
Abstract
With staggering progress on genetic manipulation strategies, Saccharomyces cerevisiae is becoming an ideal cell factory for the de novo biosynthesis of lipid compounds. However, due to their hydrophobicity, lipids tend to be accumulated within intracellular spaces and cause a high burden on cell activity and induce product inhibition effect, which ultimately restricted the lipids biomanufacturing for industrial application. Herein, an oleic acid stress (OAS) model was applied for the long-time domestication of BY4741 cells, and a subclone of A-22 was obtained through a series of acclimation (0.1% glucose and 0.2% oleic acid), showing increased accumulation of both biomass and intracellular lipid droplets compared to WT. Comparative transcriptome analysis indicated that compared to fatty acid metabolism, most transcripts enriched in the pathways of glucose catabolism (glycolysis and citrate cycle) and lipid synthesis (phospholipid and sterol) were down-regulated under OAS. While interestingly, most the above transcripts tended to be ‘restored’ in adapted strain A-22. In addition, for physical adaptation, significant increase of phosphatidylcholines was identified by lipidomic analysis, which probably caused the subsequent subcellular expansion of peroxisomes and lipid droplets as observed in the adapted strain, since phosphatidylcholines are the major constituent of their membranes. The present study systematically investigated both the phenotype change and molecular mechanism on adaptation of S. cerevisiae towards oily environment. Detailed information on functional transcripts may provide novel rational modification targets to reinforce the hydrophobic lipids biosynthesis within S. cerevisiae engineered cell factory.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lipids are a group of diverse and ubiquitous hydrophobic compounds with important biological functions, including as structural components of cell membranes, energy storage sources and vital participation in signal pathways [1]. These chemical features are present in a broad range of molecules such as fatty acids, phospholipids, sterols, sphingolipids, triacylglycerols, terpenes and other, estimated to have a thousand or more lipid species, which have been widely applied in the industry of food, medicine, fuel, cosmetics and other fields [2,3,4]. Nowadays, with the staggering advance in synthetic biology, high-yield of lipid compounds has been achieved in metabolically engineered microorganisms [5, 6], offering a promising approach in green and sustainable manner [7,8,9].
The well-characterized yeast Saccharomyces cerevisiae, aided by its GRAS (generally recognized as safe) status, has served as an attractive biosynthetic platform due to the inherent abilities such as ease of genetic editing, short generation time, and robustness and tolerance to harsh fermentation conditions. For de novo biosynthesis of lipids, the glycolytic fluxes to fatty acid biosynthesis, specifically to enhance the supply of precursors in S. cerevisiae has been considered the main strategy for metabolic engineering [10, 11], and high yields of fatty acids with different chain lengths, or its derivatives such as fatty alcohols, fatty acid esters or others have also achieved [4, 12,13,14]. In addition, the mevalonate (MVA) pathway presented in yeast as typically existed in most eukaryotic cells, providing the de novo biosynthetic route of high value terpenoids from acetyl-CoA [2], including sterols (stigmasterol, sitosterol, cholesterol), lycopene [15], β-carotene [16], arteannuic acid [17], taxene [18], α-santalene [19], ginsenoside [20], geraniol [21], etc., that can be obtained in S. cerevisiae cell factory fermented from simple carbon source.
Despite the advanced synthetic metabolic engineering strategies now available for detailed reinforcement of target lipids biosynthesis, developing new cell factories that meet the economic requirements for industrial scale production is still challenging [2, 22]. As a hydrophobic molecule, lipids tend to be accumulated within intracellular spaces rather than extracellular secretion, and the accumulated intermediates not only directly affect cell activity, but also induce product inhibitory effects [23]. Under normal physiological condition, the intracellular lipids homeostasis of S. cerevisiae is dynamically and precisely regulated by the inner membrane system, where key steps such as ester synthesis, storage and hydrolysis of lipids take place [15, 24,25,26]. Especially, enzymes involved in the post-ergosterol pathway are mainly located in the endoplasmic reticulum membrane for correct scaffolding and effective electronic transfer [27]. The accumulated ergosterol was attend to be stored in lipid droplets, a typical storage organelles at the centre of lipid and energy homeostasis that was composed of a monolayer phospholipid membrane using phosphatidylcholine as the main component [28]. Recently, genetic modification of some key molecules involved in the regulation of lipid droplets [26, 29, 30], cell wall structure [31] and extracellular secretion [32] manifested positive enhancement in lipid accumulation within S. cerevisiae. However, due to the unveiled molecular mechanism of lipids homeostasis especially for those S. cerevisiae cell factories that harboring overburdened lipids components, effective and direct cellular modification strategy to alleviate “lipids toxicity” and to increase the specific productivity per unit cell is still limited.
As the most abundant fatty acid species of the yeast cells [33], oleic acid (C18:1n-9) takes important role in the lipid homeostasis of S. cerevisiae. Accordingly, “oleic acid stress (OAS)” has been used as a classical model to monitor the phenotype change when cells exposed to the oleic acid instead of glucose, and the yeast cells exhibited induced fatty acid β-oxidation along with expansion of the peroxisomal compartment, which was based on the transcriptional induction of target genes with the oleate response element (ORE) by the transcription factor Pip2p–Oaf1p [34]. Speculating that the regulation of lipid hemostasis is a complicated and multiple-stage process that involved in extensive regulation and complex interactions between metabolic pathways as well as intracellular membrane re-organization, systematical in-depth investigation is therefore needed. Moreover, so far, the related research has mainly focused on the passive phenotypic change in S. cerevisiae when suddenly exposed to oleic acid, however, the initiative tolerance of oleic acid accumulation within S. cerevisiae has not been addressed [34, 35]. With the rapid advance in the integrated approach for the detection, quantitation and pathway reconstruction of lipids and related genes at a systems-biology level is becoming a new trend [35,36,37], such as comparative transcriptome and lipidome analysis, detailed information involved in lipids homeostasis regulation is respected to be fulfilled.
In present study, an OAS adapted strain of S. cerevisiae A-22 was obtained after a serial process of domestication in YPO medium. To provide comprehensive understanding of the intracellular lipids regulation mechanism, differentially expressed genes as well as lipid components between the original stain S. cerevisiae BY4741 and the adapted stain A-22 were respectively monitored by transcriptome and lipidome analysis under OAS, essential genes involved in the significant metabolic change as well as inner-membrane reorganization were specially noted, providing detailed information of S. cerevisiae lipids homostasis regulation, which may offer further regional modification strategy of the lipid tolerated cell factory for enhanced biosynthesis of hydrophobic components.
Materials and methods
Yeast strains, culture condition and oleic acid-stress adaptation
The BY4741 wild-type (WT) strain of S. cerevisiae (MATa, his3Δ, leu2Δ, met15Δ, ura3Δ) was obtained from Euroscarf (Institute for Molecular Biosciences, Frankfurt, Germany) (http://www.euroscarf.de/) grown in YPD medium (2% peptone, 1% yeast extract, 2% glucose). For oleic acid stress, cells were transferred into YPO medium (0.5% peptone, 0.3% yeast extract, 0.5% KH2PO4, 0.2% Tween 80, 0.1% glucose, 0.2% oleic acid), and changes in both growth behavior and inner membrane organization was respectively monitored by measuring OD600 value and fluorescent staining. For oleic acid environment adaptation, S. cerevisiae BY4741 (WT) was propagated by successive batch refreshments maintained constantly in YPO for 22 generations with 2% inoculum fermented at 180 rpm at 30 °C every 24 h as one round of domestication, and the subpopulation of the domesticated culture solution was obtained and named as A-22, and the original culture solution after 1 round of YPO treatment was named as A-1. Reagents including yeast extract (Oxoid Ltd, England), peptone (Becton, Dickinson and Company, France), glucose (Aladdin Corp., China) and oleic acid (Aladdin Corp., China) was used in present study.
Growth curve determination
1 ml of culture solutions from YPO or YPD were collected at indicated time of fermentation and the corresponding OD600 value was measured with a spectrophotometer (AOE instrument, Shanghai, China). Cells cultured in YPO were obtained by centrifugation, washed with 10% Tween 80 to remove oleic acid, and resuspended by deionized water to determine the OD600 value.
Nile red staining and fluorescence microscopy
Cell staining was typically performed with 1 ml of cell suspension. Fluorescence dyes, Nile Red (Aladdin Corp., China), was added from stock solutions in DMSO at a final concentration of 2 μg/ml. Fluorescence microscopy was performed after 20 min of incubation at 30 °C without subsequent washing of cells. Lipid droplets were observed under the RFP fluorescent cube (Ex = 531/40; Em = 593/40) by a fluorescence microscope (Carl Zeiss Microscopy LLC, USA).
Spectro-fluoremetric quantification of lipid content
As in a previous study [38], spectro-fluoremetric quantification of lipid content within yeast cell was performed by nile red staining and the fluorescence intensity was measured on a spectrofluorometer (Tecan Austria GmbH Untersbergstr. 1A A-5082, Austria). Cells were cultured in YPD or YPO media for 24 h and collected by centrifugation, washed with 10% Tween 80 to remove oleic acid combined with deionized water for three times to completely remove the oleic acid within YPO. The OD600 of the different culture solution was respectively adjusted to 1.0 with sterile water and cells were stained with 2 μg/L of nile red. The fluorescence intensity of RFP (Ex = 531/40; Em = 593/40) was quantified after 20 min of incubation at 30 °C, and the fold change of lipid content in different group was correspondingly compared.
RNA-seq analysis
Three groups of experiment samples including BY4741 cells of WT, A-1 and A-22 were collected by centrifugation after 24 h of fermentation respectively in YPD, YPO and YPO. Collected cells were washed twice with ice-cold ddH2O and the total RNAs were extracted with RNA Prep Pure Cell/Bac-teria Kit (DP430) (TIANGEN, Beijing, China) according to manufacturer’s instructions and prepared for transcriptomics analysis. The RNA amount and purity of each sample was quantified using NanoDrop ND-1000 (NanoDrop, Wilmington, DE, USA) and the RNA integrity was assessed by Agilent 2100. A total amount of 3 μg RNA per sample was used for the sample preparations. The RNA sequencing was performed on an Illumina Hiseq platform (NEB, USA) according to the manufacturer’s instructions by Lianchuan Bioinformation Technology Co., Let (Hangzhou, China) as previously reported [39]. For detailed analysis of read counts, the following software was used for trimming, mapping, and determination: Cutadapt software was used to remove the reads that contained adaptor contamination. And after removed the low quality bases and undetermined bases,we used HISAT2 software to map reads to the genome. The mapped reads of each sample were assembled using StringTie with default parameters. Then, all transcriptomes from all samples were merged to reconstruct a comprehensive transcriptome using gffcompare software. After the final transcriptome was generated, StringTie and ballgown were used to estimate the expression levels of all transcripts and perform expression level for mRNAs by calculating FPKM. The differentially expressed mRNAs were selected with fold change ≥ 2 or fold change ≤ 0.5 and p value ≤ 0.05 by R package edgeR or DESeq2, and then analysis GO enrichment and KEGG enrichment to the differentially expressed mRNAs.
Lipidomic analysis
Yeast cells for BY4741 WT, A-1 and A-22 was collected by centrifugation after 24-h fermentation in shake-flask bottle at 30 °C in YPD, YPO and YPO medium, respectively. Six independent repetitions were executed for the extraction and subsequent analysis process. The collected samples were thawed on ice, and metabolites were extracted from 20 µL of each sample using 120 µL of precooled 50% methanol buffer. Then the mixture of metabolites was vortexed for 1 min and incubated for 10 min at room temperature, and stored at − 20 °C overnight. The mixture was centrifugated at 4000g for 20 min, subsequently the supernatant was transferred to 96‐well plates. The samples were stored at − 80 °C prior to the LC‐MS analysis. Pooled quality control (QC) sample were also prepared by combining 10 μL of each extraction mixture. All chromatographic separations were performed using an ultra-performance liquid chromatography (UPLC) system (SCIEX, Cheshire, UK). An ACQUITY UPLC BEH Amide column (100 mm × 2.1 mm, 1.7 µm, Waters, UK) was used for the reversed phase separation according to a previous report [40]. A high-resolution tandem mass spectrometer TripleTOF5600plus (SCIEX, Cheshire, UK) was used to detect metabolites eluted from the column. The combined retention time (RT) and m/z data were used to identify each ion. KEGG pathway (https://www.kegg.jp/kegg/pathway.html) and HMDB (http://www.hmdb.ca/) databases were used to perform level-one and level-two identification and annotation. Screening and quantitative analysis for differential metabolites were conducted using metaX software (http://metax.genomics.cn/).
Results and discussion
Oleic acid stress induced significant growth inhibition and lipid droplets accumulation in S. cerevisiae
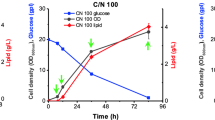
Saccharomyces cerevisiae BY4741 was extensively used as a basic cell model in previous study. Herein, changes in the phenotype of both growth behavior and inner membrane organization was respectively investigated after OAS exposure. As shown in Fig. 1A, after 16 h, the consumption of glucose induced significant growth inhibition in YPO medium, and after 48 h of fermentation, no further accumulation of biomass of A-1 was observed in YPO (OD600 = 0.35) compared with WT in YPD (OD600 = 4.32), and the content of oleic acid (0.2%) within YPO medium remained un-utilized. Distinct from the serious growth inhibition after OAS, the biomass of the adapted strain A-22 increased uniformly by 2.5-folds during the initial 16 h and the final biomass was 1.17-folds higher compared with the original strain (Fig. 1B). A-22 also had slightly higher utilization of oleic acid than A-1, although not obviously. The effect of OAS on the cellular inner-membrane organization as well as the neutral lipids content in S. cerevisiae was further analyzed by the probe nile red (Fig. 1C). Cells demonstrated remarkable enlargement of lipid droplets under OAS, and the phenotype was even dominant in the adapted strain A-22 both by the number and volume of accumulated lipid droplets. The total content of neutral lipids in each strain was subsequently quantified by the fluorescence intensity of nile red. In line with the cellular lipid droplets accumulation, strain A-22 exhibited strongest intensity per unit cell, which was 1.45-fold and 3.67-fold higher compared with A-1 and WT stain, respectively, demonstrating that long time exposure to OAS induced significant lipid droplets accumulation in the domesticated strain A-22 (Fig. 1D).

Changes in growth behavior and cell-membrane organization of BY4741 under oleic acid stress. (A). Growth curve of A-1 and WT and concentrations of glucose; (B). Growth curve of A-1 and A-22 and concentrations of glucose and oleic acid; (C). Fluorescence microscope of lipid droplets within A-1, A-22 and WT strain by nile Red staining; (D). Comparison of fluorescence intensity of nile red between A-1, A-22 and WT strain. Three independent experiments were carried out and the data were presented with standard derivations
Pathways of lipid metabolism revealed significant enrichment by RNA-seq comparison assay
To explore the detailed information on molecular mechanism of S. cerevisiae adaptation under OAS, three strains of WT, A-1 and A-22 was respectively transcriptome sequenced. Under the OAS, S. cerevisiae BY4741 showed a series of changes in KEGG pathway, among which, pathways of ribosome (ko03010), proteasome (ko03050) and nitrogen metabolism (ko00910) were noted significantly changed, perhaps due to the serious growth inhibition under OAS. In addition, pathways involved in the carbonhydrate metabolism, lipids biosynthesis including fatty acid metabolism (ko01212), terpenoid skeleton metabolism (ko00900), glycolysis/gluconeogenesis (ko00010) were found most significantly changed (p ≤ 0.05) (Table 1). Correspondingly, the number of significantly changed genes in relevant KEGG pathway was listed in Fig. 2, most of which was related with lipid metabolism or glucose metabolism and most of the transcript involved in the above pathways were down-regulated when cells initially stressed by oleic acid (A-1 vs. WT), but after the cells adapted to the oleic acid environment, these transcripts within the inhibited pathways showed significant up-regulation (A-22 vs. A-1).

The number of significantly changed genes in the relevant KEGG pathway of (A). A-1 vs. WT; (B). A-22 vs. A-1. The paths represented by the numbers on the abscissa are as follows: 1. Glycolysis / Gluconeogenesis; 2. citrate cycle (TCA cycle); 3. pentose phosphate pathway; 4. fatty acid biosynthesis; 5. fatty acid elongation; 6. fatty acid degradation; 7. biosynthesis of unsaturated fatty acids; 8. fatty acid metabolism; 9. steroid biosynthesis; 10. ubiquinone and other terpenoid-quinone biosynthesis; 11. terpenoid backbone biosynthesis; 12. sesquiterpenoid and triterpenoid biosynthesis; 13. inositol phosphate metabolism; 14. glycosylphosphatidylinositol(GPI)-anchor biosynthesis; 15. glycerophospholipid metabolism; 16. phosphatidylinositol signaling system; 17. ether lipid metabolism; 18. sphingolipid metabolism; 19. protein processing in endoplasmic reticulum; 20. peroxisome; 21. oxidative phosphorylation. These paths are classified into five groups: sugar metabolism (paths 1–3), fatty acid metabolism (paths 4–8), sterol and terpenoid metabolism (paths 9–12), phospholipid metabolism (paths 13–16), and other pathways (paths 17–21). They are represented by different colors in Fig. 2
The expansion of peroxisomes under oleic acid environment cannot be ignored due to its close relationship with β-oxidation [41]. The significantly differentially expressed with peroxisome-related function genes were listed in Table S1, most of which were remarkably up-regulated and main function in detoxification, fatty acid oxidation and the metabolism of nitrogen-containing substances, such as POX1, POT1 (Peroxisomal Oxoacyl Thiolase 1), SPS19 (SPorulation-Specific) and FAA2 (Fatty Acid Activation 2) were up-regulated by 25.2, 9.5, 10.3, and 3.2 times respectively compared A-1 to WT. In addition, obvious upregulation of PEX (PEroXin) family genes that involved in the biogenesis of peroxisomes were also observed. Especially, PEX11, involved in the proliferation of peroxisomes, defined as an increase in size and/or number of peroxisomes[42], was up-regulated 3.6 times in strain-A-1 relative to WT, and was further up-regulated by 1.5 times in strain-A-22, supporting that S. cerevisiae accumulated peroxisomes under oleic acid pressure.
Lipid metabolism was partially restored in strain A-22 after oleic acid adaption
To provide further information on metabolic reprogramming under OAS of S. cerevisiae, transcriptome sequencing was carried out and differentially expressed transcripts between A-1 vs. WT and A-22 vs. A-1 was respectively compared. Accordingly, the corresponding enrichment pathways were especially depicted with specific genes and their transcription levels were noted in different color. As shown in Fig. S1, under OAS condition, most genes involved in glucose metabolism, such as PGK1 (3-PhosphoGlycerate Kinase 1), PDA1 (Pyruvate Dehydrogenase Alpha 1), PDB1 (Pyruvate Dehydrogenase Beta subunit 1), and PDC1 (Pyruvate DeCarboxylase 1), were significantly down-regulated by 8.4, 2.8, 2.0, and 7.2 times in strain A-1 compared with WT. However, there were still some transcripts up-regulated, such as PFK1 (PhosphoFructoKinase 1) and FBA1 (Fructose-1,6-Bisphosphate Aldolase 1) respectively increased by 2.0 and 4.5 times, both involved in the synthesis of glycerone-P from D-glucose, an important intermediate for the synthesis of PA (Phosphatidic acid). Furthermore, the down-regulated transcripts including PGK1, PDA1, and PDC1 were notably up-regulated by 4.8, 4.1, and 4.6 times, respectively in the adapted strain of A-22 compared with A-1, which were even higher than WT-group as shown in Table S2. Considering that the glucose metabolism pathway is not only a way for cells to use glucose as a carbon source to supply energy, but also produces many important intermediates, the increase of key enzyme transcription may provide the molecular foundation of A-22 for the long-time adaptation to OAS.
Pathway enrichment results also demonstrated that lipids metabolism was significantly enhanced under OAS (Table 1), which was further confirmed in the adapted strain A-22. As shown in Fig. S2, due to the change of carbon source, the energy required by A-1 and A-22 cells was mainly come from the oxidation of fatty acids under OAS. In line with previous reports [34], main genes that involved in fatty acid oxidation were significantly up-regulated. For example, The FAA family member of FAA1, FAA2, FAA4 were respectively up-regulated by 2.1, 3.2, and 2.1-folds, and combined with an obvious induction of POX1, POT1 and INO1 (INOsitol requiring 1) respectively by 25.2, 9.5 and 12.1-folds that participate in fatty acids oxidation in peroxisomes. Compared with A-1, the transcript level of most genes in A-22 remained no obvious change in fatty acid oxidation pathway. However, the expression level of several genes were notably recovered in A-22 compared with A-1. ETR1 (Enoyl-acyl-carrier-protein reductase 1), which functions in the elongation of fatty acids [43], was significantly down-regulated in A-1while it was up-regulated by 3.8 times in A-22, indicating that the catabolic pathway of fatty acid was slightly biased towards biosynthesis. In addition, three transcripts of FAS1 (Fatty Acid Synthetase 1), FAS2, and HTD2 (Hydroxyacyl-Thioester Dehydratase) involved in the oxidation of fatty acids in the cytoplasm, mitochondria and plastids, were down-regulated under sudden OAS by 2.9, 1.4, and 1.2-fold in A-1, but were respectively up-regulated by 2.3, 4.0, and 2.7-fold in A-22. As shown in Table S3, fold-changes of genes related with the fatty acid oxidation pathway also showed the “restoration” performance in A-22, which may also support its adaptation to the oleic acid environment.
An obvious reduction of transcripts (up to 90%) was observed in steryl ester (SE) biosynthesis pathway as shown in Fig. 3. It is worth mentioning that the originally down-regulated ERG (Ergosterol biosynthesis) genes were significantly up-regulated in the adapted A-22 after long time of domestication, and detailed information of their fold-change was shown in Table S4. It seems that the inhibition of key enzymes in the ergosterol synthesis pathway led to the accumulation of some intermediate products, and ARE1 (Acyl-coenzyme A: cholesterol acyl transferase-Related Enzyme 1) acylated them to prevent their accumulation from causing toxicity [22]. Correspondingly, the down-regulation of ARE1 and up-regulation of ARE2 as found in A-22 strain further supported the tendency of sterol synthesis restoration.

Changes in steryl ester biosynthetic pathway of A-1 vs. WT and A-22 vs. A-1, respectively. Changes in related genes in pathway were represented by FC values, from significantly down-regulated (blue) to significantly up-regulated (red). The color on the left of each gene represents A-1 vs. WT, and the color on the right represents A-22 vs. A-1. Some specific metabolic intermediates are represented by boxes
Physiological adaptation of strain A-22 by enhanced phosphatidylcholine biosynthesis and lipid droplets expansion
Results from comparative transcriptome assay exhibited that the phospholipid synthesis pathway was significantly enriched and most of the transcripts were obviously up-regulated (Fig. 4). To provide the direct evidence, comparative lipidome analysis was further performed to compare changes in lipid components within yeast cells after adapting to the oleic acid environment. The principal component analysis of lipids between the three strains showed significant differences in the PCA diagram (Fig. 5B). The differentially expressed lipids (DELs) between the A-1 vs. WT group and the A-22 vs. A-1 group are shown in the volcano maps (Fig. 5C), in which 3570 (A-1 vs. WT, FC ≥ 2.0, Q value ≤ 0.05) and 1843 (A-22 vs. A-1, FC ≥ 2.0, Q value ≤ 0.05) substances were significantly up-regulated, and 1461 (A-1 vs. WT, FC ≤ 0.5, Q value ≤ 0.05) and 501 (A-22 vs. A-1, FC ≤ 0.5, Q value ≤ 0.05) substances are significantly down-regulated. Heat maps of DELs in A-1 vs. WT group and A-22 vs. A-1 group were drawn to show the distribution of DELs (Fig. 5D). The “restoration” of A-22 from A-1 can also be seen through the heat map. We also conducted pathway enrichment analyses of the DELs based on the KEGG database to explore the changes of metabolic pathways in A-22 vs. A-1 as shown in Table 2. Pathways related to glycerophospholipid metabolism, sphingolipid metabolism, linoleic acid metabolism were the most significantly enriched.. The DELs are mainly phosphatidylcholines, phosphatidylethanolamines, phosphatidylinositols, phosphatidylserines, phosphatides and other phospholipid related compounds. Fold changes of related genes in phospholipid synthesis pathway were listed in Table S5. A joint analysis of both transcriptome and lipidome of the phospholipid metabolism pathway demonstrated the linkage between changes in gene expression levels with the changes in lipid metabolite levels, revealing that the biosynthesis of some types of PC was up-regulated. Among them, fatty acids are mainly 16–20 carbon chain lengths, especially oleic acid and linoleic acid. This is not surprising when oleic acid was the only carbon source at the late stage of OAS. And related genes such as EKI1 (Ethanolamine KInase 1), OPI3 (OverProducer of Inositol 1) and CHO2 (CHOline requiring) related to the formation of PC (Phosphatidylcholine) from PE (Phosphatidylethanolamine), were up-regulated by 2.1 times, 3.8 times and 6.8 times, respectively, resulted in the significant accumulation of PCs [35]. In addition, It is noted that PSD1 (PhosphatidylSerine Decarboxylase 1) and PSD2 in the path from PS to PE were respectively down-regulated by 2.2 and 2.3 times that may cause PE (Phosphatidylserine) reduction. The linkage analysis of the changes in gene expression levels with the changes in lipid metabolite levels in A-22 revealed that, during adaptation to the oleic acid environment, the yeast cells further increased the PC accumulation and restored PI (Phosphatidylinositol) synthesis, which is necessary for the enrichment of membrane components [44]. Accordingly, increase in LPCs (Lysophophatidylcholines), PC and PI-Cer (Ceramide phosphoinositol) were associated with overexpression of some key genes such as OPI3, CPT1 (CholinePhosphoTransferase 1), CKI1 (Choline KInase 1) and PIS1 (Phosphatidyl Inositol Synthase 1). Therefore, the changes in the phospholipid composition of A-22 indicated that PC was important during the adaptation to the oleic acid environment, which was corresponded to the results from comparative transcriptional analysis.

Comparative transcriptome phospholipid synthesis pathway of A-1 vs. WT and A-22 vs. A-1, respectively. Changes in related genes in pathway were represented by FC values, from significantly down-regulated (blue) to significantly up-regulated (red). The color on the left of each gene represents A-1 vs. WT, and the color on the right represents A-22 vs. A-1. Some specific metabolic intermediates are represented by boxes

Comparative analysis of the significantly different lipid metabolites ( A1 vs. WT and A22 vs. A1, respectively) under oleic acid stress. (A) HMDB Super class of metabolites involved in different pathways. (B) PCA score plot for QC(blue), WT(purple), A1 (red) and A22 (green). (C) Volcano plots of the significantly different metabolites in group of WT vs. A1 (left) and A22 vs. A1 (right) based on the following criteria: log2 fold change > 1 and adjusted P-value < 0.05. The red indicates significantly up-regulated metabolites, and the green represents significantly downregulated metabolites. (D) Cluster diagram of the significantly different metabolites. Each column in the heatmap represents a sample and each row represents a metabolite
Lipid droplets are the most important lipid storage organelles in yeast cells with the monolayer phospholipid membrane coating that well separates the internal hydrophobic compounds from the external water [45, 46]. Combining the above results of the strengthened PC synthesis pathway and the expansion of lipid droplets, thereby the storage of hydrophobic compounds in yeast was subsequently enhanced [25]. To provide in-depth exploration on functional genes involved in the formation of lipid droplets, GO pathway enrichment assay was conducted and the most differentially expressed transcripts were listed in Table 3. Moreover, key steps participated in lipid droplets formation were demonstrated in Fig. 6. The results showed that a series of genes involved in lipid droplet formation experienced down-regulation in A-1 and further “restoration” in A-22. Lipid droplets are reported to form from ER where TAGs (Triacylglycerols), SEs (Steryl esters) and related proteins were aggregated [47]. In yeast, the activity of SEI1p (SEIpin 1 protein) is inseparable from Ldb16p (Low Dye Binding 16 protein) [48] (Fig. 6A). The two form LDB16/SEI1 complex and bind to the specific ER area as the origin of lipid droplets formation, then both the lipid droplet content and lipid droplet-related protein aggregated and helped lipid droplet formation in a correct direction [48]. Transcriptome results showed that the transcription levels of LDB16 and SEI1 were down-regulated by 3.3 and 2.4 times respectively in A-1. This seems to be in contradiction with the phenotype of lipid droplets expansion under OAS, which was probably due to the large amounts of TAG produced in A-1 the Ldb16p/SEI1p complex that become no longer essential for the formation of lipid droplets. Because large amounts of TAGs do not require the Ldb16p/SEI1p complex to aggregate to form LDs [49]. After all, lipid droplets could still be produced in SEI1-knocked out strain, although the size of the lipid droplets was not uniform [50,51,52]. Moreover, LDB16 and SEI1 were significantly up-regulated in A-22 strain, which further increased the intracellular lipid droplets accumulation (Fig. 1D). In addition, Ldo16p (LD Organization protein of 16kD) and Ldo45p (Lipid Droplet Organization protein of 45 KD) could form a complex and combine with Ldb16p/SEI1p. Among them, Ldo45p promoted the aggregation of neutral lipids and lipid droplet proteins, and Ldo16p was related to lipid droplet lipophages [53, 54], both of which were synchronized with the Ldb16p/SEI1p complex at transcription level. With the enrichment of lipid droplet contents on the ER membrane, the "lens" structure was formed [44], and more PLs (Phospholipids) were also recruited. At this time, Scs3p (Suppressor of Choline Sensitivity 3 protein) and Yft2p would also bind here [50, 55] (Fig. 6B) to facilitate the formation of "lens". In A-1, YFT2 and SCS3 were up-regulated by 1.1 and 2.2 times, respectively, which ensured the occurrence of lipid droplets. In A-22, SCS3 was adjusted-down by 1.2 times and YFT2 was adjusted-up by 1.8 times to cope with the down-regulation of SCS3. Although the two are homologous proteins, they seemed to have different expression levels at different stages. In addition, they also have complex relationships with a series of gene networks, such as pathways for the synthesis of sphingolipids, phospholipids and inositol phosphates. Recent studies have also shown their function as acyl–coenzyme A diphosphatase and play an important role in ER homeostasis. However, the detailed mechanisms, including whether they complement or interchangeable each other, have not yet been resolved [55, 56]. Lipid droplets will remain in contact with the ER membrane after maturation to achieve a real-time lipid exchange with the ER membrane [57] (Fig. 6C). At this time, lipid droplets would also have active contact with other organelles that depends on the kind of lipid droplet protein, which can vary greatly depending on the growth state. Some related genes not mentioned above were shown in Fig. 6. In addition, the significantly differential transcripts with lipid droplet-related functions were also listed in Table 3 and Table S6, all of which were involved in the morphology of lipid droplets (ENV9) [58], cell detoxification (YIM1) [59], metabolism of lipid droplet contents (ERG gene [2] and FAA gene [60]). These genes often showed opposite trends in different comparison groups, such as AYR1, ERG27, TDH3, etc., which were down-regulated in A-1 vs. WT group and significantly up-regulated in A-22 vs. A-1 group. Most recently, rational modulation of lipid droplets is becoming a new research hot spot to reinforce the hydrophobic component storage within yeast cells [47]. With the regard that the lipid droplets are extremely active organelles [28, 61,62,63], the above noted transcripts may provide new modification targets.

Changes of related gene involved in lipid droplet formation process. Genes were represented by FC values, from significantly down-regulated (blue) to significantly up-regulated (red), and the changes of A-1 vs. WT and A-22 vs. A-1 are shown in the left and right regions of the gene, respectively. (A) The lipid droplet contented TAG and SE, which accumulated in the lumen of the ER membrane, while the LDB16/SEI1 complex bound to the ER membrane and promoted the aggregation of the contents and lipid droplet protein. The required phospholipids were synthesized by phospholipid synthesis-related proteins; (B) with the increase of TAG and SE, SCS3p and YFT2p helped to form "lens", while more proteins were added to promote the formation of lipid droplets; (C) after the lipid droplets matured, they still kept in touch with the ER to realize the real-time exchange of neutral lipids
Conclusion
In present study, both the transcriptome and lipidome was respectively conducted during the oleic environment adaptation of S. cerevisiae BY4741. Comparative data indicated that WT cells underwent significant metabolic reprogramming when exposed to sudden oleic acid stress. While interestingly, pathways involved in lipid metabolism were partially restored in the adapted strain A-22 after long time of domestication. Besides, strain A-22 demonstrated significant expansion of cell inner-membrane with an obvious PC accumulation, enrichment of the functional genes involved in the biosynthesis of both lipid droplets and peroxisomes were specially noted. The above data systematically fulfilled molecular mechanism on the multi-stage regulation of lipids biosynthesis, transportation and storage, the differentially expressed genes as noted in our manuscript provides the potential targets of genetic modification to facilitate the biosynthesis of PC, enlarge the lipid storage organelles such as lipid droplets and peroxisomes in addition to the solely reinforcement in lipid metabolic engineering, all of which may provide new ideas on regional design of lipid tolerated cell factory for enhanced de novo biosynthesis of hydrophobic products.
Data availability statement
The authors declare that all data supporting the findings of this study are available within the article and its supplementary information files.
References
Fahy E, Cotter D, Sud M, Subramaniam S. Lipid classification, structures and tools. Biochim Biophys Acta. 2011. https://doi.org/10.1016/j.bbalip.2011.06.009.
Liu JF, Xia JJ, Nie KL, Wang F, Deng L. Outline of the biosynthesis and regulation of ergosterol in yeast. World J Microbiol Biotechnol. 2019. https://doi.org/10.1007/s11274-019-2673-2.
Guo H, Wang H, Huo YX. Engineering critical enzymes and pathways for improved triterpenoid biosynthesis in yeast. ACS Synth Biol. 2020. https://doi.org/10.1021/acssynbio.0c00124.
Hu Y, Zhu Z, Gradischnig D, Winkler M, Nielsen J, Siewers V. Engineering carboxylic acid reductase for selective synthesis of medium-chain fatty alcohols in yeast. Proc Natl Acad Sci USA. 2020. https://doi.org/10.1073/pnas.2010521117.
Lu R, Cao L, Wang K, Ledesma-Amaro R, Ji XJ. Engineering Yarrowia lipolytica to produce advanced biofuels: Current status and perspectives. Bioresour Technol. 2021. https://doi.org/10.1016/j.biortech.2021.125877.
Patra P, Das M, Kundu P, Ghosh A. Recent advances in systems and synthetic biology approaches for developing novel cell-factories in non-conventional yeasts. Biotechnol Adv. 2021. https://doi.org/10.1016/j.biotechadv.2021.107695.
Zhu YL, Huang W, Ni JR, Liu W, Li H. Production of diosgenin from Dioscorea zingiberensis tubers through enzymatic saccharification and microbial transformation. Appl Microbiol Biotechnol. 2010. https://doi.org/10.1007/s00253-009-2200-8.
Ren Y, Chen Y, Hu B, Wu H, Lai F, Li X. Microwave-assisted extraction and a new determination method for total steroid saponins from Dioscorea zingiberensis C.H. Wright. Steroids. 2015. https://doi.org/10.1016/j.steroids.2015.09.008.
Cravens A, Payne J, Smolke CD. Synthetic biology strategies for microbial biosynthesis of plant natural products. Nat Commun. 2019. https://doi.org/10.1038/s41467-019-09848-w.
Bergenholm D, Gossing M, Wei Y, Siewers V, Nielsen J. Modulation of saturation and chain length of fatty acids in Saccharomyces cerevisiae for production of cocoa butter-like lipids. Biotechnol Bioeng. 2018. https://doi.org/10.1002/bit.26518.
Fernandez-Moya R, Da Silva NA. Engineering Saccharomyces cerevisiae for high-level synthesis of fatty acids and derived products. FEMS Yeast Res. 2017. https://doi.org/10.1093/femsyr/fox071.
Ageitos JM, Vallejo JA, Veiga-Crespo P, Villa TG. Oily yeasts as oleaginous cell factories. Appl Microbiol Biotechnol. 2011. https://doi.org/10.1007/s00253-011-3200-z.
Klug L, Daum G. Yeast lipid metabolism at a glance. FEMS Yeast Res. 2014. https://doi.org/10.1111/1567-1364.12141.
Runguphan W, Keasling JD. Metabolic engineering of Saccharomyces cerevisiae for production of fatty acid-derived biofuels and chemicals. Metab Eng. 2014. https://doi.org/10.1016/j.ymben.2013.07.003.
Ma T, Shi B, Ye Z, Li X, Liu M, Chen Y, et al. Lipid engineering combined with systematic metabolic engineering of Saccharomyces cerevisiae for high-yield production of lycopene. Metab Eng. 2019. https://doi.org/10.1016/j.ymben.2018.11.009.
Wu T, Li S, Ye L, Zhao D, Fan F, Li Q, et al. Engineering an artificial membrane vesicle trafficking system (AMVTS) for the excretion of beta-carotene in Escherichia coli. ACS Synth Biol. 2019. https://doi.org/10.1021/acssynbio.8b00472.
Lindahl AL, Olsson ME, Mercke P, Tollbom O, Schelin J, Brodelius M, et al. Production of the artemisinin precursor amorpha-4,11-diene by engineered Saccharomyces cerevisiae. Biotechnol Lett. 2006. https://doi.org/10.1007/s10529-006-0015-6.
Dejong JM, Liu Y, Bollon AP, Long RM, Jennewein S, Williams D, et al. Genetic engineering of taxol biosynthetic genes in Saccharomyces cerevisiae. Biotechnol Bioeng. 2006. https://doi.org/10.1002/bit.20694.
Scalcinati G, Knuf C, Partow S, Chen Y, Maury J, Schalk M, et al. Dynamic control of gene expression in Saccharomyces cerevisiae engineered for the production of plant sesquitepene alpha-santalene in a fed-batch mode. Metab Eng. 2012. https://doi.org/10.1016/j.ymben.2012.01.007.
Dai Z, Liu Y, Zhang X, Shi M, Wang B, Wang D, et al. Metabolic engineering of Saccharomyces cerevisiae for production of ginsenosides. Metab Eng. 2013. https://doi.org/10.1016/j.ymben.2013.10.004.
Liu J, Zhang W, Du G, Chen J, Zhou J. Overproduction of geraniol by enhanced precursor supply in Saccharomyces cerevisiae. J Biotechnol. 2013. https://doi.org/10.1016/j.jbiotec.2013.10.017.
Jacquier N, Schneiter R. Mechanisms of sterol uptake and transport in yeast. J Steroid Biochem Mol Biol. 2012. https://doi.org/10.1016/j.jsbmb.2010.11.014.
Espenshade PJ, Hughes AL. Regulation of sterol synthesis in eukaryotes. Annu Rev Genet. 2007. https://doi.org/10.1146/annurev.genet.41.110306.130315.
Korber M, Klein I, Daum G. Steryl ester synthesis, storage and hydrolysis: a contribution to sterol homeostasis. Biochim Biophys Acta Mol Cell Biol Lipids. 2017. https://doi.org/10.1016/j.bbalip.2017.09.002.
Zhao Y, Zhang Y, Nielsen J, Liu Z. Production of beta-carotene in Saccharomyces cerevisiae through altering yeast lipid metabolism. Biotechnol Bioeng. 2021. https://doi.org/10.1002/bit.27717.
Qian YD, Tan SY, Dong GR, Niu YJ, Hu CY, Meng YH. Increased campesterol synthesis by improving lipid content in engineered Yarrowia lipolytica. Appl Microbiol Biotechnol. 2020. https://doi.org/10.1007/s00253-020-10743-4.
Hu Z, He B, Ma L, Sun Y, Niu Y, Zeng B. Recent Advances in Ergosterol Biosynthesis and Regulation Mechanisms in Saccharomyces cerevisiae. Indian J Microbiol. 2017. https://doi.org/10.1007/s12088-017-0657-1.
Olzmann JA, Carvalho P. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol. 2019. https://doi.org/10.1038/s41580-018-0085-z.
Peng H, He L, Haritos VS. Metabolic engineering of lipid pathways in Saccharomyces cerevisiae and staged bioprocess for enhanced lipid production and cellular physiology. J Ind Microbiol Biotechnol. 2018. https://doi.org/10.1007/s10295-018-2046-0.
Teixeira PG, David F, Siewers V, Nielsen J. Engineering lipid droplet assembly mechanisms for improved triacylglycerol accumulation in Saccharomyces cerevisiae. FEMS Yeast Res. 2018. https://doi.org/10.1093/femsyr/foy060.
Son SH, Kim JE, Oh SS, Lee JY. Engineering cell wall integrity enables enhanced squalene production in yeast. J Agric Food Chem. 2020. https://doi.org/10.1021/acs.jafc.0c00967.
Demessie Z, Woolfson KN, Yu F, Qu Y, De Luca V. The ATP binding cassette transporter, VmTPT2/VmABCG1, is involved in export of the monoterpenoid indole alkaloid, vincamine in Vinca minor leaves. Phytochemistry. 2017. https://doi.org/10.1016/j.phytochem.2017.04.019.
Rattray JB, Schibeci A, Kidby DK. Lipids of yeasts. Bacteriol Rev. 1975. https://doi.org/10.1128/br.39.3.197-231.1975.
Gurvitz A, Rottensteiner H. The biochemistry of oleate induction: transcriptional upregulation and peroxisome proliferation. Biochim Biophys Acta. 2006. https://doi.org/10.1016/j.bbamcr.2006.07.011.
Grillitsch K, Connerth M, Kofeler H, Arrey TN, Rietschel B, Wagner B, et al. Lipid particles/droplets of the yeast Saccharomyces cerevisiae revisited: lipidome meets proteome. Biochim Biophys Acta. 2011. https://doi.org/10.1016/j.bbalip.2011.07.015.
Li P, Fu X, Chen M, Zhang L, Li S. Proteomic profiling and integrated analysis with transcriptomic data bring new insights in the stress responses of Kluyveromyces marxianus after an arrest during high-temperature ethanol fermentation. Biotechnol Biofuels. 2019. https://doi.org/10.1186/s13068-019-1390-2.
Yan GL, Duan LL, Liu PT, Duan CQ. Transcriptional comparison investigating the influence of the addition of unsaturated fatty acids on aroma compounds during alcoholic fermentation. Front Microbiol. 2019. https://doi.org/10.3389/fmicb.2019.01115.
Petschnigg J, Wolinski H, Kolb D, Zellnig G, Kurat CF, Natter K, et al. Good fat, essential cellular requirements for triacylglycerol synthesis to maintain membrane homeostasis in yeast. J Biol Chem. 2009. https://doi.org/10.1074/jbc.M109.024752.
Sun J, Yan J, Yuan X, Yang R, Dan T, Wang X, et al. A computationally constructed ceRNA interaction network based on a comparison of the SHEE and SHEEC cell lines. Cell Mol Biol Lett. 2016. https://doi.org/10.1186/s11658-016-0022-0.
Li Y, Fang J, Qi X, Lin M, Zhong Y, Sun L, et al. Combined analysis of the fruit metabolome and transcriptome reveals candidate genes involved in flavonoid biosynthesis in Actinidia arguta. Int J Mol Sci. 2018. https://doi.org/10.3390/ijms19051471.
Ohdate T, Inoue Y. Involvement of glutathione peroxidase 1 in growth and peroxisome formation in Saccharomyces cerevisiae in oleic acid medium. Biochim Biophys Acta. 2012. https://doi.org/10.1016/j.bbalip.2012.05.004.
Thoms S, Erdmann R. Dynamin-related proteins and Pex11 proteins in peroxisome division and proliferation. FEBS J. 2005. https://doi.org/10.1111/j.1742-4658.2005.04939.x.
Airenne TT, Torkko JM, Van den plas S, Sormunen RT, Kastaniotis AJ, Wierenga RK, et al. Structure–function analysis of enoyl thioester reductase involved in mitochondrial maintenance. J Mol Biol. 2003. https://doi.org/10.1016/s0022-2836(03)00038-x.
Meyers A, Weiskittel TM, Dalhaimer P. Lipid droplets: formation to breakdown. Lipids. 2017. https://doi.org/10.1007/s11745-017-4263-0.
Tauchi-Sato K, Ozeki S, Houjou T, Taguchi R, Fujimoto T. The surface of lipid droplets is a phospholipid monolayer with a unique fatty acid composition. J Biol Chem. 2002. https://doi.org/10.1074/jbc.M207712200.
Fujimoto T, Parton RG. Not just fat: the structure and function of the lipid droplet. Cold Spring Harb Perspect Biol. 2011. https://doi.org/10.1101/cshperspect.a004838.
Renne MF, Klug YA, Carvalho P. Lipid droplet biogenesis: a mystery “unmixing”? Semin Cell Dev Biol. 2020. https://doi.org/10.1016/j.semcdb.2020.03.001.
Wang CW, Miao YH, Chang YS. Control of lipid droplet size in budding yeast requires the collaboration between Fld1 and Ldb16. J Cell Sci. 2014. https://doi.org/10.1242/jcs.137737.
Thiam AR, Ikonen E. Lipid droplet nucleation. Trends Cell Biol. 2021. https://doi.org/10.1016/j.tcb.2020.11.006.
Choudhary V, Ojha N, Golden A, Prinz WA. A conserved family of proteins facilitates nascent lipid droplet budding from the ER. J Cell Biol. 2015. https://doi.org/10.1083/jcb.201505067.
Thiam AR, Foret L. The physics of lipid droplet nucleation, growth and budding. Biochim Biophys Acta. 2016. https://doi.org/10.1016/j.bbalip.2016.04.018.
Thiam AR, Beller M. The why, when and how of lipid droplet diversity. J Cell Sci. 2017. https://doi.org/10.1242/jcs.192021.
Eisenberg-Bord M, Mari M, Weill U, Rosenfeld-Gur E, Moldavski O, Castro IG, et al. Identification of seipin-linked factors that act as determinants of a lipid droplet subpopulation. J Cell Biol. 2018. https://doi.org/10.1083/jcb.201704122.
Teixeira V, Johnsen L, Martinez-Montanes F, Grippa A, Buxo L, Idrissi FZ, et al. Regulation of lipid droplets by metabolically controlled Ldo isoforms. J Cell Biol. 2018. https://doi.org/10.1083/jcb.201704115.
Moir RD, Gross DA, Silver DL, Willis IM. SCS3 and YFT2 link transcription of phospholipid biosynthetic genes to ER stress and the UPR. PLoS Genet. 2012. https://doi.org/10.1371/journal.pgen.1002890.
Becuwe M, Bond LM, Pinto AFM, Boland S, Mejhert N, Elliott SD, et al. FIT2 is an acyl-coenzyme A diphosphatase crucial for endoplasmic reticulum homeostasis. J Cell Biol. 2020. https://doi.org/10.1083/jcb.202006111.
Jacquier N, Choudhary V, Mari M, Toulmay A, Reggiori F, Schneiter R. Lipid droplets are functionally connected to the endoplasmic reticulum in Saccharomyces cerevisiae. J Cell Sci. 2011. https://doi.org/10.1242/jcs.076836.
Siddiqah IM, Manandhar SP, Cocca SM, Hsueh T, Cervantes V, Gharakhanian E. Yeast ENV9 encodes a conserved lipid droplet (LD) short-chain dehydrogenase involved in LD morphology. Curr Genet. 2017. https://doi.org/10.1007/s00294-017-0702-y.
Ouyang Y, Li Q, Kuang X, Wang H, Wu J, Ayepa E, et al. YMR152W from Saccharomyces cerevisiae encoding a novel aldehyde reductase for detoxification of aldehydes derived from lignocellulosic biomass. J Biosci Bioeng. 2021. https://doi.org/10.1016/j.jbiosc.2020.09.004.
Narita T, Naganuma T, Sase Y, Kihara A. Long-chain bases of sphingolipids are transported into cells via the acyl-CoA synthetases. Sci Rep. 2016. https://doi.org/10.1038/srep25469.
Schuldiner M, Bohnert M. A different kind of love—lipid droplet contact sites. Biochim Biophys Acta Mol Cell Biol Lipids. 2017. https://doi.org/10.1016/j.bbalip.2017.06.005.
Walther TC, Chung J, Farese RV Jr. Lipid droplet biogenesis. Annu Rev Cell Dev Biol. 2017. https://doi.org/10.1146/annurev-cellbio-100616-060608.
Henne M, Goodman JM, Hariri H. Spatial compartmentalization of lipid droplet biogenesis. Biochim Biophys Acta Mol Cell Biol Lipids. 2020. https://doi.org/10.1016/j.bbalip.2019.07.008.
Acknowledgements
This research was supported by the National Key Research and Development Project of China (2019YFA0905300).
Author information
Authors and Affiliations
Contributions
YS: investigation, methodology, data curation, formal analysis, original draft and writing. XK: data curation, formal analysis, writing—review and editing. Z-HP: data curation, methodology and writing. L-SC: methodology, investigation. Z-QL: conceptualization, funding acquisition, resources, project administration, supervision. Y-GZ: funding acquisition, supervision.
Corresponding author
Ethics declarations
Conflict of interest
Authors are required to disclose financial or non-financial interests that are directly or indirectly related to the work submitted for publication.
Consent for publication
All authors agree to publication.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Shen, Y., Ke, X., Pan, ZH. et al. Comparative transcriptomic and lipidomic analysis of oleic environment adaptation in Saccharomyces cerevisiae: insight into metabolic reprogramming and lipid membrane expansion. Syst Microbiol and Biomanuf 4, 112–126 (2024). https://doi.org/10.1007/s43393-022-00098-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43393-022-00098-4