
Abstract
Groundnut (Arachis hypogaea L.) is an oilseed crop largely grown throughout the world. Ten isolates of S. rolfsii collected from groundnut grown areas were investigated for their morphological and genetic variability study. Based on morphological variability study, all isolates were divided in six groups based on sclerotia distribution pattern. Isolates produced number of sclerotia in range of 115–375 in light dark brown to dark brown colour while weight of hundred sclerotia and sizes of sclerotia was in range of 85.54–145.13 mg and 0.32–0.68 mm. Genetic diversity was studied through ITS amplification that produced rDNA amplicon size of 650–700 bp which confirmed that all isolates belong to Sclerotium rolfsii. RAPD primers confirmed genetic variation among the isolates. Highest numbers of amplicons (13) were observed in the OPA-9 primer. 102 polymorphic and 3 monomorphic bands were produced by RAPD primers. Isolates SrA-SrH (0.575), SrBKN-SrA (0.384) and SrBKN-SrBKL (0.330) exhibited highest similarity coefficient. Based on dendrogram generated by UPGMA, SrBKN, SrA, SrH, SrJPF, SrJPP and SrT isolates were considered in Group I. SrBKL showed highest (51%) diversity in Group II while Churu (SrC) isolate showed 49% diversity in Group III. Dendrogram and cluster analysis cleared that Group I was more genetically diverse among the isolates. Morphological and molecular diagnosis of the pathogen has a pivotal role in the implementation of chemical (fungicide) and biological (bio-agents) methods.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Groundnut (Arachis hypogaea L.) is an oilseed crop, commonly called peanut, goober pea, and monkey nut [33]. In India, it is growing extensively in Gujarat, Rajasthan, A.P., Tamil Nadu and Karnataka. India occupied about 5.75 million ha area with an annual production of 10.11 million tons and productivity of 1759 kg ha−1 while, Rajasthan occupied 0.80 million ha area with an annual production of 1.70 million tons and productivity of 2131 kg ha−1 [4].
Groundnut is more prone to bacterial, fungal, and viral diseases, among the diseases; S. rolfsii is a devastating soil-borne pathogen that has a wide host range on agricultural and horticultural crops [13, 29]. According to Chohan [11] and Mehan et al. [22], S. rolfsii causes stem rot, wilt (sclerotial), and stem and pod rot in plants. Production of mycelium and sclerotia on infected plants are the main diagnostic symptoms [23, 40]. Initial infection of S. rolfsii was mostly seen on younger plants which later resulted 15–70% yield losses with combined infection of rust and leaf spots [3].
In India, this disease prevails more than 5 lakh ha of groundnut growing areas. In Rajasthan, stem rot mainly appears in the Kharif season either alone or with other pathogens in complex form and causes severe yield losses. It is the most devastating, predominant, and yield-reducing factor in the north western zone of Rajasthan. The disease progresses in distinct forms, stem-rot (S. rolfsii), collar rot (A.niger), root-rot (M. phaseolina = R. bataticola), along with aflatoxin contamination (Aspergillus flavus) in the arid and semiarid region [36].
In the present scenario, there are many strains of S. rolfsii existing in various geographical regions. Morphological and molecular diagnosis of the pathogen has a pivotal role in the implementation of chemical (fungicide) and biological (bio-agents) methods. Present information on morphological and genetic variability is very scanty and work done on genetic variability has been very limited in arid and semi-arid regions [14]. Molecular markers play a key role in analyzing genetic variation among fungal pathogens [2, 30]. Among the primers and molecular markers, ITS primers and RAPD markers are still choices for genetic variation study due to the existence of high-level polymorphism [24]. Stem rot disease is predominant factors in groundnut growing area especially in arid and semiarid regions of Rajasthan and India. In groundnut growing areas, farmers are also growing vegetables crops in which this pathogen is predominant and causes yield losses in vegetable as well as in groundnut. Initially infection of this pathogen is confused with collar rot diseases in groundnut and farmers are not able to take appropriate decision on management strategy. In such situation, morphological and molecular study helpful to farmers for proper identification of disease causing pathogen. The present study would be helpful in understanding genetic diversity study within the species, and development of management strategy against the pathogen.
2 Materials and methods
2.1 Isolation and identification
Stem rot infected plants showed symptoms and sclerotia were collected from Bikaner, Jaipur, Churu, and Sikar districts of Rajasthan, Gujarat, Andhra Pradesh, and Telangana and carried in advance laboratory of plant pathology, college of agriculture, Bikaner (Rajasthan). Infected portion of stem was thoroughly washed, cut into small pieces then sterilized with 0.1 percent mercuric chloride (1 g lit−1) solution for 1 to 2 min and aseptically transferred into 20 ml PDA containing Petri dishes. All plates were incubated in BOD for 7 days at 28 ± 2 °C for growth. The pure culture of S. rolfsii was purified by the hyphal tip technique [32]. Purified isolates designated as SrBKN, SrBKL, SrJPP, SrJPF, SrC, SrS, SrG, SrT, SrA and SrH, respectively. Morphological characters are identified as per key points described in “Illustrated Genera of Imperfect Fungi” [9].
2.2 Morphological variability
For estimation of different morphological variability characteristics like, distribution pattern of sclerotia, the number of sclerotia, color, growth, diameter, and weight, 5 mm mycelial disc of each respective isolate was inoculated on PDA media and then incubated in BOD at 28 ± 2 °C for 15 days. Observations of mycelial growth and sclerotial distribution patterns on Petri plates for each isolate were recorded and categorized according to the scattering at the periphery region irregularly on one side of the Petri plate, irregularly all over the plate, and at the centre region [26]. Similarly, the total number of sclerotia per Petri plate, the color of sclerotia like light–dark/brown, brown and dark, the diameter of individual sclerotium (mm), and the average weight (mg) of 100 sclerotia was also calculated.
2.3 Genetic variability
Virulent isolates of S. rolfsii were grown on PDB at 28 ± 2 °C for 7 days in Erlenmeyer flasks. The mycelial mat of each isolate was harvested, filtered by cheesecloth, and removed of excess moisture by using tissue paper. 200 mg mycelial mat was macerated in liquid nitrogen to obtain a fine powder. Homogenized fine powder of each isolate was taken for isolation of total DNA as per protocol given by Nucleo-pore gDNA Fungal/Bacterial Mini Kit of Genetix Biotech Asia Pvt. Ltd. The intactness and integrity of DNA were judged through gel electrophoresis. DNA quantification was accomplished with a UV–VIS spectrophotometer (model IMPLEN P-300) on 260 nm and 280 nm wavelengths. Quantified DNA was diluted into a final working stock of 25 ng/µl in TE buffer (10 mM Tris HCL, 1 mM EDTA, pH 8.0).
2.4 Amplification of DNA with ITS primer
The Internal Transcribed Spacer (ITS-1 and ITS-4) rDNA was used for amplification of distinct isolates [39] (Table 1). The final PCR reaction was run in a 25 μl reaction mixture containing 12.5 μl of 2X Dream Taq Green PCR Master Mix (Genetix Biotech Asia Pvt. Ltd), 1.0 μl of ITS-1 forward (0.6 picomolar/μl), 1.0 μl of ITS-4 reverse primer (0.6 picomolar/μl), and 7.5 μl of nuclease-free sterile PCR water (Thermo Scientific, USA) and 3 μl (75 ng) of DNA sample. Amplification was performed in a Px2 thermal cycler (Thermo Scientific, USA). Initial denaturation run at 94 °C for 4 min before cycling start, PCR cycling conditions consisted of 35 cycles, of which denaturation run at 94 °C for 1 min, annealing run at 56 °C for 1 min, extension at 72 °C for 1.5 min, and final extension run at 72 °C for 6 min after cycling. After amplification of the target DNA, the product was visualized on 1.5% (Hi-media) agarose gel. The gel was prepared in 0.5X TBE buffer. Similarly, 3 μl of the ethidium bromide was also added then amplified products subsequently electrophoresis at 120 V for 2.5–3.0 h. PCR product size was evaluated with a 100 bp DNA ladder. ITS-PCR product banding pattern was put on record by the usage of the Syngene gel documentation system.
2.5 Analysis of genetic diversity by the usage of RAPD primers
Thirteen OPA series random primers were screened using one selected isolate of S. rolfsii. Based on the banding pattern, ten primers of the OPA series (Table 1) were obtained from Operon Technologies (Inc. Alameda, California) and selected for Random Amplification of Polymorphic DNA (RAPD). PCR analysis was done in a 20 μl reaction containing 2.0 μl of 10X Dream Taq buffer, 0.2 unit of Dream Taq DNA polymerase, 0.5 μl of 10 mM each dNTPs (Thermo Scientific, USA), 1.0 μl of 25 mM MgCl2 (Thermo Scientific, USA), 3.0 μl of 10 pmols per reaction of random primers (Operon Technologies), Nuclease free water and 75 ng of template DNA. RAPD PCR was performed in a Px2 thermal cycler. PCR conditions consisted of 40 cycles, of which denaturation run at 94 °C for 1 min, annealing run at 35 °C for 1 min, while, extension run at 72 °C for 2 min with an initial denaturation at 94 °C for 5 min before cycling and final extension run at 72 °C for 15 min after cycling. After the amplification of the target DNA, the product was loaded on 1.5% (Hi-media) agarose gel. The gel was prepared in 0.5X TBE buffer. Similarly, 3 μl of the ethidium bromide was also added then amplified products subsequently electrophoresis at 120 V for 2.5–3.0 h. When DNA bands were separated, the gel PCR product was viewed and the photograph was taken under the gel Syngene gel documentation system. RAPD bands were counted based on the length of an amplified polynucleotide. The molecular size of each primer's product was judged with a 1 kb DNA ladder. RAPD bands were scored as 0 and 1 based on the absence and presence of bands. Completely faint DNA bands were not counted but major bands parallel to faint bands were scored. For confirmation and reproducibility, all RAPD primers were replicated thrice. Jaccard’s similarity coefficient equation was used for the calculation of pair-wise association coefficients from the quantitative data matrix. Similarly, cluster analyses for genetic distance among the species were differentiated by UPGMA (Un-weighted Pair Group Method with Arithmetic Mean) clustering method. The dendrogram was constructed after genetic distances were obtained from cluster analysis and relationships among the strains were depicted using the computer program NTSys pc version 2.02 [34].
3 Results and discussion
3.1 Isolation and identification
Based on reports of previous authors [7, 38] PDA was the best media for S. rolfsii growth and production of sclerotia. Here we also conducted our study by uses of PDA media. In petri plates white fluffy mycelium growth of fungus was observed which showed different morphological variation. Similar mycelium morphological variation was reported by earlier workers (Deepthi and Reddy [12]; Kumar et al. [19]; Maddu and Ravuri [21] that also supported to this study. The pure culture of fungus from infected plant tissues was isolated by using of tissue segment method. Bagwan [8] and Maddu and Ravuri [21], Arunasri et al. [6] and Deepthi and Reddy [12] isolated S. rolfsii using the tissue segment method and infected tissue groundnut which is supported to this finding. The fungus produced different colour of sclerotial bodies and distribution pattern and identified as Sclerotium rolfsii Sacc based on “Illustrated Genera of Imperfect Fungi” [9]. Genetic variation can lead to differences in morphology, including variations in sclerotial size, shape, color, and texture. These differences can arise from genetic mutations, recombination among different strains of the fungus. Other factors such as temperature, humidity, light, nutrient availability, complex life cycle, adaptive advantages and host plant interactions can influence the development and appearance of sclerotia. Different environmental conditions can result in variations in sclerotial characteristics. Similar characteristics (sclerotial body formation and morphological variation) have been reported by earlier workers [10, 19, 31] that also corroborated our finding.
3.2 Morphological variability
Results from (Table 2 and Fig. 1) revealed substantial variations in the sclerotial distribution pattern among the isolates. Based on the arrangement and distribution pattern of sclerotia, S. rolfsii isolates were divided into six groups. SrBKN and SrS isolate produced sclerotia in irregular patterns scattered all over the surface included in the first group while SrBKL and SrJPF isolates produced sclerotia irregularly towards the periphery comprises in the second group. Similarly, SrJPP produced sclerotia towards the center of media considered in the third group. SrC and SrT produced sclerotia in an irregularly scattered pattern towards one side included in the fourth group; while sclerotia of SrG and SrA spread irregularly in the periphery added in the fifth group. SrH isolate produced sclerotia all over the surface was considered in group six. Similar variations in sclerotia distribution patterns have been reported by [19]. The maximum sclerotia (375) per plate was produced by SrBKL while the lowest number was produced by SrJPP (115) isolate. These results are in agreement with Ansari and Agnihotri [5] who presumed that media that highly supported pathogens produced extensive growth and more sclerotia. Le et al. [20] collected various isolates of S. rolfsii and observed 79 to 1080 per plate sclerotia which also corroborated our study. Results revealed considerable variation in sclerotia colour which was later categorized into four groups. SrBKN, SrS, and SrA isolates produced light dark brown sclerotia colour that was kept in the first group while SrBKL and SrJPF produced dark brown colour sclerotia which were included in the second group. Likewise, SrJPP and SrT produced light–dark colour sclerotia fall under the third group whereas the rest groups are considered in the fourth group (Table 2 and Fig. 1). The present finding conforms to the study of [15, 18]. The diameter of sclerotia produced by distinct isolates varied from 0.32 mm to 0.68 mm. Sclerotia diameter is categorized into two groups i.e. group-I (< 0.5 mm) and group-II (0.5 to 1.0 mm). Diameter of sclerotia (0.41, 0.43, 0.49, 0.32 and 0.46 mm) produced by SrBKL, SrC, SrS, SrG and SrH were considered in first group while diameter of sclerotia (0.62, 0.68, 0.56, 0.61 and 0.58 mm) produced by SrBKN, SrJPP, SrJPF, SrT, and SrA isolates placed in second group (Table 2). Okereke and Wokocha [25] observed a minimum of 0.8 mm and a maximum of 1.4 mm size of sclerotia in tomato isolate of S. rolfsii. The highest weight of sclerotia (145.13 mg) was observed in SrJPP followed by (135.19, 127.78, 125.03, and 123.12 mg) in SrBKN, SrT, SrA, and SrS isolates compared to the rest of the isolates (Table 2). Hussain et al. [16] reported 57.8, 49.0, and 48.5 μm sclerotia sizes in RW-2, AT-3, and SR-1 isolates which were greater than others. Kumar et al. [19] presented geographical variability between S. rolfsii isolates that is also corroborated by our study.

Mycelial and sclerotial growth pattern of different Sclerotium rolfsii isolates on potato dextrose broth medium
3.3 Genetic variability
Detection with ITS primer: ITS-1 and ITS-4 primer amplified DNA of all isolates and generated 650 to 700bp band size after compared with 100bp ladder. Amplicon size confirmed that all collected isolates belong to the genus Sclerotium rolfsii (Fig. 2). Our investigations are supported by Adandonon et al. [1] studied genetic variation after obtaining an amplification fragment of 700bp to S. rolfsii. Durgaprasad et al. [14] observed genetic variation with RAPD, ITS1, and ITS4 primers generated 650 to 700bp with rDNA amplification in ten isolates S. rolfsii. Jebaraj et al. [17] studied S. rolfsii genetic variability with ITS, RAPD, and ISSR markers. Amplification of rDNA with ITS-1 and ITS-4 primers generated 650 to 700bp amplicon size of all 22 isolates. Poornima et al. [28] studied the genetic variability among the 24 isolates of S. rolfsii by using molecular markers like ITS-PCR and RAPD primers. Amplification of ITS region of rDNA with specific ITS1 and ITS4 universal primers produced approximately 650 to 700 bp in all the isolates of the fungus confirmed that all the isolates obtained are Sclerotium rolfsii. These studies are also in conformity with our results.

Molecular characterizations of Sclerotium rolfsii isolates by ITS PCR. Lane 1: SrBKN, Lane 2: SrBKL, Lane 3: SrJPP, Lane 4: SrJPF, Lane 5: SrC, Lane 6: SrS, Lane 7: SrG, Lane 8: SrT, Lane 9: SrA, Lane: 10: SrH indicates DNA bands of S. rolfsii isolates; Lane 11: Negative control; M-100 bp DNA ladder
3.4 Analysis of genetic diversity by using RAPD markers
Ten random OPA series RAPD primers were used to detect genetic variation and to divide all isolates into similar groups. Polymorphic and monomorphic bands of all isolates were produced after DNA amplification (Figs. 3 and 4). Amplicon numbers produced with RAPD primers varied from 8 (OPA-17) to 13 (OPA-8 and OPA-9) at an annealing temperature of 35 °C (Table 3). The highest number of polymorphic bands (13) was obtained in OPA-8 followed by in OPA-9 (12) primer while the lowest number of the polymorphic band (8) was observed in OPA-17 primer. Similarly, 3 numbers of monomorphic bands were obtained in OPA-9, OPA-10, and OPA-20 primers. The rest of the primers did not show any monomorphic bands. Maximum polymorphic bands (cent percent) were observed in OPA-3, OPA-5, OPA-6, OPA-7, OPA-8, OPA-12, and OPA-17 primers. The highest monomorphic bands (10%) were recorded in the OPA-20 primer followed by 8.33 and 7.69% in OPA-10 and OPA-9 primers. Most of the scorable bands ranged from 200 to 3000 bp in size.

RAPD bands of Sclerotium rolfsii Isolates using OPA-09. Isolates numbers are shown in Lane 1: SrBKN, Lane 2: SrBKL, Lane 3: SrJPP, Lane 4: SrJPF, Lane 5: SrC, Lane 6: SrS, Lane 7: SrG, Lane 8: SrT, Lane 9: SrA, Lane 10: SrH and Lane-11, Negative control: M-1 KB DNA ladder

Brands of sclerotium rolfsii isolates using OPA-10 primer. Isolates numbers are shown in Lane 1: SrBKN, Lane 2: SrBKL, Lane 3: SrJPF, Lane 4: SrJPF, Lane 5: SrC, Lane 6: SrS, Lane 7: SrG, Lane 8: SrT, Lane 9: SrA and Lane 10: SrH, M-1 KB DNA ladder
The characteristic features of RAPD primers were assessed in the IMEC calculator. The heterozygosity index (H) was in the range of 0.282 to 0.615. Polymorphic information content (PIC) values were in ranged from 0.242 to 0.535. The highest PIC value of primer revealed maximum genetic information among the genotypes. The highest effective multiplex ratio (EMR) was (10.21) in OPA-12 primer. The highest marker index (MI) 0.615 and 0.576 was shown in OPA-17 and OPA-20 primers. Due to higher (D) power OPA-9 and OPA-10 primers, unique RAPD bands were generated for all isolates and detected by a single primer (Table 3).
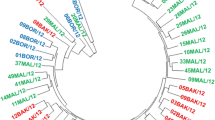
Pair-wise genetic similarity coefficient was estimated based on RAPD banding patterns in the form of a dendrogram after UPGMA-based cluster analysis. Maximum similarity coefficient in numbers (0.575) showed in SrA-SrH followed by SrBKN-SrA (0.384) isolates and found highly significant while isolates SrJPP-SrG (0.013) and SrBKL-SrG (0.016) showed least similarity and revealed non-significant (Table 4). According to clustering analysis, Group I included Bikaner (SrBKN), Anantapur (SrA), Hyderabad (SrH), Fhagi (SrJPF) and (SrJPP), Tripuati (SrT) isolates while Sikar (SrS), Gujarat (SrG) and Loonkarnsar (SrBKL) isolate comprehend in Group II. Diversity for group I was 58% while it was 51% in group II. Both the groups were genetically separated at 55% of diversity (Fig. 5). Group III comprises only one outlier i.e. Churu (SrC) which showed 49% of genetic diversity. Saude et al. [35] reported genetic relatedness and diversity between closely related strains of S. rolfsii with RAPD. Similarly, Kokub et al. [18] studied genetic variation by RAPD-PCR. Parvin et al. [27] listed genetic variation in virulent isolates of S. rolfsii obtained from eggplant and tomato using RAPD that demonstrated 20 bands ranging from 100-1500bp. Among the 20 bands, 9 bands were (45%) polymorphic and 11 bands (55%) monomorphic in eight isolates. Jebaraj et al. [17] showed genetic variability in virulent isolates of S. rolfsii with ITS, RAPD, and ISSR markers. 121 and 123 scorable and reproducible polymorphic bands were observed in ranges of 100 to 2500bp and 250 to 2000bp with ten RAPD and ISSR primers. Srividya et al. [37] studied the genetic variability in S. rolfsii by used of RAPD markers and they found that a total of 254 reproducible and scorable polymorphic bands ranging from 200 to 2000 bp were observed with twenty nine RAPD primers. The RAPD banding pattern reflected the presence of greater variability among the isolates and grouped the isolates into two clusters. The isolates, CSR 18 and 20, formed the cluster II revealing their distantness from other isolates at molecular level. The isolate, CSR 14, formed a separate sub cluster in the cluster I indicating its distant association with other isolates of this cluster that also supported our investigation. Genetic variation can lead to differences in morphology, including variations in sclerotial size, shape, color, and texture. These differences can arise from genetic mutations, recombination among different strains of the fungus. Other factors such as temperature, humidity, light, nutrient availability, complex life cycle, adaptive advantages and host plant interactions can influence the development and appearance of sclerotia. Different environmental conditions can result in variations in sclerotial characteristics. In our study, all isolates collected from different arid and semiarid regions like Bikaner (SrBKN), Anantapur (SrA), Hyderabad (SrH), Fhagi (SrJPF) and (SrJPP), Tripuati (SrT), Sikar (SrS), Gujarat (SrG) and Loonkarnsar (SrBKL) where weather factors and cropping patterns are varies and due to this genetic and morphological variations are disparate. In our findings, it is observed that RAPD primers distinguished genetic variability among the isolates of S. rolfsii that could be employed for further detection and management of other S. rolfsii isolates. The present study would help in understanding the pathogenicity and ecological parameters of S. rolfsii.

Dendogram generated by UPGMA analysis based on RAPD data showing genetic relationship among ten isolates of Sclerotium rolfsii
4 Conclusion
Different morphological parameters showed considerable variations among the isolates that could be used for further identification of S. rolfsii strains. Based on ITS primers, all isolates cluster belongs to genus Sclerotium, which is essential for the initial diagnosis and identification of pathogen for proper integrated disease management. Similarly, results obtained from RAPD distinguished isolates of S. rolfsii at the genetic level that would also be helpful in management of disease.
Availability of data and materials
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Adandonon A, Aveling TAS, Merwe NA, Sanders G. Genetic variation among Sclerotium isolates from Benin and South Africa, determined using mycelial compatibility and ITS rDNA sequence data. Aus Pl Pathol. 2005;34:19–25.
Adhipathi PA, Nakkeeran SE, Chandrasekaran AN. Morphological characterization and molecular phylogeny of Colletotrichum capsici causing leaf spot disease of turmeric. Bioscan. 2013;8:331–7.
Adiver SS. Influence of organic amendments and biological components on stem rot of groundnut. In: Adiver SS, editor. National seminar on stress management in oilseeds for attaining self-reliance in vegetable oil. Hyderabad: Indian Society of Oilseeds Research, Directorate of Oilseeds Research; 2003. p. 15–7.
Anonymous. Directorate of Economics and Statistics, Department of Agriculture and farmer welfare of India. 2021–22.
Ansari MM, Agnihotri SK. Morphological, physiological and pathological variations among Sclerotium rolfsii isolates of Soybean. Indian Phytopathol. 2000;53:65–7.
Arunasri P, Chalam TV, Eswara Reddy NP, Tirumala Reddy S, Ravindra RB. Investigations on fungicidal sensitivity of Trichoderma spp. and Sclerotium rolfsii (collar rot pathogen) in crossandra. Int J Appl Bio Pharm Tech. 2011;2:290–3.
Ayed F, Jabnoun-Khiareddine H, Aydi Ben Abdallah R, Daami-Remadi M. Effect of temperatures and culture media on Sclerotium rolfsii mycelial growth, sclerotial formation and germination. J Plant Pathol Microbiol. 2018;9:446. https://doi.org/10.4172/2157-7471.1000446.
Bagwan NB. Morphological variation in Sclerotium rolfsii Sacc. isolates collected from Maharashtra and Gujarat States causing stem rot in groundnut (Arachis hypogaea L.). Int J Plant Protec. 2011;4:68–73.
Barnett HL, Hunter BB. Illustrated genera of imperfect fungi. 4th ed. St Paul: APS Press; 1998. p. 218.
Chaurasia S, Chaurasia A. Pathological studies of Sclerotium rolfsii causing foot-rot disease of brinjal (Solanum melongena Linn.). Int J Pharm Life Sci. 2014;5:3257–64.
Chohan JS. Recent advances in disease of groundnut in India. Curr Trend Plant Pathol. 1974;10:171–84.
Deepthi KC, Reddy ENP. Stem rot disease of groundnut (Arachis hypogaea L.) induced by Sclerotium rolfsii and its management. Int J Life Sci Biotech Pharma Res. 2013;2:26–38.
Domsch KH, Gams W, Anderson TH. Compendium of soil fungi. London: Academic Press; 1980.
Prasad SD, Basha ST, Reddy NPE. Molecular variability among the isolates of Sclerotiāum rolfsii causing stem rot of groundnut by RAPD, ITS-PCR and RFLP. Eur Asian J Biosci. 2010;4:80–7.
Flieger MM, Kantorova T, Benesova S, Pazoutova VJ. Kinetics of soluble glucan production by Claviceps viridis. Folia Microbiol. 2003;48:633–8.
Hussain A, Iqbal M, Ghafoor A, Ayub N. Biochemical variability among the isolates of Sclerotium rolfsii of chickpea in Pakistan. Pak Phytopathol. 2010;22:34–9.
Jebaraj MD, Aiyanathan KEA, Nakkeeran S. Virulence and genetic diversity of Sclerotium rolfsii Sacc., infecting groundnut using nuclear RAPD and ISSR markers. J Environ Biol. 2017;38:147–59.
Kokub D, Azam F, Hassan A, Ansar M, Asad MJ, Khanumand A. Comparative growth, morphological and molecular characterization of indigenous Sclerotium rolfsii strains isolated from different locations of Pakistan. Pak J Bot. 2007;39:1849–66.
Kumar MR, Santhoshi MVM, Giridhra TK, Reddy KR. Cultural and morphological variability Sclerotium rolfsii isolates infecting groundnut and its reaction to some fungicidal. Int J Curr Microbiol App Sci. 2014;3:553–61.
Le CN, Mendes R, Kruijt M, Raaijmakers JM. Genetic and phenotypic diversity of Sclerotium rolfsii in groundnut fields in Central Vietnam. Plant Dis. 2012;96:389–97.
Maddu S, Ravuri JM. Physiological changes in groundnut (Arachis hypogaea L.) plants inoculated with Sclerotium rolfsii and Trichoderma species. Int J Sci Eng Res. 2015;6:135–8.
Mehan VK, Mayee CD, Brenneman TB, McDonald D (1995) Stem and pod rots of groundnut. Information Bulletin no 44. 1995; pp. 28.
Mehrotra RS, Aneja KR. An introduction to mycology. New Delhi.: Wilson Eastern Limited; 1990. p. 776.
Moulin MM, Rodrigues R, Goncalves LSA, Sudre CP, Pereira MG. A comparison of RAPD and ISSR markers reveals genetic diversity among sweet potato landraces (Ipomoea batatas (L.) Lam.). Acta Scient. 2012;2:139–47.
Okereke VC, Wokocha RC. In vitro growth of four isolates of Sclerotium rolfsii Sacc., in the humid tropics. Afr J Biotech. 2007;6:1879–81.
Palaiah P, Adiver SS. Morphological and cultural variability in Sclerotium rolfsii Sacc. Karnataka J Agri Sci. 2006;19:146–8.
Parvin N, Bilkiss M, Nahar J, Siddiqua MK, Meah MB. RAPD analysis of Sclerotium rolfsii isolates causing collar rot of eggplant and tomato. Int J Agri Res Innov Tech. 2016;6:47–57.
Prasad SD, Basha ST, Reddy NP. Molecular variability among the isolates of sclerotium rolfsii causing stem and pod rot of groundnut collected from Karnataka India. Int J Curr Microbiol App Sci. 2018;7(5):2925–34.
Priya RS, Chinnusamy C, Manicaksundaram P, Babu C. A review on weed management in groundnut (Arachis hypogaea L.). Int J Agri Sci Res. 2013;3:163–72.
Punja ZK, Sun LJ. Genetic diversity among mycelial compatibility groups of Sclerotium rolfsii (teleomorph Athelia rolfsii) and S. delphenii. Mycol Res. 2001;105:537–46.
Rakholiya KB, Jadeja KB. Morphological diversity of Sclerotium rolfsii caused stem and pod rot of groundnut. J Mycol Plant Pathol. 2011;41:500–4.
Rangaswami G, Mahadevan A. An agar blocks technique for isolating soil micro-organisms with special reference to pythiaceous fungi. Sci Cult. 1999;24:85–85.
Rathnakumar AL, Singh R, Parmar DL, Misra JB. Groundnut: a crop profile and compendium of varieties notified in India. Gujarat: Art India Press; 2013.
Rohlf FJ. NTSYS-pc: numerical taxonomy and multivariate analysis system, version 2.02i. New York: Exeter Softw; 1998.
Saude C, Melouk HA, Chenault KD. Genetic variability and mycelial compatibility groups of Sclerotium rolfsii. Phytopathology. 2004;94:592.
Sharma P, Saini MK, Deep S, Kumar V. Biological controls of groundnut root rot in farmer fields. J Agri Sci. 2012;4:1916–25.
Srividya PV, Ahamed Lal M, Ramana JV, Ahammed KS. Studies on diversity of Sclerotium rolfsii causing collar rot in chickpea using morphological and molecular markers. Legum Res. 2022;45(1):82–9. https://doi.org/10.18805/LR-4199.
Vaniya GR, Singh P, Deshmukh AJ. Effect of different cultural media on growth of different isolates of Sclerotium rolfsii Sacc causing stem rot of Indian bean. Pharma Innov J. 2022;11(4):14–9.
White TJ, Bruns T, Lee S, Taylor J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetic. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ, editors. PCR protocols: a guide to methods and applications. San Diego: Academic Press; 1990. p. 315–22.
Wilson C. Preventing the diseases of peanut. Washington D. C: United State Department of Agriculture, Year Book; 1953. p. 448–54.
Acknowledgements
The authors are grateful to the Head, Department of Plant Pathology, College of Agriculture, SKARU, Bikaner, and Director, ICAR- Central Research Institute for Jute and Allied Fibres, Barrackpore, Kolkata, 700 120, India for support to undertake the investigation.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Both authors equally contributed to the execution of experiments and preparation of manuscripts.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Yes.
Competing interests
The authors declare that they have no known competing financial interests that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Meena, P.N., Meena, A.K. & Ram, C. Morphological and molecular diversity in Sclerotium rolfsii Sacc., infecting groundnut (Arachis hypogaea L.). Discov Agric 1, 3 (2023). https://doi.org/10.1007/s44279-023-00003-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44279-023-00003-0