
Abstract
A density functional theory and time-dependent density functional theory approaches were used to understand the structure–property relationships of a series of Ir(III) complexes Ir(x-NHC)(y-ppy)2 [where NHC = 2,3-dihydro-1-methyl-3-phenyl-1H-imidazole, ppy = 2-phenylpyridine, x = Cl, y = H (1a); x = Cl, y = Cl (1a-Cl); x = Cl, y = F (1a-F); x = Cl, y = CN (1a-CN); x = Cl, y = CF3 (1a-CF 3 ); x = F, y = CF3 (2-CF 3 )]. The investigations on the electronic structures in the ground and lowest triplet excited states, the frontier molecular orbitals, the absorption and emission spectra, as well as charge injection and transport of these Ir complexes provided a good understanding of the structure–property relationships. Furthermore, the full details of the metal character in the phosphorescent spectra(3MLCT %), triplet energy (E T1), the singlet–triplet splitting energy (ΔE S1–Tn), 3MLCT–3MC energy gap, as well as d orbitals splitting revealed that quantum yield was effectively enhanced by introducing CN and CF3 groups on the ppy ligands. The designed complexes 1-CN, 1-CF 3 , and 2-CF 3 are expected to be highly efficient phosphorescent materials in organic light-emitting diodes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Phosphorescent iridium(III) complexes have attracted considerable attention for their greatly potential application as highly efficient electroluminescent (EL) emitters in organic light-emitting diodes (OLEDs) [1–3]. Greater success has been achieved in developing the Ir(III) type emitters with 2-phenylpyridine (ppy) ligand [4–6]. For example, the green light emitting tris(2-phenylpyridine) iridium [Ir(ppy)3] exhibits very bright phosphorescence with 90 % efficiency and fast charge injection and transport, which makes Ir(ppy)3 highly attractive for application in OLEDs [4, 7, 8]. In order to meet the requirements of red–green–blue (RGB) full-color displays, enormous efforts have been devoted to modifying the chemical structure of cyclometalated ligands [9–14].
Most recently, String et al. [15] synthesized a series of blue emitters with two ppy groups in combination with N-heterocyclic carbenes (NHC) unit. It was found that the introduction of the electron-withdrawing (Cl, F) and electron-donating (OMe) groups in the NHC ligand exerted a great influence on the photoluminescence properties. Although the previous study [16] has qualitatively analyzed the effect of F atoms on the metal-to-ligand charge transfer (MLCT) and radiative decay rate (k r), detailed analyses about the nonradiative decay rate (k nr), which is another important factor to influence the quantum efficiency, are still scarce. Also, to our knowledge, the effect of Cl atoms at NHC on the phosphorescence quantum yield (another efficient blue emit Ir(Cl-NHC)(ppy)2 synthesized by String et al. [15]) has not been revealed. Therefore, to shed light on the natures of the high quantum efficiency and provide valuable information for new emitters design, complexes 1a-Cl, 1a-F, 1a-CN, 1a-CF 3 , and 2-CF 3 (see Fig. 1a) have been designed and investigated theoretically. Detailed investigations on the electronic structures, frontier molecular orbitals, absorption and emission spectra, as well as charge injection and transport abilities are performed to estimate the photophysical properties of these materials. Furthermore, the analyses of participation of 3MLCT, the singlet–triplet splitting energy (ΔE S1–T1), transition electric dipole moment in the S0–S1 transition (μ S1), d orbital splitting, as well as the energy difference between the 3MLCT/π–π and 3MC d–d state for these Ir complexes are estimated the k r and k nr values to explore the reason of the differences in the quantum yields. We hope this work can provide an in-depth understanding of the optical and electronic properties of this type of complexes, thus providing valuable information for design and synthesis of new emitters with high phosphorescence quantum efficiency.

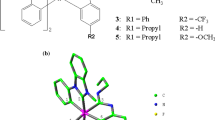
The (1) schematic structures and (2) optimized ground state geometry of studied Ir complexes
2 Computational methods
Density functional theory calculations (DFT) using the mPW1PW91 functional [17] combined with 6-31G(d) basis set for the non-metal atoms and LANL2DZ for Ir atom [18–20] was used to optimize the ground state and ion state geometries of studied complexes, which had been proved to be a reasonable and credible method to predict the Ir(III) complexes with ppy and NHC chelating ligands [16]. Vibrational frequencies were further calculated at the same level to confirm that each of ground geometry was a minimum on the potential energy surface. At the optimized ground geometries, TDDFT calculations using mPW1PW91 functional with the same basis set were carried out to obtain the absorption spectra. To analyze the nature of absorption, the hole and electron distributions were calculated by Multiwfn software [21]. The singlet state (S1) and lowest triplet (Tn) n = 1–3 excited states geometries of studied complexes were calculated at TD-mPW1PW91 method with LANL2DZ basis sets to analyze the S1 and Tn energy gap (ΔE S1–Tn). M06-2X functional [22] was adopted to predict the emission spectra of these complexes, which had been proved to be a reliable method [16]. In addition, the electronic configurations of the triplet metal centered (3MC) d–d states were optimized according to the methodology suggested in Thummel’s work [23]. For a better analysis of the nature of the excited states involved in the emission process, natural orbital transition (NTO) analyses [24] were performed via TD-DFT approach. All the calculations were made with the Gaussian09 software package [25].
3 Results and discussion
3.1 Geometries in ground and lowest-lying triplet excited states
The schematic structures and optimized ground state geometries of six complexes are shown in Fig. 1, together with the number of some key atoms. The metal–ligand bond lengths and bond angles of investigated complexes are collected in Table 1.
Table 1 shows that the changes of these bond angles are minor from complex 1a to these designed materials. As shown in Fig. 1, all these studied complexes adopt a distorted octahedral geometry, which are caused by the d 6 configuration of the Ir(III) center. Besides, the calculated results show that the Ir–C1 (2.075 Å) and Ir–C2 (2.111 Å) bond lengths in complex 1a are shortened by 0.012 and 0.005 Å, respectively, in comparison with those of F-substituted NHC complex 1 (2.087 Å for Ir–C1 and 2.116 Å for Ir–C2) [16]. Clearly, the Cl substitutions in NHC moiety have great influence on the metal–ligand relative to F substitutions, which will increase the probability of charge transfer from the metal to the ligand. In addition, the calculated metal–ligand bond lengths Ir–C3, Ir–C4, Ir–N1, and Ir–N2 on ppy ligands of complex 1a are much shorter than those on NHC ligand (Ir–C1 and Ir–C2 bond lengths), suggesting a strong interaction between Ir metal center and the ppy ligands. Therefore, the introduction of the F, Cl, CN, and CF3 substitutions on the ppy ligands could be efficient strategies to tune the geometry and photophysical properties. As collected in Table 1, the Ir–C3, Ir–N1, Ir–C4, Ir–N2 bonds for 1a-Cl (2.038, 2.048, 2.056, and 2.054 Å, respectively) are much shortened compared to those for 1a (2.043, 2.055, 2.063, and 2.062 Å, respectively). This means that the interactions between the metal and ppy ligands are strengthened by importing the Cl atoms. Similar shortening for the Ir–C and Ir–N bonds (by ca. 0.01 Å) also observed for 1a-F, 1a-CN, 1a-CF 3 , and 2-CF 3 . Normally, the strengthened metal–ligand bond may increase the probability of charge transfer from the metal center to ppy ligands, which will be further testified in the following section.
In order to gain insight into the geometric relaxations upon excitation, the geometries parameters at the lowest-lying triplet excited states (T1) of studied complexes are also collected in Table 1. As listed in Table 1, all the metal–ligand bond lengths of complex 1a at the T1 state are pulled closed to the metal compared with S0 state, especially the bond lengths of Ir–C4 and Ir–N2, which are shortened by 0.014 and 0.032 Å, respectively, upon excitation to T1 state. The strengthened metal–ligand bond is useful to increase the charge transfer from the metal to ligand (MLCT). Furthermore, the Ir–N1 for 1a-Cl and 1a-F is contracted by 0.045 and 0.052 Å, respectively, upon S0–T1 excitation. The slightly elongated Ir–C3 for 1a-Cl and 1a-F can be easily rationalized by the stronger Ir–N1 bond interaction, which weakened the Ir–C3 at its trans disposition. Similarly, for 1a-CN, 1a-CF 3 , and 2-CF 3 , the Ir–C4 in the T1 state is significantly shortened (0.022, 0.045, and 0.036 Å for 1a-CN, 1a-CF 3 , 2-CF 3 , respectively) compared to the S0 state. Therefore, it can be concluded that the ppy ligand is pulled in closer to the metal by introducing these substitutions, which may facilitate the metal center to ppy ligand charge transfer, eventually improving the phosphorescence quantum efficiency.
3.2 Frontier molecular orbitals
To reveal the photophysical properties of these complexes, we will discuss in detail the frontier molecular orbital (FMO) energy levels and components. The main FMO energy level and the contour plots of the HOMO and LUMO are drawn in Fig. 2. The details of FMO compositions of studied complexes are shown in Table S1–S6 in Supporting Information.

The orbital energy level (eV), energy gap (eV), and contour plot of the HOMO and LUMO for studied complexes
Figure 2 shows the HOMO (−5.27 eV) and LUMO (−1.19 eV) values of complex 1a with Cl substitutions at NHC ligand are much lower than the experimental synthesized complex with no substitutions at NHC ligand (HOMO = −5.02 eV and LUMO = −1.00 eV for 1 [16]) and with F substitutions at NHC ligand (HOMO = −5.17 eV and LUMO = −1.12 eV for 1′ [16]). We also noticed that the 1a-CF 3 has decreased LUMO (−2.02 eV) energy level compared with that of 2-CF 3 (−1.97 eV). It is obvious that a type of attracting electron transporting materials can be obtained by introducing the Cl atoms on NHC ligand instead of F atoms. In addition, both of HOMO and LUMO energy levels of these Ir complexes are dramatically stabilized by introducing the electron-withdrawing substitutions on ppy ligand compared to applying them on the NHC ligand, which agrees with our previous report [16]. For example, for 1a-F and 1a-Cl, the LUMO energy levels are decreased by 0.19 and 0.50 eV, respectively, compared with complex 1a. This means that the electron injection ability can be effectively tuned by introducing F and Cl atoms at ppy ligands. Moreover, the LUMO energy of 1a-CN, 1a-CF 3 , and 2-CF 3 are dramatically decreased by 1.44, 0.83, 0.78 eV, respectively, compared with that of complex 1a, which may be attributed to extended conjugated plane (see Fig. 2) between the ppy and NHC in 1a-CN, 1a-CF 3 , and 2-CF 3 . Because of the different degree of decrease in the HOMO and LUMO energy level, the HOMO–LUMO energy gap is decreased in the following order: 1a-F (4.31 eV) > 1a-Cl (4.15 eV) > 1a (4.08 eV) > 1a-CF 3 (3.98 eV) > 2-CF 3 (3.94 eV) > 1a-CN (3.80 eV). It is worth noting that the small HOMO–LUMO gaps of 1a-CF 3 , 2-CF 3 , and 1a-CN might render a red-shift in the emission spectra compared with 1a. Besides the HOMO and LUMO energy, we also notice that inner molecular orbital energies have been increased relative to their corresponding HOMO levels. The energy gap between HOMO-1 and HOMO is calculated to be 0.24, 0.25, 0.12, 0.19 eV for 1a-Cl, 1a-F, 1a-CN, 1a-CF 3 , respectively, which is much smaller than those of 1a (0.39 eV) and 2-CF 3 (0.79 eV). This indicates that the probabilities of electron transition from inner occupied MOs are increased.
As shown in Fig. 2 and Table S1–S6, all these Ir complexes have similar HOMO and LUMO distribution, i.e., the HOMO is composed of ca. 40 % d(Ir), while LUMO is predominantly localized on the π*(ppy) ligands. But the contributions from the ligands in these orbitals are different. For example, the HOMO of complex 1a, 1a-F, and 1a-Cl is mainly composed of ca. 55 %π*(ppy), while for 1a-CN, 1a-CF 3 , and 2-CF 3 , their HOMOs are contributed by 25 %π*(Cl-NHC) + 33 %π*(CN-ppy), 9 %π*(Cl-NHC) + 48 %π*(CF3-ppy), 20 %π*(F-NHC) + 43 %π*(CF3-ppy), respectively. Therefore, the well-conjugated ligands in 1a-CN, 1a-CF 3 , and 2-CF 3 are useful for leading to better electron delocalization over the whole molecule. This would be helpful to understand the trend of the decreased orbital energy levels. In addition, it is also noticed that the contribution of Ir(d) to the occupied frontier orbitals are different by adding the substitutions on ppy ligands (see Table S1–S6), all these different distributions will result in different electronic transition characters upon excitation, which will be further discussed in following section.
3.3 Absorption spectra
On the basis of the optimized ground state geometry, TD-mPW1PW91 method was used to calculate the absorption properties of these Ir complexes. The simulated absorption spectra for these complexes are presented in Fig. 3. The calculated absorption properties including the absorption wavelength spectra, excitation energies, oscillator strength, main configurations, as well as assignment are collected in Table S7–S12. As depicted in Fig. 3, complex 1a displays intense high-energy absorption band (230–300 nm) and weaker band in the region of 300–350 nm, which can reproduce the experimental data [15]. Furthermore, the calculated main absorption peaks of complex 1a are at 394.5, 334.5, 268.5, 256.0, and 237.4 nm (see Table S7), which are well consistent with the experimental values 401, 368, 273, 244, and 215 nm [15], respectively. Thus, the calculations obtained by TD-mPW1PW91 method are reliable in understanding the electronic transition of studied complexes.

Simulated absorption spectra of studied complexes
In order to analyze the nature of absorption, the hole and electron distributions are calculated by Multiwfn software [21]. The hole and electron distributions for the main absorption peaks of 1a are drawn in Fig. 4, while the hole and electron distributions for other complexes are given in Figure S1 in supporting information. Figure 4 reveals that the S0–S1 and S0–S32 transitions for 1a mostly process metal-to-ligand charge transfer (MLCT)/intraligand charge transfer (ILCT) transition. Because the holes of S1 and S32 states are mainly contributed by Ir(III) atom and ppy moiety, while the electrons are only localized on ppy moiety. For S0–S5, S0–S26, S0–S47 transitions of 1a, the distribution of their holes is focused on Ir(III) atom, NHC, and ppy moiety, while the electrons are localized on ppy ligands (except for S47 state, the electrons are localized on NHC ligand), and as a result the mixed MLCT/LLCT/ILCT transition occur. According to the orbital component analysis, the differences in the contributions of the Ir(III) atom on hole and electron distribution (ΔIr) dramatically decrease for S26 (18.7 %), S32 (13.7 %), S47 (7.3 %) relative to S1 (40.9 %) and S5 (40.9 %) of 1a. Thus the mount of MLCT of high-lying absorption band (S0–S26, S0–S32, and S0–S47) of 1a slightly decreases relative to low-lying absorption band (S0–S1 and S0–S5).

The distributions of electron and hole for 1a at the S1, S5, S26, S32, and S47 state. The blue and green isosurfaces represent hole and electron distributions, respectively. The differences in the contributions of the Ir(III) atom on hole and electron distribution (ΔIr, %) are collected
As drawn in Fig. 3, the absorption intensity of these designed Ir complexes is larger than 1a (except for 2-CF 3 ). Therefore, the probability of the intersystem crossing (ISC) from singlet and triplet state may be well tuned by the introduction of these substituent groups at ppy ligands. The calculated lowest-lying absorption band (S1) of 1a-Cl, 1a-F, 1a-CN, 1a-CF 3 , and 2-CF 3 are at 385.9, 369.8, 422.0, 405.8, and 409.7 nm, respectively. Compared with that of 1a (394.5 nm), 1a-Cl and 1a-F complexes are blue-shifted (by 8.6 and 25.5 nm, respectively), while 1a-CN, 1a-CF 3 , and 2-CF 3 are red-shifted (by 27.5, 11.3, 15.2 nm, respectively). This is well consistent with the trend of the HOMO–LUMO energy gaps (see Sect. 3.2), because the excitation of HOMO → LUMO is responsible for the lowest-lying absorption peaks. Based on the hole and electron distributions analysis (Figure S1), all the S1 states for these complexes can be characterized as MLCT/ILCT transition. As expect, the amount of ΔIr in the S1 of 1a-Cl (43.0 %), 1a-F (42.4 %), 1a-CN (46.9 %), 2-CF 3 (45.3 %), and 1a-CF 3 (45.5 %) are large relative to 1a (40.9 %), which means the participation of MLCT in the absorption spectra can be well tuned by introduction of the electron-withdrawing substituent at ppy ligands. Besides, Figure S1 also illustrates all the main absorption peaks for the complexes can be characterized as MLCT/ILCT or MLCT/LLCT transitions. Especially, the high-energy absorption peak of 1a-Cl (268.4 nm), 1a-CN (256.2 nm), 2-CF 3 (237.3 nm) has 35.6, 32.9, and 32.3 % ΔIr contribution, respectively. Obviously, the participation of MLCT in the absorption spectra can be increased by introduction of the Cl, CN, and CF3 substituent at ppy ligands. The increased amount of MLCT might be efficient to collect light energy participation of metals and increase the spin-orbital coupling effect. Meanwhile, the intensities of the absorption bands of 1a-Cl, 1a-CN, and 1a-CF 3 are much larger than those of 1a, which may result in high intensity for the triplet excited states through the ISC procedure, and hence increasing the phosphorescence efficiency.
3.4 Phosphorescence properties
As reported in our previous work [16], the TDDFT method with M06-2X functional is reasonable to predict the emission wavelength of this type of compounds. Thus, the M06-2X functional is applied to calculate the emission spectra of all the studied molecules. The calculated emission wavelengths, main configurations, as well as assignment are summarized in Table 2, together with the available experimental data [15]. The molecular orbital compositions which are responsible for the emission spectra are summarized in Table 3.
The phosphorescence 496.9 nm of complex 1a agrees well with the experimental observed value 507 nm, which is contributed by the excitations of HOMO → LUMO and HOMO-1 → LUMO. Table 4 shows that the HOMO and HOMO-1 has 39 %Ir(d) +56 %π(ppy) and 41 %d(Ir) + 26 %π(ppy) + 33 %π(Cl-NHC), respectively, while LUMO is 96 %π*(ppy) type orbital. Thus, the emission at 496.9 nm can be contributed from 16 % 3MLCT, 3LLCT, and 3ILCT characters. The calculated emission spectra are at 495.3, 479.7, 551.5, 538.2, and 553.2 nm for 1a-Cl, 1a-F, 1a-CN, 1a-CF 3 , and 2-CF 3 , respectively. Compared with complex 1a, the chlorine and fluorine substituted ppy ligand in 1a-Cl (1.6 nm) and 1a-F (17.2 nm), respectively, lead to slightly blue shift, which can be easily understood by the widening HOMO–LUMO gap. Combining Tables 3 and 4, the emission of 1a-Cl and 1a-F can be described as the 3{d(Ir) + π[(Cl-NHC) + Cl-ppy)] → π*(ppy)} and 3{d(Ir) + π[(Cl-NHC) + F-ppy)] → π*(ppy)}excited state, respectively, which can be described as 3MLCT/3LLCT/3ILCT character. For 1a-CN, 1a-CF 3 , and 2-CF 3 , there is a dramatically large red-shifted (by 54.6, 41.3, 56.3 nm, respectively) compared with that of complex 1. This indicates that the emitting color of 1a is efficiently tuned by introducing CF3 and CN substitutions. As seen from Table 3, the emission of 1a-CN, 1a-CF 3 , and 2-CF 3 at 551.5, 538.2, and 553.2 nm, respectively, is also mainly contributed by LUMO → HOMO transition. Due to the significant composition of the d(Ir) orbital in HOMOs, the phosphorescence spectra of 1a-CN, 1a-CF 3 , and 2-CF 3 are mixed characters of 3MLCT/3LLCT/3ILCT.
In order to gain a better insight into the nature of the triplet state, the calculated natural transition orbital NTO is depicted in Fig. 5. The NTO eigenvalues (λ) of these Ir complexes (1a: 0.9616; 1a-Cl: 0.9462; 1a-F: 0.9413; 1a-CN: 0.9766; 1a-CF 3 : 0.9887; 2-CF 3 : 0.9884) are close to 1. It indicates that the hole and electron wave functions dominate the description of S0–T1 transition. From Fig. 5, all these studied complexes have similar hole and electron wave functions. The hole wave function is mainly localized in ppy ligand and Ir(III) atom, while the electron wave function is focused on the same ppy ligand. Obviously, the T1 state of these Ir complexes corresponds to localized transition (LE). In addition, Table 4 shows the distance between centroid of hole and electron (D) of 1a, 1a-Cl, 1a-F, 1a-CF 3 , and 2-CF 3 are 1.332, 1.513, 1.519, 0.997, 0.822 Å, respectively; therefore, it is expected that these complexes probably have a charge transfer excitation (CT) character. Furthermore, as listed in Table 4, the Ir(III) atom composition from hole to electron of these Ir complexes dramatically decreases (12.17, 11.78, 12.17, 14.16, 23.12, and 19.34 % for 1a, 1a-Cl, 1a-F, 1a-CN, 1a-CF 3 , and 2-CF 3 , respectively), which indicates that the MLCT dominates the description of S0–T1 transition.

The hole and electron wave functions of natural transition orbitals (NTO) of Ir complexes for the T1 state
3.5 The phosphorescence quantum yields
To increase the quantum yield (Φ PL) from the emissive excited state to the ground state, a large radiative (k r) value and a small nonradiative rate (k nr) are necessary, as shown in the Eq. 1 [26, 27]
Theoretically, the k r is approximated by the Eq. 2. [28–30]
where n, h, ε 0 are the refractive index, Planck’s constant, and permittivity in vacuum, respectively. The E T1 is the emitting energy and μ S1 denotes transition electric dipole moment in the S0–S1 transition. The 〈Φ S1|H SOC|Φ T1〉 is SOC matrix element, which is considered as a key factor in determine the k r [31]. It is well accepted that the increased contribution of 3MLCT in the T1 state or a minimal ΔE ST is beneficial to enhance the SOC and transition probability [32, 33]. Furthermore, the ISC rate can be performed as a function of the energy separation between the singlet S1 and the closest triplet Tn, [34] which means the minimal ΔE S1–Tn is beneficial to enhance the ISC rate and thus might increase k r [35]. In addition, the d orbital splittings are usually in determining the phosphorescence efficiency [36, 37]. The Δddocc is the energy differences between the two highest occupied molecular d orbitals, while the Δdd* is the splitting between the highest lying occupied and lowest unoccupied d orbitals. Therefore, the calculated E T1, 3MLCT, μ S1, ΔE S1–T1, Δddocc, and Δdd* are collected in Table 5.
As listed in Table 5, the E T1 value of 1a is similar with that of 1′ [16] and 1 [16]; however, the k r value of 1a is obviously different from 1′ and 1, indicating the E T1 value is not a crucial factor to k r. Furthermore, it is interesting to find that the participation of 3MLCT of 1a (16.0 %) [16] is much larger than that of 1′ (13.4 %) [16], and the ΔE S1–T1 of 1a (0.35 eV) [16] is much smaller than that of 1′ (0.67 eV) [16]; however, their k r values are still close to each other. It suggests that other factors may also play an important role in the quantum yield. The larger μ S1 value of 1′ (2.51D) [16] relative to 1a (1.46D) might be one of the main reasons. In addition, the smaller Δddocc values at both the S0 and T1 states of 1′ (0.29 eV for S0 and 0.38 eV for T1) compared with 1a (0.39 eV for S0 and 0.48 eV for T1) may also result in a larger Hsoc matrix element and a faster the radiative decay rate constant k r [37]. Therefore, it can be concluded that the largest 3MLCT and μ S1, as well as the lowest ΔE S1–T1 and Δddocc values are necessary to judge the k r. Furthermore, the excitation energy levels of T2/T3 of designed molecules obtained by M06-2X method based on optimized T2/T3 geometry are shown in Fig. 6. Figure 6 demonstrates that T2 and T3 states of studied complexes are closer to corresponding T1 state; thus, the ΔE S1–T1 is an important parameter to explain the differences in ISC rate as well as the k r values. As shown in Fig. 6, the ΔE S1–T1 is calculated to be 0.24, 0.09, and 0.18 eV for 1a-CN, 1a-CF 3 , and 2-CF 3 , respectively, which is much smaller than experimental synthesized 1′ (0.67 eV) and 1 (0.35 eV). The minimal ΔE S1–T1 will definitely overcome competitive non-radiative decay pathways, therefore, leading to an increased k r. Especially for 1a-CF 3 , its ΔE S1–T1 is close to the most efficient thermally activated delay fluorescence (TADF; ΔE S1–T1 ≤ 0.1 eV) [38, 39] as reported by Adachi’s group. Accordingly, our designed 1a-CF 3 may have high phosphorescence quantum efficiency. Besides, 1a-CN has larger 3MLCT contribution (18 %), μ S1 value (1.22D), and smaller Δddocc (at both S0 and T1 state) than 1a-CF 3 and 2-CF 3 complexes, suggesting that the introducing CN might increase the possibility of enhancement of k r value.

The calculated excitation energy levels (eV) of studied complexes
According to Eq. 1, it is necessary to provide some insight into k nr. The 3MC d–d state is considered as one of the most important deactivation pathways for the emission, the separation between the 3MLCT/π–π and 3MC d–d states is believed to be closely related to nonradiative pathway [40]. Thus, the energy differences between these states are shown in Fig. 7, along with the spin density of theirs 3MC d–d state. As depicted in Fig. 7, the spin density is mainly localized on the Ir atom. The 3MC d–d excited state is lying at a relatively higher energy than that of the 3MLCT/π–π excited states, indicating that the emitting phosphors are the most thermodynamically stable ones. The energy difference between the 3MLCT/π–π and 3MC d–d state of 1a is close to that of 1 and 1′, indicating the nonradiative pathway of 1a is similar with 1 and 1′. Thus k r of these complex [1a (1.6 × 105 s−1), 1 (2.3 × 105 s−1), and 1′ (2.5 × 105 s−1)] are close to each other. By introducing these substitutions, 1a-CN (13.7 kcal/mol), 1a-CF 3 (10.3 kcal/mol), 2-CF 3 (11.4 kcal/mol) have larger energy gaps between 3MLCT/π–π and 3MC d–d states than experimental synthesized molecules, thus leading to a lower k nr. Furthermore, the calculated Δdd* of 1a-CF 3 , 1a-CN, and 2-CF 3 at theirs S0 and T1 state have larger value than 1a, which may result in an effective thermal unaccessible MC and reduce the non-radiative quenching [37].

The energy level diagram of the theoretical designed and experimental synthesized complexes [15] in the T1 and 3MC excited states, together with the contour plots of the spin density distribution in the 3MC state. Complexes 1 [16] and 1′ [16] are the synthesized complexes with F substitutions and no substitutions at NHC ligand, respectively
From the analysis of the participation of 3MLCT, ΔE S1–T1, μ S1, d orbital splitting, as well as the energy difference between the 3MLCT/π–π and 3MC d–d state, it can be founded that compounds 1a-CN, 1a-CF 3 and 2-CF 3 have relatively larger kr values and smaller k nr values compared with experimental synthesized and other theoretical designed molecules. Thus, 1a-CN, 1a-CF 3 and 2-CF 3 may be potential emitting materials with high efficiency.
3.6 Performance of OLEDs
The charge injection, transport, as well as balance are important for the device performances of OLEDs. The ionization potential (IP) and electron affinity (EA) are calculated to evaluate the energy barrier of injection of holes and electrons. A small IP value (large EA) indicates the ease of holes injection (electrons) from the charge injection layer into the emitting materials. The IP and EA are calculated both the vertical excitation (v; at the geometry of neutral molecule) and adiabatic excitation (a: optimized structure for both the neutral and charged molecule). The calculated vertical IP(v), vertical EA(v), adiabatic IP(a), and adiabatic EA(a) of these compounds are listed in Table 6. Table 6 shows both the IP and EA values (both vertical and adiabatic) of 1a are larger than those of 1′ [16] and 1 [16]. Similarly, 1a-CF 3 gives much larger EAs compared with 2-CF 3 . Clearly the electron injection ability of this type of complex can be enhanced by introducing Cl atoms at NHC ligand instead of F atoms, which is consistent with the analyses in their LUMO energies. In addition, it can be seen that the calculated IP values (both vertical and adiabatic) decrease in the order: 1a-CN > 1a-CF 3 > 2-CF 3 > 1a-Cl > 1a-F > 1a, suggesting the ability of hole injection gradually increases. This tendency is consistent with the trend of HOMO energies in Sect. 3.2. For EA values (both vertical and adiabatic), the introduction of CN, CF3, Cl, and F has a positive influence on increasing the EA values. The calculated EA values (both vertical and adiabatic) decrease in the following order: 1a-CN > 1a-CF 3 > 2-CF 3 > 1a-Cl > 1a-F > 1a, indicating that these designed complexes accept an electron more easily than 1a. This trend is well consistent with the order of the LUMO energy.
The mobility of the hole and electron is investigated based on the Marcus theory [41]. As well known, for efficient charge transport, the reorganization energy is required to be small. The hole/electron reorganization energy (λ h/λ e) is calculated as described in previous work [42], and the calculated values of λ h and λ e are also listed in Table 5. From the calculated data in Table 5, 1a has smaller λ e (0.20 eV) than its λ h (0.49 eV). It means that 1a is an excellent electron transport material, which is similar with its derivatives 1′ [16] and 1 [16]. After introducing the electron-withdrawing (F, Cl, and CN) substitutions at ppy ligands, 1a-F, 1a-Cl, and 1a-CN have similar λe and λh values with those of 1a. Their λ h values are almost two times larger than theirs λ e values, suggesting that 1a-Cl, 1a-F, and 1a-CN still have excellent electron transport ability. With respect to 1a-CF 3 and 2-CF 3 , the λ e and λ h values become larger, indicating that they have relatively poor charge transport rate comparison with those of 1a. It is also found that the energy differences between λ e and λ h (Δλ h–e) for all these designed complexes are ca. 0.30 eV, implying that these materials are not suitable to be used as emitting-layer materials in OLEDs because of their unbalanced hole and electron transport.
4 Conclusion
We have employed DFT and TDDFT investigations on the electronic structures, photophysical properties, and phosphorescence quantum yields of a series of Ir complexes coordinated with 2-phenylpyridine (ppy) and N-heterocyclic carbenes (NHC) ligands. It is found that the introduction of F, Cl, CN, and CF3 substitutions on the ppy ligands can strengthen the metal–ligand bond, stabilize the LUMO energy levels, as well as increase the absorption intensities and participations of MLCT in the absorption band. Through the analyses of participation of 3MLCT, ΔE S1–T1, μ S1, d orbital splitting, as well as the energy difference between the 3MLCT/π–π and 3MC d–d state, it can be inferred that compounds 1a-CN, 1a-CF 3 , and 2-CF 3 have larger k r values and smaller k nr values compared with other molecules. In addition, these designed complexes are suitable to be used as electron transport materials because of better electron injection and transfer rates. Theses structure–property relationships are hoped to provide valuable information to obtain highly efficient phosphorescent emitters.
References
Lamansky S, Djurovich P, Murphy D, Abdel-Razzaq F, Lee HE, Adachi C, Burrows PE, Forrest SR, Thompson ME (2001) J Am Chem Soc 123:4304–4312
Zhou G, Wong W-Y, Yao B, Xie Z, Wang L (2007) Angew Chem Int Ed 46:1149–1151
Zhao YY, Gao HF, Wang XM, Qi HL (2015) Inorg Chem 54:1446–1453
Hofbeck T, Yersin H (2010) Inorg Chem 49:9290–9299
Kim DH, Cho NS, Oh HY, Yang JH, Jeon WS, Park JS, Suh MC, Kwon JH (2011) Adv Mater 23:2721–2726
Tian N, Aulin YV, Lenkeit D, Pelz S, Mikhnenko OV, Blom PWM, Loi MA, Holder E (2010) Dalton Trans 39:8613–8615
Finkenzeller WJ, Yersin H (2003) Chem Phys Lett 377:299–305
Baldo MA, Lamansky S, Burrows PE, Thompson ME, Forrest SR (1999) Appl Phys Lett 75:4–6
Sajoto T, Djurovich PI, Tamayo A, Yousufuddin M, Bau R, Thompson ME, Holmes RJ, Forrest SR (2005) Inorg Chem 44:7992–8003
Wu Y, Wu SX, Li HB, Geng Y, Su ZM (2011) Dalton Trans 40:4480–4488
Ma MS, Zou LY, Li Y, Ren AM, Feng JK (2015) Org Electron 22:180–190
Ren XF, Kang G-J, Zhang S-F, Ren A-M, Wong W-Y, Zhou GJ, Liu Y-L (2015) J Photochem Photobiol A Chem 311:85–94
Deaton JC, Young RH, Lenhard JR, Rajeswaran M, Huo S (2010) Inorg Chem 49:9151–9161
Wang L, Wu Y, Geng Y, Wu J, Zhu DX, Su ZM (2014) J Phys Chem A 118:5058–5067
Stringer BD, Quan LM, Barnard PJ, Wilson DJD, Hogan CF (2014) Organometallics 33:4860–4872
Kang GJ, Ren XF, Bai SY (2015) J Organomet Chem 785:44–51
Adamo C, Barone V (1998) J. Chem. Phys. 108:664–675
Hay PJ, Wadt WR (1985) J Chem Phys 82:270–283
Wadt WR, Hay PJ (1985) J Chem Phys 82:284–298
Hay PJ, Wadt WR (1985) J Chem Phys 82:299–310
Lu T, Chen FW (2012) Multiwfn: a multifunctional wavefunction analyzer. J Compd Chem 33:580–592
Zhao Y, Truhlar DG (2008) J Theor Chem Acc 120:215–241
Bark T, Thummel RP (2005) Inorg Chem 44:8733–8739
Martin RL (2003) J Chem Phys 118:4775–4777
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09, Revision B.04. Gaussian, Inc, Wallingford
You Y, Park SY (2009) Dalton Trans 8:1267–1282
Siddique ZA, Yamanoto Y, Ohno T, Nozaki K (2003) Inorg Chem 42:6366–6378
Turro N (1991) Modern molecular photochemistry. University Science Books, Palo Alto
Haneder S, Como ED, Feldmann J, Lupton JM, Lennartz C, Erk P, Fuchs E, Molt O, Münster I, Schildknecht C, Wagenblast G (2008) Adv Mater 20:3325–3330
Minaev B, Baryshnikov G, Agren H (2014) Phys Chem Chem Phys 16:1719–1758
Si YL, Sun XB, Liu YQ, Qu XC, Wang Y, Wu ZJ (2014) Dalton Trans 43:714–721
Sprouse S, King KA, Spellane PJ, Watts RJ (1984) J Am Chem Soc 106:6647–6653
Shang XH, Han DM, Zhan Q, Zhang G, Li DF (2014) Organometallics 33:3300–3308
Kozhevnikov DN, Kozhevnikov VN, Shafikov MZ, Prokhorov AM, Bruce DW, Gareth JA (2011) Williams. Inorg Chem 50:3804–3815
Burin AL, Ratner MA (1998) J. Chem. Phys. 109:6092–6102
Yersin H, Finkenzeller WJ (2008) In: Yersin H (ed) Highly efficient OLEDs with phosphorescent materials. Wiley-VCH, Weinheim, pp 1–97
Tong GSM, Che CM (2009) Chem Eur J 15:7225–7237
Uoyamal H, Goushi K, Shizu K, Nomura H, Adachi C (2012) Nature 492:234–238
Nakanotani H, Higuchi T, Furukawa T, Morimoto K, Numata M, Tanaka H, Sagara Y, Yasuda T, Adachi C (2014) Nat Commun 5:4016–4023
Alary F, Heully JL, Bijeire L, Vicendo P (2007) Inorg Chem 46:3154–3165
Marcus RA (1993) Rev Mod Phys 65:599–610
Ren XF, Ren AM, Feng JK, Sun CC (2009) J Photochem Photobiol A Chem 203:92–99
Acknowledgments
Financial supports from NSFC (Nos. 21243006 and 51304193), the Basic Research Program of Jiangsu Province (No. BK20130172), the Fundamental Research Funds for the Central Universities (No. 2013QNA14). A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions. We are grateful to the High Performance Computing Center of China University of Mining and Technology for the award of CPU hours to accomplish this work.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ren, XF., Kang, GJ., He, QQ. et al. A theoretical analysis of the effects of electron-withdrawing substitutions on electronic structures and phosphorescent efficiency of a series of Ir(III) complexes with 2-phenylpyridine ligands. Theor Chem Acc 135, 24 (2016). https://doi.org/10.1007/s00214-015-1773-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-015-1773-9