
Abstract
Enzymes are considered as functional proteins, also known as biocatalysts, which are required for normal cellular function. The commercial production of enzymes with distinct features always remains the top priority of researchers for industrial biocatalysis. However, the insufficient yield, low stability, low activity, formation of by-products, complex purification process, and many other factors are challenging for the enzyme production industry. Novel advancements in protein engineering, especially the combination of multidisciplinary techniques including post-translational enzyme modification, structured assisted protein tailoring and computational modeling approaches, open a new horizon to more efficient production of biocatalysts. In this review, we focus on the development process of enzyme modification design strategies, such as rational design, semi-rational design and de novo design. Selected examples for each strategy, which are particularly useful for novel enzyme design, are given. We hope this review is helpful for the development of biocatalysts for industrial application.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Enzymes produced by living cells are molecular machines, which catalyze chemical reactions. They are tools of nature and are specific in their action with high efficiency under mild conditions [1]. What is more, enzymes can carry out reactions which are challenges for chemical synthesis. Because of these characteristics described above, functional enzymes have been commercially applied in many fields, such as textile, food, detergent, paper, and medical industries [2,3,4,5,6]. Important industrial enzymes and their application conditions are shown in Table 1.
Microorganisms are the primary source of industrial enzymes. Enzymes can be obtained by different microorganisms, such as bacteria, fungi, and also from animals and plants. In industrial production, the majority of enzymes are produced by a very limited number of microbial species, of which fungi and bacteria, especially Aspergillus, Trichoderma, Bacillus and yeast, dominate the chief position. For example, amylases, which catalyze starch hydrolysis, can be produced by various microorganisms, but in commercial applications, Bacillus spp. are commonly used, such as B. stearothermophilus, B. licheniformis, and B. amyloliquefaciens [17]. Fungi, bacteria, yeast, and Streptomyces contribute 60%, 24%, 4%, and 2%, respectively [18].
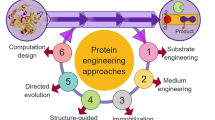
The industrial application prospects of enzyme are very attractive, not only due to their high efficiency and specificity, but also due to their environmental-friendly features, which can minimize waste generation. However, microorganisms used to produce industrial enzymes are mainly obtained through screening from natural environments. The performance of enzymes produced in the natural environment usually cannot meet the needs of the industry, and their properties have to be optimized. Thus, there are still some problems limiting the application of industrial enzymes, such as thermal stability, pH stability and organic solvent tolerance. Classical strategies such as immobilization, additives or process engineering have been used to overcome these problems (Fig. 1), but these techniques could provide only limited improvement. The recent advancements of different technologies and methods have given enormous guidances to speed up the process. Among these techniques and methods, DNA recombinant technology and protein engineering play vital roles in efficient enzyme production [19]. Common strategies used in molecular engineering of industrial enzymes include direct evolution, site-directed mutagenesis, terminal fusion and truncation [20,21,22,23]. However, each of these has its own disadvantages. For example, the mutation library of direct evolution is so large that it results in low efficiency of screening; to do site-directed mutagenesis, we need to know the structure–function relationships of target enzymes [20].

Schematic diagram of the biocatalytic process with multiple approaches. This figure shows the enzyme modification approaches including process engineering, enzyme immobilization and protein engineering
Nowadays, the increase of computational power makes the biomolecular simulations a useful tool to understand enzyme mechanisms and create improved or novel enzymes [24, 25]. Computational engineering has advanced our knowledge of protein function and afforded new perspectives to functional enzyme design. These additional knowledge can help us to design or evolve enzymes with high activity, environmental tolerance, specific catalysis abilities and stereo/enantioselectivity (Fig. 2). Commonly used enzyme modification design strategies include rational design and semi-rational design, and de novo design is a newly developed strategy to create enzymes catalyzing novel reactions, which will be a trend in the future (Table 2). This review aims to summarize the current advances of protein design methods in molecular engineering of industrial enzymes and discuss their possible future applications.

The process of designing or evolving enzymes with high activity, environmental tolerance, specific catalyze abilities and stereo/enantioselectivity. This figure shows a flow diagram of enzyme design from structure analysis to hot-point screening and then getting the novel enzymes with enzyme design strategies
Rational enzyme design
Enzymes selected and evolved in natural environments cannot meet the requirements of artificial production environment, such as high catalytic activity, substrate specificity and environmental adaptability. Most enzymes used as catalysts need to be modified and optimized before they are applied in industrial production. Wei et al. used an immobilization method to enhance the activity and stability of multiple enzymes, which offers excellent biocompatibility. They introduced a facile co-immobilization of glucose oxidase (GOx) and horseradish peroxidase (HRP) in the matrix of Cu (copper) and guanosine 5-monophosphate (GMP), which increased 40% of the stability at pH 3 and more than 70% at 90 °C [41]. Ribosomally synthesized and posttranslationally modified peptides (RiPPs) are a class of natural products with diverse structural features and biological activities. Mitchell et al. demonstrated that ranthipeptide-forming rSAM enzyme PapB is intolerant to expansion and contraction of the thioether motifs and defined its intolerant acceptor position through a mass spectrometry-based activity assay. Then they increased the activity of PapB by in vitro recombination, which provided insights into ranthipeptide biosynthesis [42]. With these deep researches of enzyme production, different enzymes with high stability and wide substrate specificity have been found, which leads to an increasing understanding of the mechanism of enzymes to maintain their stability and specificity. Rational design is based on understanding of protein structures and functions of enzymes. According to the structural information of protein and the sequence comparison of homologous protein, amino acid residues are rationally selected as the target. Combined with site-specific mutation technology, key amino acids are mutated to screen the target mutants. Currently, the widely used rational design methods include: design strategy based on protein conformation and design strategy based on computer simulation.
Analysis of different binding domains of enzymes based on their structure, function and catalytic mechanisms can give effective guidances in modification of enzyme catalytic activity, substrate specificity and stability [43]. In the early 1980s, Winter et al. used recombinant tools, such as oligonucleotide synthesis, DNA amplification and site-specific cutting and pasting, to modify native enzymes successfully for the first time [44]. These recombinant tools enabled researchers to deliberately and precisely substitute specific amino acid residues [45]. Arnold raised the idea of directed evolution of enzyme, and proposed remodeling the natural enzyme by replacing amino acid residues in 1993 [46]. Recently, many enzymes have been modified to enhance their activity and stability by rational design. For example, Jakobrinnert et al. [47] analyzed the protein structure (PDB: 1r37) of carbonyl reductase (CPCR2) of Candida parapsilosis. They found that A275 was located at the dimer interface, which was close to the active center and contained a subunit hydrogen bond inside. Then, a mutant strain was obtained by site-directed mutagenesis at A275, which improved its enzyme activity and stability in the two-phase medium by 1.5 times. In addition to site-directed mutagenesis, there were also reports of rational truncation, truncation or replacement of a flexible loop to improve the stability of enzymes. Hauer et al. [48] analyzed the structure of NCR reductase, and obtained a loop with high B-factor value. B-factors (atomic displacement parameters) from crystal structures were analyzed by the B-FIT method and were considered as an important index to reflect the structural flexibility and dynamics of proteins, and thus were applied to predict the flexibility and thermal stability of proteins [48]. After four amino acids were cut off, the activity of mutant enzyme at 45 °C was 84.6% higher than that of wild type, and the stability in organic solvent 2-methyl-2-pentenal was increased by 54.1%. By analyzing the structure of phospholipase D, Iwasaki et al. [49] found a highly flexible loop on the surface of the enzyme. The thermostability of the mutant enzyme at 70 °C was increased by 11.7 times by cutting off the whole loop. Simulation analysis showed that the removal of the loop ring improved the conformational stability of the enzymes. On the other hand, loop replacement was also reported to improve the stability of the enzyme. Bornscheuer et al. [50] replaced the “cap region” of thermostable esterase BsteE with the corresponding region of its homologous thermostable esterase BsubE, and increased the dissolution temperature of BsteE by 4 °C. Thus, the stability and catalytic properties of enzymes can be effectively improved by rational design based on protein conformation.
The above rational design is usually easily possible to remodel the active site. However, due to the complexity of enzymes’ structures, deeper understanding of the conformation of enzymes is needed to help researchers to predict their effects. With the development of X-ray crystal diffraction technology, more and more protein structures have been resolved. The total number of resolved enzymes is still small, which greatly limits the further development of rational design. In recent years, the development of computer simulation technology has made up for this deficiency. Homologous structure can be used as a template when there is no information about its three-dimensional structure. Molecular simulation technology, to predict structures of enzymes, has been gradually applied to the rational design of enzymes [51]. Gao et al. [52] simulated the three-dimensional structure of thermophilic xylanase AoXyn11A and its homologous thermophilic enzyme EvXyn11TS by molecular dynamics, and then 31 amino acids at the N-terminal were compared, which showed large difference in B-factor value. After the N-terminus of AoXyn11A from Ser1 to Gln37 was replaced by the corresponding region of EvXyn11TS from Asn1 to Asn 42, the total energy value of modified AoXyn11A was reduced from − 611.2 to − 663.2 kJ mmol−1 via molecular dynamics simulation. This result proved that the conformation of modified AoXyn11A was more stable. What is more, the enzyme activity of AoXyn11A after modification was increased by 197 times under 70 °C. Similarly, using homologous modeling and molecular docking technology, Chao et al. [53] found that the 192, 155 and 208 amino acids of cinnamyl coenzyme reductase (CCR) of Populus tomentose formed the substrate binding region. Then a mutant F155y, obtained by site-directed mutagenesis, showed 4.7 times higher catalytic efficiency than that of wild type. Thus, enzymes could also be modified through the computer simulation-based rational design strategy when the three-dimensional structure of the target enzyme protein is not clear enough. However, due to the limitation of delivery efficiency, it may be difficult to evaluate the phenotype of the enzyme variants obtained from traditional protein engineering methods, which made the construction of screening methods particularly important for the rational design of enzymes [54]. Recently, the CRISPR/Cas9 technology has achieved great progresses and has been applied for the engineering of microbial strains [55, 56]. Due to its simplicity and efficiency, CRISPR/cas9 is an ideal tool, which can integrate gene variants of the target enzyme into the chromosomes of the production strain quickly and effectively [57]. A protein engineering method combining CRISPR/cas9-facilitated engineering with growth-coupled and sensor-guided in vivo screening (CGSS) was proposed by Zeng et al. [58]. They used lambda-red recombination coupled with the CRISPR/Cas9 system to integrate the mutation library of the target gene into the chromosome with high efficiency. This growth-coupled and sensor-guided library screening methods could be combined with in vivo targeted mutagenesis [59, 60], which may trigger the development of more advanced methods.
Semi-rational enzyme design
Due to the large number of mutants in the constructed library of direct evolution, the workload of screening is extremely huge. Therefore, researchers begin to pay more attention to semi-rational design methods combining the crystal structures and catalytic properties of enzymes [61]. Semi-rational design is to randomly mutate some “hot spots” or specific areas on the enzyme molecules that play an important role in the enzyme properties to quickly obtain the mutant enzyme with changed enzyme properties. On the premise of not affecting the mutation effect, this method can reduce the size of the mutation library and significantly improve the efficiency of molecular engineering.
The combination of active site saturation test (CAST) is particularly suitable for optimizing the stereoselectivity of enzyme catalysts or expanding the substrate spectrum. The basic idea of CAST is to determine the active center of enzyme catalysis by analyzing the three-dimensional structure or homologous modeling structure of the target enzyme. Then, some amino acid residues within the determined range are selected to construct the mutation library. This method was first applied to the enantioselective modification of epoxide hydrolase from Aspergillus niger [62], in which 15 amino acid residues around the active pocket were selected, and 6 mutation libraries were constructed by CAST method to obtain a high E-value mutant. CAST requires a certain understanding and analysis of the three-dimensional structure and function of the enzyme to identify the amino acids that make up the substrate binding pocket. At the same time, a more accurate high-throughput screening method is also needed to screen the mutation library. Compared with random mutation, the size of the mutation library of CAST is greatly reduced [33]. This is one of the trends of protein evolution: building a small and precise mutation library.
In recent years, great progress has emerged in high speed and intelligent tools for excavating the detailed structural and functional roles of proteins, such as InterProScan5 [63] and PFAM search [64], which greatly improves our ability to define “hot spots” or specific areas of certain enzymes. To further reduce the screening workload and improve the modification efficiency, researchers have developed a series of new methods based on amino sequences or three-dimensional structure, such as single codon saturation mutagenesis (SCSM) [34] or triple codon saturation mutagenesis (TCSM) [35]. The constructed mutation libraries with these new methods are smaller, which improves the screening efficiency. Three codon saturation mutagenesis is to select three kinds of amino acids as the target amino acids of saturation mutagenesis by analyzing the sequence and structure information of protein. Recently, the TCSM methods have been successfully applied to the modification of the enantioselectivity, catalytic activity and stability of cytochrome P450 monooxygenase, limonene epoxide hydrolase and alcohol dehydrogenase. Alsao, the co-evolution of enantioselectivity, activity and stability of limonene epoxide hydrolase was realized by using TCSM strategy, which increased EE value to 94% (s, s) and thermal stability to 5–10 °C [35].
Molecular dynamics (MD) of proteins play an important role in the process of enzyme catalysis, which affects the key steps of substrate recognition and binding, allosteric regulation, enzyme–substrate complex formation and product release [65, 66]. Molecular dynamics simulation is a method that uses Newtonian mechanics to simulate the motion of atoms and molecules in relative space, and obtains the motion track of atoms through calculation and analysis. RMSF (root mean square fluctuation) is usually used to express the freedom degree of each atom in the molecule and then represent the conformational change of a certain part of the protein. NAMD [37], AMBER [67] and GROMACS [68] are commonly used molecular dynamics simulation software, which can be used to simulate the molecular dynamics of biological macromolecules such as proteins, sugars and nucleic acids. With the development of computer technology, MD simulations have become an effective method to improve the enantioselectivity and activity of enzymes [69].
De novo enzyme design
Recent advances in protein structure prediction, evolutionary information, protein folding [70] and computational modeling [71, 72] have strongly promoted the development of de novo protein design [73]. De novo design is a newly developed methodology to create enzymes catalyzing novel reactions. Different from the design strategy described above, de novo design aims to create a new direction that has not been discovered in the evolutionary process. The core problem of de novo design can be divided into two subclasses: the de novo design of new function, and the redesign of existing enzymes [74].
Using the program Dezymer to design a His3-Fe–O2 metal active site to obtain a superoxide dismutase-like enzyme was the first time to attempt computational protein design (CPD) techniques into enzyme design [75, 76]. Due to the complexity of molecular structure simulation, the method of computer simulation is used in protein structure design and sequence prediction. It mainly relies on the initial skeleton structure and heuristic algorithm to produce a large number of possible results. Rosetta is a set of software for protein structure calculation, modeling and analysis [72], which has been widely used in the de novo design of proteins and plays an important role in the design and synthesis of enzymes [77]. In addition, Ludwiczak et al. [78] combined Rosetta with molecular dynamics to enhance protein structure design and corresponding sequence selection. The combination method generated 20–30% more sequences than the existing method, which improved the diversity of sequence prediction and the similarity with the natural sequence. In the last 5 years, over 80 methods have been developed to solve protein design problems with Rosetta, including the prediction of protein structures; modeling protein–protein complexes; docking small-molecule ligands into proteins; modeling and designing antibodies and immune system proteins; designing new proteins and functions; designing interfaces between proteins and interaction partners; symmetric protein assemblies modeled using parametric design; modeling peptides and peptidomimetics; modeling membrane proteins; adding carbohydrates to the modeling process [79]. The above prediction ability could not only be used to analyze the existing data, but also to provide information for experiments, hence stimulate the engineering of industrial enzymes and enable the synthesis of new biomaterials [79].
Naturally existing enzymes often have some special structural characteristics, which play a decisive role in some specific functions. Therefore, how to design and synthesize enzymes with these special structural features from scratch has a great influence on the application of de novo design. Huang et al. [39] synthesized a TIM barrel protein with high-temperature resistant and reversible folding ability by a de novo design strategy. The model structure and the natural structure showed high similarity. The successful synthesis of this structure provided a new pathway for enzyme customization. Thomas et al. [80] designed a α-helix barreled protein (α-HB), which can adjust the size of the hydrophobic channel to adapt to the binding of specific target molecules. At the same time, due to the existence of thermal stability and low complex repeat sequence in its structure, the protein can withstand more mutations in its hydrophobic channel, which shows that it can be used as a potential carrier for small-molecule recognition carrier. Recently, Jiang et al. designed and constructed a pathway for the synthesis of acetyl-CoA (SACA) by recombinant glyoxalase synthase and acetyl-phosphatase synthase. They designed and engineered a glycolaldehyde synthase by de novo design to condense two molecules of formaldehyde into one glycolaldehyde with more then 70-fold higher activity [81]. Boyken et al. created a de novo six-helix bundle, which was a protein complex consisting of identical alpha helices [40]. To create a protein that could be energetically stable in more than one conformation, Backer et al. transformed an IgG-binding protein with a 4β + α fold into a 3-α fold protein with albumin-binding ability [82]. This work pushed the boundaries of de novo design by building on top of a synthetic protein [83].
Conclusion and perspectives
Herein, we have reviewed the current advance of protein design strategies in molecular engineering of industrial enzymes. From rational design to de novo design, we can find a clear logic line from the development process: initially, enzymes were isolated from nature and were used for their natural activities. Rapidly, enzymes produced in natural environment usually could not catch up with the needs of industrial production and their properties have to be optimized. Therefore, protein engineering techniques, such as site-directed mutagenesis, directed evolution, saturation mutagenesis, truncation, and terminal fusion, has become a research hotspot of researchers. At that time, due to the lack of information on the catalytic mechanism and active sites of enzymes, random mutation was the commonly used technique to modify enzymes, which required a large workload to screen the target enzymes from mutation libraries. With the development of DNA sequence and protein structure analysis techniques, the deeper understanding of the relationship between enzyme function and structure made it possible to use rational design strategy on enzyme modification. The emergence of rational design greatly alleviated many problems, such as reducing the number of mutants and providing a new method for enzyme–substrate-specific modification. In recent years, with the information of enzyme gene sequence becoming available, spatial structure and catalytic mode have been understood gradually. To further reduce the workload and dependence on high-throughput screening methods, semi-rational design strategy was developed to guide the design of smaller and more focused libraries of protein mutants. This strategy combined the information from protein structure databases and the advanced computational algorithms to make researchers focus on amino acid residues in or near the active site to effectively guide protein engineering. The newest goal for enzyme design was “you get what you want”. De novo design allowed researchers to create enzymes that can catalyze novel reactions. Moreover, the enzymes produced by the combination of computational design and protein engineering techniques now can be used in new metabolic pathways. However, one of the basic problems encountered in redesigning natural proteins to provide new functions (such as catalytic sites) is that changing a large number of amino acid residues to introduce functions will inevitably change aspects of the structure, which may influence the stability of enzymes. Also, protein designed by this method is expected to be stable in calculation, but its expression in the host cannot be guaranteed. Thus, this method requires future improvement of the energy calculation and requires the screening of more suitable criteria for protein design.
With the rapid increase of the sequencing flux and the structural data, how to dig and analyze the biological data as well as to provide a new idea and platform technology for the rational design of enzymes is the key problem that needs to be solved. Recently, machines can be trained to predict key factors for enzyme evolution by collecting data in larger amounts with better quality via more advanced experimental techniques, such as next-generation sequencing, high-throughput screening, and microfluidics. Looking forward to the future, artificial intelligence and other advanced computing tools will play an increasingly important role in the prediction of enzyme structure and its corresponding characteristics and functions, which will make the design of artificial enzyme more reasonable and accurate.
References
Ferrer S, Ruiz-Pernía J, Martí S, et al. Hybrid schemes based on quantum mechanics/molecular mechanics simulations. Adv Protein Chem Struct Biol. 2011;85:81–142.
Bhavsar K, Gujar P, Shah P, et al. Combinatorial approach of statistical optimization and mutagenesis for improved production of acidic phytase by Aspergillus niger NCIM 563 under submerged fermentation condition. Appl Microbiol Biotechnol. 2013;97(2):673–9.
Juturu V, Wu J. Microbial xylanases: engineering, production and industrial applications. Biotechnol Adv. 2012;30(6):1219–27.
Ling Z, Liu Y, Teng S, et al. Rational design of a novel propeptide for improving active production of Streptomyces griseus trypsin in Pichia pastoris. Appl Environ Microbiol. 2013;79(12):3851–5.
Lu X, Liu S, Zhang D, et al. Enhanced thermal stability and specific activity of Pseudomonas aeruginosa lipoxygenase by fusing with self-assembling amphipathic peptides. Appl Microbiol Biotechnol. 2013;97:9419–27.
Plagemann I, Zelena K, Arendt P, et al. Loxpsa1, the first recombinant lipoxygenase from a basidiomycete fungus. J Mol Catal B Enzym. 2013;87:99–104.
Patil P, Bari S. Nitrogen heterocycles as potential monoamine oxidase inhibitors: synthetic aspects. Arab J Chem. 2014;7(6):857–84.
Almeida J, Alnoch R, Souza E, et al. Metagenomics: is it a powerful tool to obtain lipases for application in biocatalysis? Biochimica et Biophysica Acta (BBA)—Proteins Proteomics. 2020;1868(2):140320.
Hur S, Lim B, Decker E, et al. In vitro human digestion models for food applications. Food Chem. 2011;125(1):1–12.
Mangiagalli M, Brocca S, Orlando M, et al. The “cold revolution”. Present and future applications of cold-active enzymes and ice-binding protein. New Biotechnol. 2020;55:5–11.
Pan S, Ding N, Ren J, et al. Maltooligosaccharide-forming amylase: characteristics, preparation, and application. Biotechnol Adv. 2017;35(5):619–32.
Rios N, Pinheiro B, Pinheiro M, et al. Biotechnological potential of lipases from Pseudomonas: sources, properties and applications. Process Biochem. 2018;75:99–120.
Siqueira J, Rodrigues C, Vandenberghe L, et al. Current advances in on-site cellulase production and application on lignocellulosic biomass conversion to biofuels: a review. Biomass Bioenerg. 2020;132:105419.
Yang S, Yang B, Duan C, et al. Applications of enzymatic technologies to the production of high-quality dissolving pulp: a review. Biores Technol. 2019;281:440–8.
Singh G, Arya S. Utility of laccase in pulp and paper industry: a progressive step towards the green technology. Int J Biol Macromol. 2019;134:1070–84.
Kaur R, Tyagi R, Zhang X. Review on pulp and paper activated sludge pretreatment, inhibitory effects and detoxification strategies for biovalorization. Environ Res. 2020;182:109094.
Barros F, Simiqueli A, de Andrade C, et al. Production of enzymes from agroindustrial wastes by biosurfactant-producing strains of Bacillus subtilis. Biotechnol Res Int. 2013;2013:1–9. https://doi.org/10.1155/2013/103960
Raveendran S, Parameswaran B, Ummalyma S, et al. Applications of microbial enzymes in food industry. Food Technol Biotechnol. 2018;56(1):16–30.
Srivastava N. Production of food-processing enzymes from recombinant microorganisms. Enzym Food Biotechnol. 2019. https://doi.org/10.1016/B978-0-12-813280-7.00043-8.
Yang H, Li J, Shin H, et al. Molecular engineering of industrial enzymes: recent advances and future prospects. Appl Microbiol Biotechnol. 2014;98(1):23–9.
Bornscheuer U, Hauer B, Jaeger K, et al. Directed evolution empowered redesign of natural proteins for the sustainable production of chemicals and pharmaceuticals. Angew Chem Int Ed. 2019;58(1):36–40.
Arnold F. Innovation by evolution: bringing new chemistry to life (nobel lecture). Angew Chem Int Ed. 2019;58(41):14420–6.
Qu G, Li A, Acevedo-Rocha G, et al. The crucial role of methodology development in directed evolution of selective enzymes. Angew Chem Int Ed. 2020. https://doi.org/10.1002/anie.201901491.
Dusan P, Shina C. Molecular modeling of conformational dynamics and its role in enzyme evolution. Curr Opin Struct Biol. 2018;52:50–7.
Musil M, Konegger H, Hon J, et al. Computational design of stable and soluble biocatalysts. ACS Catal. 2019;9(2):1033–54.
Ghislieri D, Green A, Pontini M, et al. Engineering an enantioselective amine oxidase for the synthesis of pharmaceutical building blocks and alkaloid natural products. J Am Chem Soc. 2013;135:10863–9.
Kille S, Zilly F, Acevedo J, et al. Regio- and stereoselectivity of p450-catalysed hydroxylation of steroids controlled by laboratory evolution. Nat Chem. 2011;3(9):738–43.
Liskova V, Bednar D, Prudnikova T, et al. Balancing the stability-activity trade-off by fine-tuning dehalogenase access tunnels. ChemCatChem. 2015;7(4):648–59.
Pavlova M, Klvana M, Prokop Z, et al. Redesigning dehalogenase access tunnels as a strategy for degrading an anthropogenic substrate. Nat Chem Biol. 2009;5(10):727–33.
Reetz T, Carballeira J, Vogel A. Iterative saturation mutagenesis on the basis of B factors as a strategy for increasing protein thermostability. Angewanate Chemie. 2006;45:7745–51.
Pokhrel S, Joo J, Yoo Y. Shifting the optimum pH of Bacillus circulans xylanase towards acidic side by introducing arginine. Biotechnol Bioprocess Eng. 2013;18(1):35–42.
Xu H, Zhang F, Shang H, et al. Alkalophilic adaptation of XynB endoxylanase from Aspergillus niger via rational design of pKa of catalytic residues. J Biosci Bioeng. 2013;115(6):618–22.
Reetz M, Bocola M, Carballeira J, et al. Expanding the range of substrate acceptance of enzymes: combinatorial active-site saturation test. Angew Chem Int Ed. 2005;44(27):4192–6.
Sun Z, Lonsdale R, Kong X, et al. Reshaping an enzyme binding pocket for enhanced and inverted stereoselectivity: use of smallest amino acid alphabets in directed evolution. Angew Chem Int Ed. 2015;54(42):12410–5.
Li G, Zhang H, Sun Z, et al. Multiparameter optimization in directed evolution: engineering thermostability, enantioselectivity, and activity of an epoxide hydrolase. ACS Catal. 2016;6(6):3679–87.
Jiménez-Osés G, Osuna S, Gao X, et al. The role of distant mutations and allosteric regulation on Lov D active site dynamics. Nat Chem Biol. 2014;10(6):431–6.
Park H, Nam S, Lee J, et al. Design and evolution of new catalytic activity with an existing protein scaffold. Science. 2006;311(5760):535–8.
Burton A, Thomson A, Dawson W, et al. Installing hydrolytic activity into a completely de novo protein framework. Nat Chem. 2016;8:837–44.
Huang P, Feldmeier K, Parmeggiani F, et al. De novo design of a four-fold symmetric TIM-barrel protein with atomic-level accuracy. Nat Chem Biol. 2016;12(1):29–34.
Boyken S, Chen Z, Groves B, et al. De novo design of protein homo-oligomers with modular hydrogen-bond network-mediated specificity. Science. 2016;352:680–7.
Memon A, Ding R, Yuan Q, et al. Coordination of GMP ligand with Cu to enhance the multiple enzymes stability and substrate specificity by co-immobilization process. Biochem Eng J. 2018;136:102–8.
Precord T, Mahanta N, Mitchell D. Reconstitution and substrate specificity of the thioether-forming radical S-adenosylmethionine enzyme in freyrasin biosynthesis. ACS Chem Biol. 2019;14:1981–8.
Garst A, Bassalo M, Pines G, et al. Genome-wide mapping of mutations at single-nucleotide resolution for protein, metabolic and genome engineering. Nat Biotechnol. 2017;35:48–55.
Winter G, Fersht A, Wilkinson A, et al. Redesigning enzyme structure by site-directed mutagenesis: tyrosyl trna synthetase and ATP binding. Nature. 1982;299(5885):756–8.
Wilkinson A, Fersht A, Blow D, et al. Site-directed mutagenesis as a probe of enzyme structure and catalysis: tyrosyl-trna synthetase cysteine-35 to glycine-35 mutation. Biochemistry. 1983;22(15):3581–6.
Chen K, Arnold F. Tuning the activity of an enzyme for unusual environments: sequential random mutagenesis of subtilisin E for catalysis in dimethylformamide. Proc Natl Acad Sci USA. 1993;90(12):5618–22.
Jakoblinnert A, Anne V, Shivange A, et al. Design of an activity and stability improved carbonyl reductase from Candida parapsilosis. J Biotechnol. 2013;165(1):52–62.
Reich S, Kress N, Nestl B, et al. Variations in the stability of NCR ene reductase by rational enzyme loop modulation. J Struct Biol. 2014;185(2):228–33.
Damnjanovic J, Nakano H, Iwasaki Y. Deletion of a dynamic surface loop improves stability and changes kinetic behavior of phosphatidylinositol-synthesizing streptomyces phospholipase D. Biotechnol Bioeng. 2014;111(4):674–82.
Gall M, Nobili A, Pavlidis I, et al. Improved thermostability of a Bacillus subtilis esterase by domain exchange. Appl Microbiol Biotechnol. 2014;98(4):1719–26.
Damborsky J, Brezovsky J. Computational tools for designing and engineering enzymes. Curr Opin Chem Biol. 2014;19:8–16.
Gao S, Wang J, Wu M, et al. Engineering hyperthermostability into a mesophilic family 11 xylanase from Aspergillus oryzae by in silico design of N-terminus substitution. Biotechnol Bioeng. 2013;110(4):1028–38.
Chao N, Li N, Qi Q, et al. Characterization of the cinnamoyl-CoA reductase (CCR) gene family in Populus tomentosa reveals the enzymatic active sites and evolution of CCR. Planta. 2017;245:61–75.
Ren C, Xu K, Segal D, et al. Strategies for the enrichment and selection of genetically modified cells. Trends Biotechnol. 2019;37(1):56–71.
Schuster A, Erasimus H, Fritah S, et al. RNAi/CRISPR screens: from a pool to a valid hit. Trends Biotechnol. 2019;37(1):38–55.
Wu Y, Chen T, Liu Y, et al. Design of a programmable biosensor-CRISPRi genetic circuits for dynamic and autonomous dual-control of metabolic flux in Bacillus subtilis. Nucleic Acids Res. 2020;48:996–1009.
Guo X, Chavez A, Tung A, et al. High-throughput creation and functional profiling of DNA sequence variant libraries using CRISPR–Cas9 in yeast. Nat Biotechnol. 2018;36:540–6.
Chen M, Chen L, Zeng A. Crispr/cas9-facilitated engineering with growth-coupled and sensor-guided in vivo screening of enzyme variants for a more efficient chorismate pathway in E. coli. Metab Eng Commun. 2019;9:e00094.
Jakociunas T, Pedersen L, Lis A, et al. CasPER, a method for directed evolution in genomic contexts using mutagenesis and CRISPR/Cas9. Metab Eng. 2018;48:288–96.
Ravikumar A, Arzumanyan G, Obadi M, et al. Scalable, continuous evolution of genes at mutation rates above genomic error thresholds. Cell. 2018;175:1946–57.
Wijma H, Floor R, Janssen D. Structure- and sequence-analysis inspired engineering of proteins for enhanced thermostability. Curr Opin Struct Biol. 2013;23(4):588–94.
Reetz M. Directed evolution of enantioselective enzymes as catalysts for organic synthesis. Adv Catal. 2006;49:1–69.
Madeira F, Park Y, Lee J, et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019;47:636–41.
El-Gebali S, Mistry J, Bateman A, et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019;47:427–32.
Boehr D, Nussinov R, Wright P. The role of dynamic conformational ensembles in biomolecular recognition. Nat Chem Biol. 2009;5(12):789–96.
Goodey N, Benkovic S. Allosteric regulation and catalysis emerge via a common route. Nat Chem Biol. 2008;4(8):474–82.
Padhi S, Fujii R, Legatt G, et al. Switching from an esterase to a hydroxynitrile lyase mechanism requires only two amino acid substitutions. Chem Biol. 2010;17(8):863–71.
Chaloupkova R, Sykorova J, Prokop Z, et al. Modification of activity and specificity of haloalkane dehalogenase from Sphingomonas paucimobilis UT26 by engineering of its entrance tunnel. J Biol Chem. 2003;278(52):52622–8.
Carterchilders M, Daggett V. Insights from molecular dynamics simulations for computational protein design. Mol Syst Des Eng. 2017;2:9–33.
Onuchic J, Luthey-Schulten Z, Wolynes P. Theory of protein folding: the energy landscape perspective. Annu Rev Phys Chem. 1997;48:545–600.
Hansen N, Van Gunsteren W. Practical aspects of free-energy calculations: a review. J Chem Theory Comput. 2014;10(7):2632–47.
Leaver-Fay A, Tyka M, Lewis S, et al. Rosetta3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487(11):545–74.
Huang P, Boyken S, Baker D. The coming of age of de novo protein design. Nature. 2016;537(7620):320–7.
Zanghellini A. De novo computational enzyme design. Curr Opin Biotechnol. 2014;29:132–8.
Hellinga H, Richards F. Construction of new ligand binding sites in proteins of known structure. I. Computer-aided modeling of sites with pre-defined geometry. J Mol Biol. 1991;222(3):763–85.
Hellinga H, Caradonna J, Richards F. Construction of new ligand binding sites in proteins of known structure. II. Grafting of a buried transition metal binding site into Escherichia coli thioredoxin. J Mol Biol. 1991;222:787–803.
Tinberg C, Khare S. Computational design of ligand binding proteins. Methods Mol Biol. 2017;1529:363–73. https://doi.org/10.1007/978-1-4939-6637-0_19
Ludwiczak J, Jarmula A, Dunin-Horkawicz S. Combining Rosetta with molecular dynamics (MD): a benchmark of the MD-based ensemble protein design. J Struct Biol. 2018;203(1):54–61.
Leman J, Weitzner B, Lewis S, et al. Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nat Methods. 2020. https://doi.org/10.1038/s41592-020-0848-2.
Lu X, Liu Y, Yang Y, et al. Constructing a synthetic pathway for acetyl-coenzyme a from one-carbon through enzyme design. Nat Commun. 2019;10:1378.
Thomas F, Dawson W, Lang E, Burton A, Bartlett G, Rhys G, Mulholland A, Woolfson D. De novo-designed α-helical barrels as receptors for small molecules. ACS Synth Biol. 2018;7(7):1808–16.
Wei K, Moschidi D, Bick M, et al. Computational design of closely related proteins that adopt two well-defined but structurally divergent folds. Proc Natl Acad Sci USA. 2020;117:7208–15.
Morcos F. Protein conformations à la carte, a step further in de novo protein design. Proc Natl Acad Sci USA. 2020;117(16):8674–6.
Acknowledgements
This study is supported by the Chinese Scholarship Council and Ministry of Education, the People’s Republic of China.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that they do not have any conflict of interest.
Rights and permissions
About this article

Cite this article
Dinmukhamed, T., Huang, Z., Liu, Y. et al. Current advances in design and engineering strategies of industrial enzymes. Syst Microbiol and Biomanuf 1, 15–23 (2021). https://doi.org/10.1007/s43393-020-00005-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43393-020-00005-9