
Abstract
Amphiregulin (AREG) stimulates human epithelial ovarian cancer (EOC) cell invasion by downregulating E-cadherin expression. YAP is a transcriptional cofactor that has been shown to regulate tumorigenesis. This study aimed to examine whether AREG activates YAP in EOC cells and explore the roles of YAP in AREG-induced downregulation of E-cadherin and cell invasion. Analysis of the Cancer Genome Atlas (TCGA) showed that upregulation of AREG and EGFR were associated with poor survival in human EOC. Treatment of SKOV3 human EOC cells with AREG induced the activation of YAP. In addition, AREG downregulated E-cadherin, upregulated Egr-1 and Slug, and stimulated cell invasion. Using gain- and loss-of-function approaches, we showed that YAP was required for the AREG-upregulated Egr-1 and Slug expression. Furthermore, YAP was also involved in AREG-induced downregulation of E-cadherin and cell invasion. This study provides evidence that AREG stimulates human EOC cell invasion by downregulating E-cadherin expression through the YAP/Egr-1/Slug signaling.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Due to metastases that are resistant to conventional therapies, epithelial ovarian cancer (EOC) is the most lethal gynecologic malignancy [1]. Amphiregulin (AREG) is an epidermal growth factor (EGF)-like growth factor that was first purified from the serum-free conditioned medium of PMA-stimulated MCF-7 human breast carcinoma cells [2]. It has been characterized that amphiregulin (AREG), similar to other EGF receptor (EGFR) ligands such as EGF and transforming growth factor-α (TGF-α), specifically binds to the EGFR rather than other ErbB receptors [3]. The tumor microenvironment provides specific conditions for supporting cancer development and progression, including human EOC [4]. Targeting EGFR signaling with small-molecule tyrosine kinase inhibitors and monoclonal antibodies has potential therapeutic effects on EOC [5]. In human ovarian follicular fluid, the concentration of EGF is very low, while AREG has been found to be the most abundant EGFR ligand [6]. In EOC tissues and cell lines, the expression levels of AREG are higher than those of EGF and TGF-α [7,8,9]. In addition, the concentration of AREG in the peritoneal fluid of EOC patients is significantly higher than the concentration of TGF-α [10]. Importantly, the upregulation of AREG expression is detected in all different histological subtypes of human EOC [11]. Given the high expression level of AREG, AREG may play a predominant role in the regulation of human EOC progression [12].
E-cadherin, localized to the surface of epithelial cells in regions of cell–cell contact known as adherens junctions, is required to maintain the epithelial cell polarity and structure [13]. The downregulation of E-cadherin is an important event in epithelial-mesenchymal transition (EMT) and is associated with epithelial tumor cell metastasis [14]. In human EOC, EMT plays a vital role in disease progression and therapy resistance [15]. The Hippo pathway was first identified in Drosophila and acts as a critical regulator of organ size [16]. In mammals, inactivation of the Hippo pathway results in the de-phosphorylation of yes-associated protein (YAP), the major downstream effector of the Hippo pathway, which allows it to stay in the nucleus and exert its transcriptional activities by interacting with the TEAD family of transcription factors [17]. Increasing evidence has demonstrated that in addition to the control of organ size, YAP is also involved in the regulation of tissue regeneration, stem cell self-renewal, and cancer development [18, 19]. In gynecological cancers, including EOC, YAP can initiate tumorigenesis by inducing cell proliferation, migration, invasion, and drug resistance [20]. Importantly, recent studies have provided evidence of cross-talk and interplay between YAP and the process of EMT in some types of cancer [21].
In a context-dependent manner, activation of EGFR by EGF activates YAP in some cell systems but fails to do so in others [22]. Early growth response protein-1 (Egr-1) is an early response gene that can be rapidly activated by many extracellular signaling molecules and plays an important role in the regulation of tumor angiogenesis and tumor growth [23, 24]. EGF-induced transcription of Egr-1 is inhibited in YAP knockout HEK293 cells, which indicates the interplay between YAP and Egr-1 [25]. In SKOV3 and OVCAR5 human EOC cell lines, we have shown that EGF induces Egr-1 expression, which subsequently binds to the Slug promoter and stimulates the expression of Slug, an E-cadherin transcriptional repressor, to downregulate the expression of E-cadherin [26]. Our previous studies have also demonstrated that AREG induces cell invasion by downregulating the expression of E-cadherin in SKOV3 and OVCAR5 cells [27,28,29,30]. Notably, although both AREG and EGF bind exclusively to EGFR, these two EGFR ligands can cause distinct downstream biological activities and act either redundantly or differentially in a cell-type-dependent manner [31]. To date, whether YAP and Egr-1 can be activated by the AREG in human EOC cells remains unclear. If it does, whether YAP/Egr-1 mediates the AREG-induced downregulation of E-cadherin needs to be defined. Given the profound inhibitory effect of AREG on E-cadherin in SKOV3 cells [26], this study used SKOV3 cells as the experimental model to explore the involvement of YAP/Egr-1/Slug signaling in AREG-induced downregulation of E-cadherin and cell invasion in human EOC cells.
Methods
Antibodies and Reagents
The E-cadherin (#610818) antibody was purchased from BD Biosciences. The Egr-1 (#4153), Slug (#9585), phospho-YAPSer127 (#13008), YAP (#12395), and Cyr61 (#14479) antibodies were purchased from Cell Signaling Technology. The EGFR (#sc-03) and α-tubulin (#sc-23948) antibodies were purchased from Santa Cruz Biotechnology. The recombinant human amphiregulin was obtained from R&D systems. The AG1478 was obtained from Sigma.
Cell Culture
The SKOV3 cell line was purchased from the American Type Culture Collection through an official distributor in China (Beijing Zhongyuan Limited). Cells were cultured in a humidified atmosphere containing 5% CO2 and 95% air at 37 °C in RPMI 1640 Medium (Gibco) supplemented with 10% fetal bovine serum (FBS) (Hyclone), 100 U/mL penicillin, and 100 μg/mL streptomycin sulfate (Boster).
Invasion Assay
The transwell cell culture inserts (8 µm pore size, 24 wells, BD Biosciences) were used to measure the cell invasiveness. Each experiment was performed with triplicate inserts. Inserts were coated with 40 µL of 1 mg/mL growth factor-reduced Matrigel (BD Biosciences) for 4 h. Cells (1 × 105 cells/insert) in 250 μL culture medium supplemented with 0.1% FBS were seeded on the top of Matrigel. In the lower chamber, 750 μL of culture medium containing 10% FBS was added. After 48 h of incubation, un-invaded cells were wiped away from the top of the membrane. Invasive cells that penetrated the Matrigel and membrane were fixed with cold methanol at -20 °C for 30 min and then stained with 0.5% crystal violet (Sigma) for 30 min at room temperature. The crystal violet was removed by washing the inserts with tap water. For the quantification, five microscopic fields in each membrane were photographed under an optical microscope, and the number of crystal violet-stained cells was counted manually.
Reverse Transcription-Quantitative Real-Time PCR (RT-qPCR)
The total RNA of treated cells was extracted with TRIzol (Invitrogen). One μg of RNA was reverse-transcribed into first-strand cDNA with the iScript Reverse Transcription Kit (Bio-Rad Laboratories). SYBR Green RT-qPCR was performed on an Applied Biosystems QuantStudio 12 K Flex system equipped with 96-well optical reaction plates. Each 20 μL qPCR reaction contained 1X SYBR Green PCR Master Mix (Applied Biosystems), 60 ng of cDNA, and 250 nM of each specific primer. The following primers were used: E-cadherin, 5'-TCC TGG GCA GAG TGA ATT TT-3' (sense) and 5'-CCG TAG AGG CCT TTT GAC TG-3' (antisense); Slug, 5’-TTC GGA CCC ACA CAT TAC CT-3’ (sense) and 5’-GCA GTG AGG GCA AGA AAA AG-3’ (antisense), and GAPDH, 5'-GAG TCA ACG GAT TTG GTC GT-3' (sense) and 5'-GAC AAG CTT CCC GTT CTC AG-3' (antisense). RNAse-free water and extracted RNA samples without RT were used as negative controls. Melting curve analysis was performed for every primer set to validate the specificity of each assay. The size of each PCR product was confirmed by agarose gel electrophoresis. All RT-qPCR results represent the mean of at least three independent experiments, and each sample was assayed in triplicate. Relative quantification of mRNA levels was conducted by the comparative Ct method with GAPDH as the internal control and using the formula 2–∆∆Ct.
Western Blot Analysis
Western blot was performed as described [32]. Briefly, after treatments, cells were washed with cold phosphate-buffered saline (PBS) and lysed in cell lysis buffer (Cell Signaling Technology) supplemented with a protease inhibitor cocktail (Sigma). Equal amounts of protein were separated by SDS polyacrylamide gel electrophoresis and transferred onto PVDF membranes. After 1 h of blocking at room temperature with 5% nonfat dry milk in Tris-buffered saline (TBS), the PVDF membranes were incubated overnight with primary antibodies diluted in 5% nonfat milk/TBS on the orbital shaker at 4 °C. Following primary antibody incubation, the PVDF membranes were washed with TBS and then incubated with appropriate HRP-conjugated secondary antibodies (Bio-Rad Laboratories). Immunoreactive bands were detected using an enhanced chemiluminescent substrate (Bio-Rad Laboratories) and imaged with a ChemiDoc MP Imager (Bio-Rad Laboratories). The Scion Image software was used to quantify the intensity of a specific band.
Small Interfering RNA (siRNA) Transfection and YAP Overexpression
To knock down endogenous EGFR, Egr-1, or YAP, cells were transfected with 50 nM ON-TARGETplus SMARTpool siRNA targeting for the specific gene (Dharmacon) using Lipofectamine RNAiMAX (Invitrogen). The siCONTROL NON-TARGETING pool siRNA (Dharmacon) was used as the transfection control. To overexpress YAP, cells were transfected with 1 µg empty pCMV vector or vector encoding a full-length constitutively active form of YAP (YAPS127A) with N-terminal Flag-tag (Addgene) using Lipofectamine 3000 (Invitrogen).
Statistical Analysis
The results are presented as the mean ± SEM of at least three independent experiments. All statistical analyses were analyzed by PRISM software. Multiple comparisons were analyzed using one-way ANOVA followed by Tukey’s multiple comparison test. A significant difference was defined as p < 0.05.
Results
Upregulations of AREG and EGFR mRNA are Associated with Poor Survival of Human EOC
To investigate the relationship between AREG and human EOC survival, we queried mRNA expression data on 489 high-grade serous carcinoma (HGSC) samples with mRNA expression z-scores relative to all samples (log microarray) that were published by the Cancer Genome Atlas (TCGA) Research Network [33]. Using cBioPortal for Cancer Genomics [34], our analysis results showed that upregulation of AREG mRNA was detected in 50 (10.2%) of 489 cases. The upregulation of AREG mRNA was not associated with overall survival (Log-rank P = 0.266), but a significant association between the upregulation of AREG mRNA and disease-free survival was observed (Log-rank P = 0.0182) (Fig. 1A). In addition to AREG, upregulation of EGFR mRNA was detected in 56 (11.4%) cases, and that is associated with poor overall (Log-rank P = 4.966e-3) but not disease-free survival (Log-rank P = 0.703) (Fig. 1B). These results indicate that elevated expression of AREG and its receptor, EGFR, may play an important role in the regulation of human EOC progression.

Elevation of AREG or EGFR mRNA levels is associated with poor survival in high-grade serous ovarian carcinomas (HGSC). A and B, The cBioPortal for Cancer Genomics was used to query 489 HGSC samples with mRNA expression data from The Cancer Genome Atlas for the upregulation of AREG (A) and EGFR (B) (z-score threshold ± 1.5). The upper panel shows the graphical summary (OncoPrint) of the samples exhibiting upregulation of AREG or EGFR. The lower panel shows overall and disease-free survival differences between unaltered samples and those with the elevation of AREG or EGFR. Results are displayed as Kaplan–Meier survival curves with P values from a Log-rank test
EGFR Mediates AREG-induced Downregulation of E-cadherin
We have shown that the concentration of AREG in the ovarian follicular fluid of women is ~ 200 ng/mL [35]. Therefore, to examine the effect of AREG on E-cadherin expression, we treated SKOV3 human EOC cells with 50, 100, and 200 ng/mL recombinant human AREG. RT-qPCR results showed that treatment with 50, 100, and 200 ng/mL induced a comparable inhibitory effect on the E-cadherin mRNA levels (Fig. 2A). Western blot results further confirmed the suppressive effect of AREG on E-cadherin protein levels in SKOV3 cells (Fig. 2B). Therefore, we used 100 ng/mL AREG for the following experiments. To examine whether EGFR is required for the AREG-induced downregulation of E-cadherin expression, an EGFR inhibitor AG1478 was used to block the function of EGFR. As shown in Fig. 2C, pretreated cells with AG1478 for 1 h blocked the suppressive effect of AREG on the E-cadherin protein levels. To further confirm this, endogenous EGFR was knocked down by transfecting cells with EGFR siRNA. As shown in Fig. 2D, transfection of EGFR siRNA significantly downregulated EGFR protein levels. In addition, the knockdown of EGFR abolished the AREG-induced downregulation of E-cadherin protein levels.

AREG downregulates E-cadherin expression in SKOV3 cells. A and B, Cells were treated with 50, 100, and 200 ng/mL AREG for 24 h. The mRNA (A) and protein (B) levels of E-cadherin were examined by RT-qPCR and western blot, respectively. C, Cells were pretreated with vehicle control (DMSO) or 10 µM AG1478 for 1 h, and then treated with 100 ng/mL AREG for 24 h. The E-cadherin protein levels were examined by western blot. D, Cells were transfected with 50 nM control siRNA (si-Ctrl) or EGFR siRNA (si-EGFR) for 48 h, and then treated with 100 ng/mL AREG for 24 h. The protein levels of E-cadherin and EGFR were examined by western blot. The results are expressed as the mean ± SEM of at least three independent experiments. Values that are statistically different from one another (p < 0.05) are indicated by different letters
AREG Activates YAP Through EGFR
Activation of EGFR by EGF treatment activates YAP by reducing its phosphorylation at Ser127 rapidly in MCF-10A cells [36]. To examine the effect of AREG on YAP activation, SKOV3 cells were treated with AREG for 10, 30, and 60 min. The activation of YAP was examined by the reduction of its phosphorylation at Ser127. As shown in Fig. 3A, treatment with AREG for 30 and 60 min resulted in a reduction of YAP phosphorylation at Ser127 which indicated its activation. The AREG-activated YAP was blocked by pretreatment with the EGFR inhibitor AG1478 (Fig. 3B). Using EGFR siRNA, we further confirmed that EGFR was required for the AREG-induced activation of YAP (Fig. 3C).

AREG activates YAP in SKOV3 cells. A, Cells were treated with 100 ng/mL AREG for 10, 30, and 60 min. The levels of phosphorylated YAP at Ser127 and the total form of YAP were determined by western blot. B, Cells were pretreated with vehicle control (DMSO) or 10 µM AG1478 for 1 h, and then treated with 100 ng/mL AREG for 1 h. The levels of phosphorylated YAP at Ser127 and the total form of YAP were determined by western blot. C, Cells were transfected with 50 nM control siRNA (si-Ctrl) or EGFR siRNA (si-EGFR) for 48 h, and then treated with 100 ng/mL AREG for 1 h. The levels of phosphorylated YAP at Ser127, the total form of YAP, and protein levels of EGFR were determined by western blot. The results are expressed as the mean ± SEM of at least three independent experiments. Values that are statistically different from one another (p < 0.05) are indicated by different letters
AREG Downregulates E-cadherin Expression by Inducing Egr-1 Expression
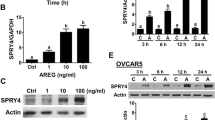
To examine whether AREG treatment induces Egr-1 expression, SKOV3 cells were treated with AREG for 1, 3, and 6 h. As shown in Fig. 4A, AREG treatment induced a transient expression of Egr-1 protein, with the maximal effect observed after 3 h of AREG treatment. The induction of Egr-1 protein expression by AREG was blocked by the inhibition of EGFR function and the knockdown of EGFR (Fig. 4B and C). Whether AREG-induced Egr-1 expression participates in the AREG-downregulated E-cadherin expression remains unknown. Therefore, Egr-1 siRNA was used to block the AREG-induced Egr-1 expression. As shown in Fig. 4D, AREG induced Egr-1 protein expression in cells transfected with control siRNA. However, this induction was not observed in cells transfected with Egr-1 siRNA. Importantly, the AREG-induced downregulation of E-cadherin protein levels was attenuated by the siRNA-mediated knockdown of Egr-1.

AREG upregulates Egr-1 expression in SKOV3 cells. A, Cells were treated with 100 ng/mL AREG for 1, 3, and 6 h. The protein levels of Egr-1 were determined by western blot. B, Cells were pretreated with vehicle control (DMSO) or 10 µM AG1478 for 1 h, and then treated with 100 ng/mL AREG for 3 h. The protein levels of Egr-1 were determined by western blot. C, Cells were transfected with 50 nM control siRNA (si-Ctrl) or EGFR siRNA (si-EGFR) for 48 h, and then treated with 100 ng/mL AREG for 3 h. The protein levels of Egr-1 and EGFR were determined by western blot. D, Cells were transfected with 50 nM control siRNA (si-Ctrl) or Egr-1 siRNA (si-Egr-1) for 48 h, and then treated with 100 ng/mL AREG for 24 h. The protein levels of Egr-1 and E-cadherin were determined by western blot. The results are expressed as the mean ± SEM of at least three independent experiments. Values that are statistically different from one another (p < 0.05) are indicated by different letters
Egr-1 Mediates AREG-Upregulated Slug Expression
Slug is a well-known E-cadherin transcriptional repressor [37]. RT-qPCR results showed that AREG upregulated Slug mRNA levels in SKOV3 cells after 3 h of treatment, and this stimulatory effect was also observed after 6 h of AREG treatment (Fig. 5A). Western blot results showed a similar stimulatory effect of AREG on Slug protein levels (Fig. 5B). In addition, AREG-induced upregulation of Slug protein levels was blocked by the siRNA-mediated knockdown of EGFR (Fig. 5C). Using Egr-1 siRNA, we revealed that AREG-upregulated Slug protein levels were partially blocked by the knockdown of Egr-1 (Fig. 5D).

AREG upregulates Slug expression in SKOV3 cells. A and B, Cells were treated with 100 ng/mL AREG for 1, 3, and 6 h. The mRNA and protein levels of Slug were determined by RT-qPCR (A) and western blot (B), respectively. C, Cells were transfected with 50 nM control siRNA (si-Ctrl) or EGFR siRNA (si-EGFR) for 48 h, and then treated with 100 ng/mL AREG for 6 h. The protein levels of Slug and EGFR were determined by western blot. D, Cells were transfected with 50 nM control siRNA (si-Ctrl) or Egr-1 siRNA (si-Egr-1) for 48 h, and then treated with 100 ng/mL AREG for 6 h. The protein levels of Slug and Egr-1 were determined by western blot. The results are expressed as the mean ± SEM of at least three independent experiments. Values that are statistically different from one another (p < 0.05) are indicated by different letters
YAP Mediates AREG-Induced Downregulation of E-Cadherin and Upregulation of Egr-1 and Slug Expression
Next, we examined whether activation of YAP is involved in AREG-downregulated E-cadherin expression. Transfecting SKOV3 cells with YAP siRNA significantly downregulated endogenous YAP protein levels. Knockdown of YAP did not affect the basal E-cadherin expression but attenuated the AREG-downregulated E-cadherin protein levels (Fig. 6A). In addition, the AREG-induced Egr-1 protein levels were inhibited by the YAP knockdown (Fig. 6B). Moreover, the knockdown of YAP attenuated the AREG-stimulated upregulation of Slug protein levels (Fig. 6C). To further confirm the role of YAP in the regulation of Egr-1, Slug, and E-cadherin, a constitutively active form of YAP, the YAP serine 127 to alanine (S127A) mutant with a Flag-tag was transfected into SKOV3 cells. Western blot results showed that overexpression of YAPS127A upregulated its well-known target Cyr61, which indicated the success of the YAPS127A overexpression. In addition, the protein levels of E-cadherin were downregulated by the overexpression of YAPS127A. Moreover, overexpression of YAPS127A upregulated the protein levels of Egr-1 and Slug (Fig. 6D). Taken together, these results indicate that AREG-activated YAP leads to the expression of Egr-1, which subsequently contributes to the Slug-mediated downregulation of E-cadherin expression.

YAP mediates AREG-induced downregulation of E-cadherin expression and upregulation of Egr-1 and Slug expression. A-C, Cells were transfected with 50 nM control siRNA (si-Ctrl) or YAP siRNA (si-YAP) for 48 h, and then treated with 100 ng/mL AREG for 24 h (A), 3 h (B), and 6 h (C). The protein levels of E-cadherin, Egr-1, Slug, and YAP were determined by western blot. D, Cells were transfected with 1 µg control vector (pCMV) or vector containing YAPS127A cDNA (pCMV-YAPS127A) for 48 h. The protein levels of E-cadherin, Egr-1, Slug, Cyr61, and YAP were examined by western blot. The results are expressed as the mean ± SEM of at least three independent experiments. Values that are statistically different from one another (p < 0.05) are indicated by different letters
YAP-Mediated Egr-1 Expression is Required for AREG-Stimulated SKOV3 Cell Invasion
Consistent with our previous study, treatment with AREG stimulated SKOV3 cell invasion. The pro-invasive effect of AREG was blocked by the inhibition of EGFR (Fig. 7A). Our previous study has shown that overexpression of E-cadherin blocked the AREG-induced EOC cell invasion [27]. Given the involvement of YAP and Egr-1 in AREG-induced downregulation of E-cadherin, we examined whether the knockdown of YAP or Egr-1 affects the AREG-stimulated cell invasion. As shown in Fig. 7B, siRNA-mediated knockdown of Egr-1 blocked the AREG-stimulated cell invasion. In addition, the AREG-stimulated cell invasion was attenuated by the knockdown of YAP. Moreover, the invasiveness was increased in SKOV3 cells that overexpressed YAPS127A (Fig. 7C).

YAP and Egr-1 are required for the AREG-stimulated SKOV3 cell invasion. A, Cells were pretreated with vehicle control (DMSO) or 10 µM AG1478 for 1 h, and then treated with 100 ng/mL AREG. B, Cells were transfected with 50 nM control siRNA (si-Ctrl), Egr-1 siRNA (si-Egr-1) or YAP siRNA (si-YAP) for 48 h, and then treated with 100 ng/mL AREG. C, Cells were transfected with 1 µg control vector (pCMV) or vector containing YAPS127A cDNA (pCMV-YAPS127A) for 48 h. The levels of cell invasiveness were examined by transwell invasion assay. The scale bar represents 50 μm. The results are expressed as the mean ± SEM of at least three independent experiments. Values that are statistically different from one another (p < 0.05) are indicated by different letters
Discussion
EGFR gene amplification, mutation, and overexpression of EGFR protein levels are frequently observed in human EOC and are associated with more aggressive clinical behavior and poor prognosis [38]. Regional dissemination of the cancer cells and metastases are hallmarks of human EOC progression [39]. Human EOC cells with low E-cadherin expression are more invasive, and the absence of E-cadherin expression is associated with poor survival [40, 41]. AREG, EGF, and TGF-α have been shown to stimulate EOC cell invasion by downregulating the expression of E-cadherin [26,27,28,29,30, 42,43,44,45]. A previous study measuring the concentrations of EGF-like ligands in ascites fluid from 43 ovarian cancer patients shows that AREG is the most abundant and generalized ligand secreted by tumors compared to EGF, TGF-α, betacellulin, and HB-EGF. Treatment of EOC cells with cisplatin increases the AREG promoter activity. In addition, the knockdown of AREG or treating an anti-AREG monoclonal antibody inhibits the tumorigenic growth of human EOC cells [9]. Moreover, AREG has been shown to increase the stemness and drug resistance of EOC cells [46]. These results indicate the critical roles of AREG in EOC development and progression and suggest that EGFR and AREG are potential therapeutic targets for human EOC [47]. In the present study, our TCGA analysis results showed that EOC patients with upregulated EGFR had lower overall survival. However, the overall survival was not significantly altered in patients with high AREG expression, although the upregulation of AREG was associated with disease-free survival. Regardless of our TCGA analysis results, a previous study shows that higher AREG protein levels correlate with the advanced stages of EOC in both clinical patients and the tissue array samples. In addition, Kaplan–Meier survival analysis of 518 EOC cases from the Oncomine data reveals that EOC patients with high AREG expression correlate with significantly shorter survival than those with low AREG expression [46]. Taken together, these results suggest that increased AREG and EGFR expression in human EOC cells contribute to poor survival of the disease progression.
In mammals, YAP is the major downstream effector of the Hippo pathway. Activation of the Hippo pathway stimulates the serine/threonine kinases MST1/2, which phosphorylate the downstream kinases LATS1/2. Phosphorylated LATS1/2 subsequently phosphorylates YAP, leading to its cytoplasmic localization and proteolytic degradation. Conversely, inactivation of the Hippo pathway results in the dephosphorylation of YAP, allowing it to stay in the nucleus and regulate gene expression [17]. It has been shown that YAP can be phosphorylated at different serine and tyrosine residues in both LATS1/2-dependent and -independent manners to regulate its activation [48,49,50]. However, the roles of these phosphorylation sites in regulating YAP function are largely undetermined. In the present study, it remains unknown whether activation of EGFR by AREG treatment phosphorylates YAP at sites other than Ser127. Therefore, investigating whether AREG can affect YAP phosphorylation at different sites and subsequently contribute to the regulation of YAP function in human EOC will be of great interest.
Many cancer-related cellular processes, such as cell proliferation, migration, invasion, differentiation, and stemness, can be regulated by YAP. Thus, YAP has been considered an oncogene, and its aberrant activation and expression are observed in several cancers, including human EOC [51]. Phosphorylation of the YAP protein at Ser127 prevents its nuclear translocation and transcription coactivator function by promoting YAP binding to cytoplasmic 14–3-3 proteins. By contrast, de-phosphorylation of YAP at Ser127 activates the YAP and increases its nuclear translocation [17]. In human EOC, although both normal and tumor tissues express YAP, the YAP protein is differentially located in the cytoplasm and the nucleus of normal and the EOC tissues, respectively. Importantly, the phosphorylation levels of YAP at Ser127 are significantly decreased in human EOC tissues when compared to normal and benign tissues. In addition, YAP mediates the lysophosphatidic acid-induced EOC cell migration and invasion by increasing the AREG expression [52]. These results indicate the aberrant activation of YAP plays a vital role in regulating EOC progression. In the human mammary epithelial cell line MCF10A, YAP was first identified to be able to induce the downregulation of E-cadherin [53]. Since this initial study, accumulating evidence has reported that the YAP acts as an important inducer of EMT in many types of human cancer [21]. Although AREG is the well-known YAP target, and activation of EGFR activates YAP, to the best of our knowledge, there is no study that directly examines the effect of AREG on YAP activation in human EOC cells. Our results showed that AREG treatment activated YAP in SKOV3 cells. The stimulatory effect of AREG on YAP activation is consistent with a previous study in human cervical cancer cells [54]. In addition, using gain- and loss-of-function approaches, we showed that YAP was involved in the AREG-induced downregulation of E-cadherin and cell invasion.
Many studies have reported how external stimuli affect YAP activity. However, the downstream target genes that mediate the biological functions of YAP remain not much explored [55]. In HEK293 cells, EGF-induced expressions of AP-1, Egr-1, and Egr-3 are significantly attenuated by the knockdown of YAP [25]. These results indicate that these transcription factors could propagate the function of YAP. We showed that Egr-1 expression was induced by the AREG treatment in SKOV3 cells. The Egr-1 was required for the AREG-induced Slug expression, which subsequently contributed to the AREG-downregulated E-cadherin. These results, together with our previous study, indicate that activation of EGFR by AREG or EGF results in the induction of Egr-1, which leads to the upregulation of Slug and the following downregulation of E-cadherin and cell invasion [26]. Importantly, our results revealed that AREG-induced Egr-1 and Slug expressions were attenuated by the knockdown of YAP. In addition, overexpressed the constitutively active form of YAP upregulated the expression of Egr-1 and Slug. These findings indicate that YAP mediates the inhibitory effect of AREG on E-cadherin expression by upregulating the expression levels of its downstream target, Egr-1,which contributes to the induction of Slug. Interestingly, the knockdown of YAP did not fully block the stimulatory effects of AREG on Egr-1 and Slug expression. These results suggest the existence of other upstream factors of Egr-1/Slug signaling that can mediate the AREG-induced downregulation of E-cadherin. In addition to the regulation of Egr-1 expression, a previous study shows that YAP can form a complex with Egr-1 and enhances its stimulatory effect on Bax expression in irradiated prostate cancer cells [56]. Whether the same is true in human EOC cells remains unknown and warrants further investigation.
Human EOCs encompass a spectrum of histologic subtypes distinguished by their unique histological, clinical, molecular, and epidemiological features. These subtypes exhibit distinct morphological and molecular characteristics that influence their clinical behavior, response to treatment, and prognosis [57]. High-rise serous carcinoma (HGSC) is the most prevalent among these subtypes. However, reports suggest that SKOV3, previously categorized within HGSC, may not accurately represent this subtype [58, 59]. We are aware that our study was conducted using a single human EOC cell line. While this approach allowed us to delve deeply into specific molecular pathways, extrapolating these findings to different subtypes of human EOC requires further investigation.
In summary, the present study reveals that the upregulation of AREG and its receptor, EGFR, are associated with poor survival in human EOC. In human EOC cells, AREG treatment downregulates E-cadherin expression and stimulates cell invasion. Mechanistically, we demonstrate that AREG activates YAP and induces Egr-1 and Slug expression. Using gain- and loss-of-function approaches, our results show that YAP is required for the AREG-induced Egr-1 and Slug expression. In addition, YAP mediates the inhibitory effect of AREG on E-cadherin expression and the stimulatory effect on cell invasion. These findings provide evidence that YAP plays an important role in mediating EGFR-regulated EMT. Our study increases the understanding of the pathological function of AREG in human EOC and provides important insights into the regulation of E-cadherin expression and cell invasion, which may help to develop new therapeutic methods for human EOC.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Code Availability
Not applicable.
References
Shaik B, Zafar T, Balasubramanian K, Gupta SP. An Overview of Ovarian Cancer: Molecular Processes Involved and Development of Target-based Chemotherapeutics. Curr Top Med Chem. 2021;21(4):329–46.
Shoyab M, McDonald VL, Bradley JG, Todaro GJ. Amphiregulin: a bifunctional growth-modulating glycoprotein produced by the phorbol 12-myristate 13-acetate-treated human breast adenocarcinoma cell line MCF-7. Proc Natl Acad Sci U S A. 1988;85(17):6528–32.
Singh B, Carpenter G, Coffey RJ: EGF receptor ligands: recent advances. F1000Res 2016;5:F1000 Faculty Rev–2270.
Nwani NG, Sima LE, Nieves-Neira W, Matei D. Targeting the Microenvironment in High Grade Serous Ovarian Cancer. Cancers (Basel). 2018;10(8):266.
Han C, Altwerger G, Menderes G, Haines K, Feinberg J, Lopez S, Manzano A, Varughese J, Santin AD. Novel targeted therapies in ovarian and uterine carcinosarcomas. Discov Med. 2018;25(140):309–19.
Inoue Y, Miyamoto S, Fukami T, Shirota K, Yotsumoto F, Kawarabayashi T. Amphiregulin is much more abundantly expressed than transforming growth factor-alpha and epidermal growth factor in human follicular fluid obtained from patients undergoing in vitro fertilization-embryo transfer. Fertil Steril. 2009;91(4):1035–41.
Yotsumoto F, Yagi H, Suzuki SO, Oki E, Tsujioka H, Hachisuga T, Sonoda K, Kawarabayashi T, Mekada E, Miyamoto S. Validation of HB-EGF and amphiregulin as targets for human cancer therapy. Biochem Biophys Res Commun. 2008;365(3):555–61.
Tanaka Y, Miyamoto S, Suzuki SO, Oki E, Yagi H, Sonoda K, Yamazaki A, Mizushima H, Maehara Y, Mekada E, et al. Clinical significance of heparin-binding epidermal growth factor-like growth factor and a disintegrin and metalloprotease 17 expression in human ovarian cancer. Clin Cancer Res. 2005;11(13):4783–92.
Carvalho S, Lindzen M, Lauriola M, Shirazi N, Sinha S, Abdul-Hai A, Levanon K, Korach J, Barshack I, Cohen Y, et al. An antibody to amphiregulin, an abundant growth factor in patients’ fluids, inhibits ovarian tumors. Oncogene. 2016;35(4):438–47.
Yagi H, Miyamoto S, Tanaka Y, Sonoda K, Kobayashi H, Kishikawa T, Iwamoto R, Mekada E, Nakano H. Clinical significance of heparin-binding epidermal growth factor-like growth factor in peritoneal fluid of ovarian cancer. Br J Cancer. 2005;92(9):1737–45.
D’Antonio A, Losito S, Pignata S, Grassi M, Perrone F, De Luca A, Tambaro R, Bianco C, Gullick WJ, Johnson GR, et al. Transforming growth factor alpha, amphiregulin and cripto-1 are frequently expressed in advanced human ovarian carcinomas. Int J Oncol. 2002;21(5):941–8.
Fang L, Sun YP, Cheng JC. The role of amphiregulin in ovarian function and disease. Cell Mol Life Sci. 2023;80(3):60.
Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer. 2004;4(2):118–32.
Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009;28(1–2):15–33.
Loret N, Denys H, Tummers P, Berx G. The Role of Epithelial-to-Mesenchymal Plasticity in Ovarian Cancer Progression and Therapy Resistance. Cancers (Basel). 2019;11(6):838.
Pan D. Hippo signaling in organ size control. Genes Dev. 2007;21(8):886–97.
Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev. 2014;94(4):1287–312.
Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130(6):1120–33.
Zhao B, Tumaneng K, Guan KL. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat Cell Biol. 2011;13(8):877–83.
Wang D, He J, Dong J, Meyer TF, Xu T. The HIPPO pathway in gynecological malignancies. Am J Cancer Res. 2020;10(2):610–29.
Akrida I, Bravou V, Papadaki H. The deadly cross-talk between Hippo pathway and epithelial-mesenchymal transition (EMT) in cancer. Mol Biol Rep. 2022;49(10):10065–76.
Park HW, Guan KL. Regulation of the Hippo pathway and implications for anticancer drug development. Trends Pharmacol Sci. 2013;34(10):581–9.
Gashler A, Sukhatme VP. Early growth response protein 1 (Egr-1): prototype of a zinc-finger family of transcription factors. Prog Nucleic Acid Res Mol Biol. 1995;50:191–224.
Fahmy RG, Dass CR, Sun LQ, Chesterman CN, Khachigian LM. Transcription factor Egr-1 supports FGF-dependent angiogenesis during neovascularization and tumor growth. Nat Med. 2003;9(8):1026–32.
Koo JH, Plouffe SW, Meng Z, Lee DH, Yang D, Lim DS, Wang CY, Guan KL. Induction of AP-1 by YAP/TAZ contributes to cell proliferation and organ growth. Genes Dev. 2020;34(1–2):72–86.
Cheng JC, Chang HM, Leung PC. Egr-1 mediates epidermal growth factor-induced downregulation of E-cadherin expression via Slug in human ovarian cancer cells. Oncogene. 2013;32(8):1041–9.
So WK, Fan Q, Lau MT, Qiu X, Cheng JC, Leung PC. Amphiregulin induces human ovarian cancer cell invasion by down-regulating E-cadherin expression. FEBS Lett. 2014;588(21):3998–4007.
So WK, Cheng JC, Liu Y, Xu C, Zhao J, Chang VT, Leung PC. Sprouty4 mediates amphiregulin-induced down-regulation of E-cadherin and cell invasion in human ovarian cancer cells. Tumour Biol. 2016;37(7):9197–207.
Cheng JC, Chang HM, Xiong S, So WK, Leung PC. Sprouty2 inhibits amphiregulin-induced down-regulation of E-cadherin and cell invasion in human ovarian cancer cells. Oncotarget. 2016;7(49):81645–60.
Qiu X, Cheng JC, Klausen C, Fan Q, Chang HM, So WK, Leung PC. Transforming growth factor-alpha induces human ovarian cancer cell invasion by down-regulating E-cadherin in a Snail-independent manner. Biochem Biophys Res Commun. 2015;461(1):128–35.
Wee P, Wang Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers (Basel). 2017;9(5):52.
Wu Z, Fang L, Yang S, Gao Y, Wang Z, Meng Q, Dang X, Sun YP, Cheng JC. GDF-11 promotes human trophoblast cell invasion by increasing ID2-mediated MMP2 expression. Cell Commun Signal. 2022;20(1):89.
Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–15.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1.
Fang L, Yu Y, Li Y, Wang S, He J, Zhang R, Sun YP. Upregulation of AREG, EGFR, and HER2 contributes to increased VEGF expression in granulosa cells of patients with OHSSdagger. Biol Reprod. 2019;101(2):426–32.
Fan R, Kim NG, Gumbiner BM. Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc Natl Acad Sci U S A. 2013;110(7):2569–74.
Xu R, Won JY, Kim CH, Kim DE, Yim H. Roles of the Phosphorylation of Transcriptional Factors in Epithelial-Mesenchymal Transition. J Oncol. 2019;2019:5810465.
Lassus H, Sihto H, Leminen A, Joensuu H, Isola J, Nupponen NN, Butzow R. Gene amplification, mutation, and protein expression of EGFR and mutations of ERBB2 in serous ovarian carcinoma. J Mol Med (Berl). 2006;84(8):671–81.
Naora H, Montell DJ. Ovarian cancer metastasis: integrating insights from disparate model organisms. Nat Rev Cancer. 2005;5(5):355–66.
Veatch AL, Carson LF, Ramakrishnan S. Differential expression of the cell-cell adhesion molecule E-cadherin in ascites and solid human ovarian tumor cells. Int J Cancer. 1994;58(3):393–9.
Darai E, Scoazec JY, Walker-Combrouze F, Mlika-Cabanne N, Feldmann G, Madelenat P, Potet F. Expression of cadherins in benign, borderline, and malignant ovarian epithelial tumors: a clinicopathologic study of 60 cases. Hum Pathol. 1997;28(8):922–8.
Cheng JC, Klausen C, Leung PC. Hydrogen peroxide mediates EGF-induced down-regulation of E-cadherin expression via p38 MAPK and snail in human ovarian cancer cells. Mol Endocrinol. 2010;24(8):1569–80.
Cheng JC, Klausen C, Leung PC. Hypoxia-inducible factor 1 alpha mediates epidermal growth factor-induced down-regulation of E-cadherin expression and cell invasion in human ovarian cancer cells. Cancer Lett. 2013;329(2):197–206.
Cheng JC, Qiu X, Chang HM, Leung PC. HER2 mediates epidermal growth factor-induced down-regulation of E-cadherin in human ovarian cancer cells. Biochem Biophys Res Commun. 2013;434(1):81–6.
Qiu X, Cheng JC, Chang HM, Leung PC. COX2 and PGE2 mediate EGF-induced E-cadherin-independent human ovarian cancer cell invasion. Endocr Relat Cancer. 2014;21(4):533–43.
Tung SL, Huang WC, Hsu FC, Yang ZP, Jang TH, Chang JW, Chuang CM, Lai CR, Wang LH. miRNA-34c-5p inhibits amphiregulin-induced ovarian cancer stemness and drug resistance via downregulation of the AREG-EGFR-ERK pathway. Oncogenesis. 2017;6(5): e326.
Ntanasis-Stathopoulos I, Fotopoulos G, Tzanninis IG, Kotteas EA. The Emerging Role of Tyrosine Kinase Inhibitors in Ovarian Cancer Treatment: A Systematic Review. Cancer Invest. 2016;34(7):313–39.
Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 2010;24(1):72–85.
Cheng Y, Mao M, Lu Y. The biology of YAP in programmed cell death. Biomark Res. 2022;10(1):34.
Sugihara T, Werneburg NW, Hernandez MC, Yang L, Kabashima A, Hirsova P, Yohanathan L, Sosa C, Truty MJ, Vasmatzis G, et al. YAP Tyrosine Phosphorylation and Nuclear Localization in Cholangiocarcinoma Cells Are Regulated by LCK and Independent of LATS Activity. Mol Cancer Res. 2018;16(10):1556–67.
Wang Y, Xu X, Maglic D, Dill MT, Mojumdar K, Ng PK, Jeong KJ, Tsang YH, Moreno D, Bhavana VH, et al. Comprehensive Molecular Characterization of the Hippo Signaling Pathway in Cancer. Cell Rep. 2018;25(5):1304-1317 e1305.
Cai H, Xu Y. The role of LPA and YAP signaling in long-term migration of human ovarian cancer cells. Cell Commun Signal. 2013;11(1):31.
Overholtzer M, Zhang J, Smolen GA, Muir B, Li W, Sgroi DC, Deng CX, Brugge JS, Haber DA. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci U S A. 2006;103(33):12405–10.
He C, Mao D, Hua G, Lv X, Chen X, Angeletti PC, Dong J, Remmenga SW, Rodabaugh KJ, Zhou J, et al. The Hippo/YAP pathway interacts with EGFR signaling and HPV oncoproteins to regulate cervical cancer progression. EMBO Mol Med. 2015;7(11):1426–49.
Totaro A, Panciera T, Piccolo S. YAP/TAZ upstream signals and downstream responses. Nat Cell Biol. 2018;20(8):888–99.
Zagurovskaya M, Shareef MM, Das A, Reeves A, Gupta S, Sudol M, Bedford MT, Prichard J, Mohiuddin M, Ahmed MM. EGR-1 forms a complex with YAP-1 and upregulates Bax expression in irradiated prostate carcinoma cells. Oncogene. 2009;28(8):1121–31.
Lheureux S, Braunstein M, Oza AM. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA Cancer J Clin. 2019;69(4):280–304.
Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun. 2013;4:2126.
Ciucci A, Buttarelli M, Fagotti A, Scambia G, Gallo D. Preclinical models of epithelial ovarian cancer: practical considerations and challenges for a meaningful application. Cell Mol Life Sci. 2022;79(7):364.
Acknowledgements
This work was supported by an operating grant (32170868) and the Research Fund for International Young Scientists (32050410302) from the National Natural Science Foundation of China to Jung-Chien Cheng. This work was also supported by the Funding for Scientific Research and Innovation Team of The First Affiliated Hospital of Zhengzhou University (QNCXTD2023011) to Lanlan Fang.
Author information
Authors and Affiliations
Contributions
J.C.C. and L.F. contributed to the study design, data analysis, interpretation of data, and manuscript writing. Q.J., H.W., and B.B. contributed to the manuscript drafting. Q.J., H.W., B.B., X.H., Y.J., and L.Z. performed the experiments and prepared the figures. A.T. contributed to the study design and provided English editing and proofreading of the manuscript.
Corresponding author
Ethics declarations
Ethics Approval
This study was carried out in accordance with the approved guidelines from the Zhengzhou University Research Ethics Board.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jia, Q., Wang, H., Bi, B. et al. Amphiregulin Downregulates E-cadherin Expression by Activating YAP/Egr-1/Slug Signaling in SKOV3 Human Ovarian Cancer Cells. Reprod. Sci. (2024). https://doi.org/10.1007/s43032-024-01673-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s43032-024-01673-x