Mattis VB, Tom C, Akimov S, Saeedian J, Østergaard ME, Southwell AL, et al. HD iPSC-derived neural progenitors accumulate in culture and are susceptible to BDNF withdrawal due to glutamate toxicity. Hum Mol Genet 2015; 24: 3257–71.
HD iPSC Consortium. Induced pluripotent stem cells from patients with Huntington's disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell 2012; 11: 264–78.
Cheng PH, Li CL, Chang YF, Tsai SJ, Lai YY, Chan AWS, et al. miR-196a ameliorates phenotypes of Huntington disease in cell, transgenic mouse, and induced pluripotent stem cell models. Am J Hum Genet 2013; 93: 306–12.
Raja WK, Mungenast AE, Lin YT, Ko T, Abdurrob F, Seo J, et al. Self-organizing 3D human neural tissue derived from induced pluripotent stem cells recapitulate Alzheimer's disease phenotypes. PLoS One 2016; 11: e0161969.
Garcez PP, Loiola EC, Costa RM da, Higa LM, Trindade P, Delvecchio R, et al. Zika virus impairs growth in human neurospheres and brain organoids. Science 2016; : aaf6116.
Ravikumar B, Duden R, Rubinsztein DC . Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet 2002; 11: 1107–17.
Malagelada C, Jin ZH, Jackson-Lewis V, Przedborski S, Greene LA . Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson's disease. J Neurosci Off J Soc Neurosci 2010; 30: 1166–75.
Zhang X, Li L, Chen S, Yang D, Wang Y, Zhang X, et al. Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy 2011; 7: 412–25.
Federico CA, Carlisle B, Kimmelman J, Fergusson DA . Late, never or non-existent: the inaccessibility of preclinical evidence for new drugs. Br J Pharmacol 2014; 171: 4247–54.
Kumarapeli ARK, Horak KM, Glasford JW, Li J, Chen Q, Liu J, et al. A novel transgenic mouse model reveals deregulation of the ubiquitin-proteasome system in the heart by doxorubicin. FASEB J Off Publ Fed Am Soc Exp Biol 2005; 19: 2051–3.
Ahmed T, Gilani AUH, Abdollahi M, Daglia M, Nabavi SF, Nabavi SM . Berberine and neurodegeneration: a review of literature. Pharmacol Rep PR 2015; 67: 970–9.
Acknowledgements
The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada through Ontario Genomics Institute[OGI-055], Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant No 115766], Janssen, Merck KGaA, MSD, Novartis Pharma AG, Ontario Ministry of Research, Innovation and Sciences (MRIS), Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and the Wellcome Trust. This work is also supported by additional funding awarded to Y.T. (Discovery Grant: RGPIN-2017-06520) from the Natural Sciences and Engineering Research Council of Canada.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Harding, R., Tong, Yf. Proteostasis in Huntington's disease: disease mechanisms and therapeutic opportunities. Acta Pharmacol Sin 39, 754–769 (2018). https://doi.org/10.1038/aps.2018.11
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2018.11
- Springer Nature Singapore Pte Ltd.
Keywords
This article is cited by
-
HAP40 modulates mutant Huntingtin aggregation and toxicity in Huntington’s disease mice
Cell Death & Disease (2024)
-
The ubiquitin thioesterase YOD1 ameliorates mutant Huntingtin induced pathology in Drosophila
Scientific Reports (2023)
-
The updated development of blood-based biomarkers for Huntington’s disease
Journal of Neurology (2023)
-
Discovery of an autophagy inducer J3 to lower mutant huntingtin and alleviate Huntington’s disease-related phenotype
Cell & Bioscience (2022)
-
An overview of PROTACs: a promising drug discovery paradigm
Molecular Biomedicine (2022)