
Abstract
We downloaded gene expression data, clinical data, and somatic mutation data of cervical squamous cell carcinoma (CSCC) patients from The Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression (GTEx) databases. Predictive lncRNAs were screened using univariate analysis and least absolute shrinkage and selection operator (LASSO) regression, and risk scores were calculated for each patient according to the expression levels of lncRNAs and regression coefficients to establish a risk model that could be a novel signature. We assessed the correlation between immune infiltration status, chemotherapeutics sensitivity, immune checkpoint proteins (ICP), and the signature. Therefore, we selected 11 immune-related lncRNAs (WWC2,AS2, STXBP5.AS1, ERICH6.AS1, USP30.AS1, LINC02073, RBAKDN, IL21R.AS1, LINC02078, DLEU1, LINC00426, BOLA3.AS1) to construct the risk model. Patients who were in the high-risk group had a shorter survival time than those in the low-risk group (p < 0.001). Risk scores in the signature were negatively correlated with macrophage M1, macrophage M2, and T cell CD8 + ; what’s more, T cell CD8 + was higher in the low-risk group. The expression levels of ICP such as PD-L1, PD-1, CTLA-4, TIGIT, LAG-3, and TIM-3 were substantially higher in the low-risk group. For chemotherapeutic agents, high-risk scores were associated with higher half-inhibitory concentrations (IC50) of cisplatin. These findings suggested that the risk model can be a novel signature for predicting CSCC patients’ prognosis, and it also can be used to formulate clinical treatment plans for CSCC patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cervical squamous cell carcinoma (CSCC) is the fourth dominant cause of cancer-related deaths in women worldwide, accounting for 80–85% of all cervical cancer diagnoses [1, 2]. With human papillomavirus (HPV) vaccination and early-stage screening, the incidence rate of CSCC has decreased significantly. Surgery, radiotherapy, and chemotherapy have been commonly used for cervical cancer patients, but the 5-year survival rate is still not satisfactory because of advanced stage, relapse, metastasis, and drug resistance [3,4,5]. Although the use of immunotherapy checkpoint inhibitors (ICIs) has been successful in the treatment of various cancers [6], the development and application of immunotherapy for CSCC patients are restricted because the positive response remains low [7]. Thus, it is crucial to provide CSCC with new therapeutic targets and to find novel biomarkers for its early diagnosis and prognosis.
Long non-coding RNAs (lncRNAs) are a class of non-coding transcripts with sequences longer than 200 nucleotides that can mediate post-transcriptional modifications [8, 9]. They play a key role in immune activation, cancer immunity, antigen presentation, and immune cell infltration [10, 11]. For example, lncRNA CamK-A was found to be highly expressed in various human cancers and could regulate Ca2+-signaling-mediated tumor microenvironment remodeling [12]. Also, in colorectal cancer cells, overexpression of HLA-F-AS1 could repress miR-375 and promote the expression of PFN1, further parting macrophages toward M2 phenotype and exacerbating tumorigenesis [13]. EGFR promoted immune escape of hepatocellular carcinoma cells and Treg cell differentiation through specific binding to EGFR protein [14]. In addition to participating in the tumor microenvironment, lncRNAs can also affect the response to immunotherapy [15], which is rarely reported in CSCC patients.
In this study, we used the RNA sequencing data of CSCC patients in the Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression (GTEx) database to identify immune-related lncRNA (irlncRNA) and established an 11-irlncRNA signature based on LASSO regression analysis. Subsequently, we explored the company between the signature of 11-irlncRNA and several disease characteristics in CSCC patients. Drug sensitivity analysis also was performed to improve drug therapy. Collectively, the present study may provide a strategy to predict prognosis and immune-related therapeutic targets in CSCC patients.
Materials and Methods
Processing Data and Obtaining Immune-Related lncRNAs
The gene expression data, clinical data, and somatic mutation data of CSCC patients were acquired from the TCGA index. The gene expression profiles of 10 normal cervical tissues were obtained from the GTEx database. The GTF file from Ensembl was used to annotate and distinguish mRNAs and lncRNAs. Gene set enrichment analysis (GSEA) was applied to screen immune-related pathways; genes in these pathways were identified as immune genes. We used R software packages “limma” and “edgeR” to filter differentially expressed lncRNAs (DElncRNAs), with |log2FC|> 2 and FDR q value < 0.05. DElncRNAs with immune gene correlation coefficients greater than 0.5 and p value < 0.001 were selected as irlncRNAs.
Construction of Risk Model
We first used univariate Cox analysis to identify irlncRNAs that were significantly associated with overall survival of CSCC patients (p < 0.05). Immediately after, we used LASSO regression analysis to further screen the irlncRNAs with prognostic value [16]. The features were chosen based on the best penalty parameter λ. We used the coefficients obtained from LASSO to calculate the risk scores:
where Coefi is the coefficients and xi is the fragments per kilobase million (FPKM) value of each irlncRNA. We can calculate the risk scores of each patient according to this equation.
Validation of the Risk Model
The receiver operating characteristics (ROC) for 1, 3, and 5 years of the model were calculated. The Akaike information criterion (AIC) value of the ROC curve was evaluated to select the maximum inflection point, which was considered to be the key point for distinguishing high-risk and low-risk groups. We used Kaplan–Meier analysis to explore the prognostic value based on the patient survival time; the survival curves were used for visualization of the model. The specific risk scores value of each sample in the model were visualized with R software. We performed principal component analysis (PCA) on all samples based on irlncRNAs, 224 DEirLncRNAs, immune genes, and whole gene expression profiles in the risk model, respectively. The PCA results were compared to observe the differences in the distribution of risk scores.
Pathways in Two Risk Groups
To figure out the significantly changed immunological pathways, we did a gene set enrichment analysis (GSEA) [17] for two risk groups. We selected significantly enriched pathways according to p value and FDR q value; they were less than 0.05 and 0.25, respectively.
Correlation Analysis of Risk Model with Clinicopathological Features
The relationship between the model and clinicopathological features of CSCC patients was evaluated with a Chi-square test. These features included age, grade, clinical stage, and TNM stage. Tumor mutational burden (TMB) and microsatellite instability (MSI) were also be analyzed. The risk score difference between the groups corresponding to these clinicopathological characteristics was calculated with the Wilcoxon rank-sum test.
Immune Cell Infiltration Analysis
For the connection between risk and immune cell characteristics, we calculated the amount of immune cell infiltration in TCGA cervical cancer samples. The immune cell data were mainly obtained from XCELL, TIMER, QUANTISEQ, MCPCOUNTER, EPIC, CIBERSORT, and CIBERSORT-ABS. We used Spearman correlation analysis to evaluate the association between the risk scores and immune infiltrating cells. According to the immune cell infiltration data in CIBERSORT, we assessed the interaction among these immune cells and also the correlation between 11 irlncRNA and immune cells.
Exploring the Immunotherapeutic Treatment of the Risk Model
The Wilcoxon’s test was used to explore the differential expression of ICP (PD-L1, PD-1, CTLA-4, TIGIT, LAG-3, and TIM-3) between two risk groups. Then we used the tumor immune dysfunction and exclusion algorithm (TIDE) to predict the possibility of immunotherapy response, which is a computational framework expanded for immunotherapy forecast, and its predictive function has been successfully verified [18].
Non-negative Matrix Factorization
To assess if any clusters were present in the CSCC cohorts, molecular subtyping was used to perform non-negative matrix factorization (NMF). The best r value was chosen according to quality factors. These factors were the first r value at which the concord coefficient began to decline and the first value at which the residual sum of squares curve appeared to have an inflection point, as well as a direct visual inspection of the consensus matrix. The association of two subtypes and risk groups was showed in Sankey diagram. We also explored the expression of immune cells and immune checkpoints proteins in two subtypes. PCA was performed with the R package ggplot2.
Exploration of Drug Therapy Targeting Risk Model
To determine if the models we constructed could be used for drug therapy in CSCC patients, we calculated the half-maximal inhibitory concentration (IC50) of common chemotherapy drugs in the CSCC dataset. Anti-tumor drugs such as cisplatin, paclitaxel, docetaxel, gemcitabine, and vinorelbine were advocated for the treatment of CSCC patients. The IC50 difference between the two risk groups was compared by Wilcoxon’s test. Results were plotted with R packages “pRRophetic” and “ggplot2.”
Establishing a Predictive Nomogram
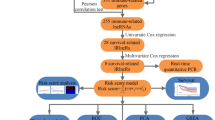
A nomogram combined various factors, and a calibration curve evaluated whether the predicted survival was consistent with certain survival in the nomogram. Calibration curves and AUC-ROC were also used to appraise the predictive ability of the prognostic model, and the clinical efficacy of the nomogram was evaluated with the decision curve analysis (DCA) (Fig. 1).
Result
Construction and Validation of the Risk Assessment Model
Data analysis of TCGA and GTEx databases produced 1,081 DElncRNAs, of which 224 were DEirlncRNAs (Fig. 2a), and 11 of those were related to prognosis in CSCC samples. These 11 DEirlncRNAs were subjected to univariate analysis (Fig. 2b) and LASSO regression analysis (Fig. 2c, d), and a risk signature including these 11 DEirlncRNAs was constructed. Eleven DEirlncRNAs were dominated by moderate or weak correlations, suggesting that they might play a role in similar biological processes (Fig. 2e). The risk scores for each patient were computed using the following:

Flow chart of this study

Screen immune-related lncRNA to construct a risk model. a Heat map showing the expression of 224 DEirLncRNAs in CSCC tissues (n = 306) and normal cervical tissues (n = 13). b Forest map of univariate Cox analysis, green indicates protection factor, and red indicates risk factor. c Distribution of the LASSO coefficients for 11 lncRNAs. d Partial likelihood deviation of the LASSO coefficient distribution. Vertical dashed lines indicate lambda. e Correlation chord diagram between 11 lncRNAs
The AUC-ROC of the risk model for OS was 0.801, 0.846, and 0.847 at 1, 3, and 5 years (Fig. 3a), respectively. Using the AIC value (Fig. 3b), we selected the maximum inflection point as the demarcation cut-off point of the 5-year ROC curve. Using this cut-off point, we obtained a high-risk group of 115 patients and a low-risk group of 189 patients. As shown by Kaplan–Meier analysis, patients in the high-risk group lived for a shorter period of time than those in the low-risk group (Fig. 3c). Figure 3d depicted the distribution of risk levels between the low-risk group and the high-risk group. Figure 3e showed the survival time of patients and the survival status in two risk groups, and Fig. 3f displayed the corresponding expression standards of the 11 irlncRNAs for each patient. This implied that the risk model had a high accuracy in predicting the prognosis of CSCC patients.

Validation of the risk model. a The 1-, 3-, and 5-year ROC of the risk model. b The maximum inflection point is the cut-off point obtained by the AIC. c Survival analysis of high- and low-risk patients in CSCC. Patients with CSCC were divided into a low-risk group (n = 189) and a high-risk group (n = 115) according to their AIC values. d Expression profile of 11 lncRNAs in the CSCC patients (n = 306). e Risk score distribution of CSCC patients (n = 306). f Survival status and survival time of CSCC patients (n = 306)
PCA Further Verifies the Grouping Ability of the Risk Model
Based on the expression profiles of 11 irlncRNAs in the risk model, 224 irLncRNA, immune genes, and whole genes, we used PCA to research the distribution patterns of the high-risk and low-risk groups in the CSCC patients (Fig. 4a–d). While based on the 11 irLncRNA, these two groups were significantly different, indicating that the risk model was best for CSCC patients.

Principal component analysis between the high- and low-risk groups. Principal component analysis based on a lncRNAs in risk model, b 224 DEirlncRNA, c immune genes, and d whole gene expression profiles
Immune-Related Pathways of the Two Risk Groups
The results of GSEA (Table 1) suggested that three of the TOP 5 pathways enriched in the low-risk group were related to immunity, including autoimmune thyroiditis, primary immunodeficiency, and intestinal immune network (Fig. 5). This indicated that patients in the low-risk group had a better immune status.

Gene set enrichment analysis. GSEA showed a significant enrichment of immune-related pathways in the low-risk group
Clinical Evaluation by the Risk Model
We investigated the relationship among risk signature, clinicopathological characteristics, and 11 DEirlncRNAs. T stage, N stage, and just were related to the risk group (Fig. 6a). Further, patients at T4 had higher risk scores than at T1 (Fig. 6e), whereas patients at stage IV had higher risk scores than at stage III (Fig. 6f). These results suggested that risk scores can be used to assess the degree of progression of CSCC, and that the higher the risk score, the more malignant the tumor.

Correlation analyses of risk scores with clinicopathological characteristics of CSCC patients. a Heatmap and clinicopathologic features of high- and low-risk groups (< 0.001 = ***, < 0.01 = ** *, and < 0.05 = *). Chi-square test acted as the statistical significance test. b Relationship between the risk scores and clinical parameters. Wilcoxon rank-sum or Kruskal–Wallis rank-sum test acted as the statistical significance test
Immune Landscape in CSCC Patients
The risk scores were negatively associated with macrophages M1, macrophages M2, myeloid dendritic cell, and CD8 + T cells, whereas it was positively associated with macrophages M0 (Fig. 7a). Meanwhile, the difference analysis also showed CD8 + T cells were significantly higher in the group of low-risk, and macrophages M0 were significantly higher in the group of high-risk (Fig. 7c). The proportion and infiltrating level of immune cells in 309 CSCC samples is shown in Fig. 7b, and CD4 + T cells made up the highest percentage (Fig. 7d). There was a weak or moderate correlation between 22 tumor immune cells (Fig. 7e). Two LncRNAs among the risk signature had better relationship with immune cells; they were LINC00426 and USP30.AS1(|correlation coefficients|> 0.4) (Fig. 7f).

Immune infiltrate landscape in CSCC patients and estimation of immunosuppressed molecules by the risk model. a Correlation between immune cells and risk scores, b relative proportion of immune cell infiltrates in CSCC patients, c content of 22 immune cells, d correlation matrix of immune cells, e comparison of the expression of immune infiltrating cells in low- and high-risk patients, f correlation of lncRNAs in risk model with immune cells, g differential expression of immune checkpoint proteins in the high- and low-risk patients, h TIDE prediction difference in the high- and low-risk patients
Six immune checkpoint proteins PD-L1, PD-1, CTLA-4, LAG-3, TIGIT, and TIM-3 were expressed highly in the low-risk group (Fig. 7g), suggesting that the immunosuppressive microenvironment led to a poor prognosis for low-risk patients. The prediction results of the TIDE algorithm showed that low-risk patients score higher than high-risk patients, which indicated that low-risk patients responded better to immunotherapy (Fig. 7h).
CSCC Sample Cluster into Two Subtypes
CSCC tumors were classified into two clusters containing 64 and 242 members. (Fig. 8a). The two subtypes identified by the NMF algorithm were confirmed by PCA (Fig. 8b). Cluster 1 mainly contained patients with low-risk scores, while the high-risk and low-risk patients in cluster2 were basically the same (Fig. 8c). This showed a good agreement in CSCC subtypes and risk groupings. PD-L1, PD-1, CTLA-4, LAG-3, TIGIT, and TIM-3 in cluster 1 had significantly higher expression than in cluster 2 (Fig. 8d). Expression of immune cells like CD8 + T cells, macrophages M1, T cells follicular helper, T cells regulatory (Tregs), and Mast cells activated expressed was higher in cluster 2, whereas that of macrophages M0 was higher in cluster 1 (Fig. 8e).

CSCC patients clustered into two subtypes based on risk mode. a Consensus clustering of CSCC samples by NMF. Correlation matrix heatmap subtypes and risk groups, d differential expression of immune checkpoint proteins in cluster1 and cluster2, e differences in the expression of immune cells between the two subtypes
The Significance of the Model in Clinical Treatment
Our study demonstrated that the low-risk group had a lower IC50 of cisplatin (p < 0.05) (Fig. 9), also implying that low-risk patients were more sensitive to cisplatin. This suggested that the low-risk patients might be better suited for chemotherapy.

Sensitivity of chemotherapy drugs in high- and low-risk groups. a cisplatin, b paclitaxel, c docetaxel, d gemcitabine, and e vinorelbine
Construction and Assessment of Predictive Nomograms
A nomogram was constructed to predict the 1-, 3-, and 5-year OS incidences (Fig. 10a). Calibration curves and ROC analyses showed good predictive performance of the nomogram in predicting patient survival (Fig. 10b, c). The decision curve showed that the nomogram has good clinical efficacy (Fig. 10d–f), suggesting that it can predict the survival of CSCC patients.

Construction and evaluation of a prognostic nomogram. a The nomogram predicts the probability of the 1-, 3-, and 5-year OS, b the AUC of risk scores and clinical factors, c the calibration plot of the nomogram predicts the probability of the 1-, 3-, and 5-year OS, d the decision curve of nomogram, and e clinical impact curve
Discussion
It has been suggested that lncRNAs play a key role in the adjustment of the immune system and the tumor microenvironment [10]. Immune-related lncRNAs are prognostic markers of various types of cancer [18] linked to immune cell infiltration and are a potential target for cancer treatment [19]. lncRNA SNHG14 has been shown to be involved in the activation of the JAK-STAT pathway in cervical tumor cells [20]. In cervical cancer HeLa cells, the STAT3 binding sequence in the enhanced region of lncRNA MALAT1 proved to be the key to the activation of the MALAT1 promoter induced by IL-6 or STAT3 [21]. In the eleven immune-related lncRNAs of the risk model, STXBP5-AS1 targets miR-96-5p/PTEN axis to drive cervical cancer cell proliferation and invasion [22], and DLEU1 promotes cervical cancer cell proliferation and invasion via the miR-381/HOXA13 axis [23].
We used the AIC value to get the best cut-off point for the model fitting. The prognostic model performed better in distinguishing between risk groups of high and low. Patients with low-risk showed a favorable prognosis, which suggested that our model may be able to stratify risk. GSEA revealed that immune-related pathways were significantly enriched in the low-risk group. We also analyzed the tumor immune infiltration and efficacy of chemotherapy drugs in the treatment of CSCC, which determined that this model worked well. In this study, consensus clustering based on NMF was used to define CSCC subtypes. The CSCC sample could be separated into two subtypes with distinct molecular profiles with differences in immune cell infiltration and checkpoint-related biomarkers. A nomogram was established to show the exact agreement between the observation rate and the predicted rate of 1-year, 3-year, and 5-year OS. The nomogram had better prediction accuracy and higher clinical efficacy, which passed calibration curve and DCA verification.
The ability of immunotherapy depends on the immunogenicity of the tumor microenvironment, so the understanding of the tumor microenvironment is the key to assessing the possibility of immunotherapy [24]. Immune cell infiltration can be a predictive biomarker of cancer immunotherapy [25]. Patients with more CD8 + T cell infiltration get a better response from pembrolizumab than those with less infiltration [26]. Macrophages suppressed T cell recruitment and regulated other aspects of tumor immunity; they can be regulators of tumor immunity and immunotherapy [27]. We used seven accepted methods to estimate the correlation between risk scores and immune cells; the methods include XCELL [28], QUANTISEQ [29], MCPcounter [30], EPIC [31], TIMER [32, 33], CIBERSORT [34], and CIBERSORT-ABS [35]. By integrating analysis, our results revealed that the risk model was negatively associated with macrophages M1, macrophages M2, and CD8 + T cells, meanwhile CD8 + T cells were significantly higher in the low-risk group. We can support that patients in the low-risk group may be more suitable for immunotherapy.
Cancer cells activate inhibitory immune checkpoint pathways to prevent the occurrence of autoimmunity [36]; therefore, suppression of the immune checkpoint approach is a promising treatment. Evidence suggests that immunotherapy is a novel therapeutic strategy for the treatment of cervical cancer [37]. Anti–CTLA-4, anti-PD-1, and anti-PD-L1 therapies are possible treatment options for cervical cancer, studies show [38,39,40]. Furthermore, in a well-defined clinical study of 115 cervical cancer patients, PD-L1 was expressed in 19% of cervical tumors, and more than 50% of tumor-infiltrating CD8 + T cells were expressed PD-1 [41]. We explored the relationship between several common immune checkpoint proteins in the treatment of cervical cancer and risk models, showing that all six immune checkpoints were higher in the group of low-risk. Thus, under this model, it suggested that patients with low-risk may benefit more from immunotherapy. The predictions of the TIDE algorithm indicated that patients with low-risk subtypes had a better response to immunotherapy, which was consistent with the above-mentioned studies. Our risk model also showed that low-risk patients were more sensitive to cisplatin, a small-molecule platinum compound that treat recurrent cervical cancer effectively [42], so we can formulate chemotherapy regimens for low-risk patients.
In our risk model, the risk scores are related to the clinical stage, and the risk scores of early cervical cancer are lower than that of the advanced stage. In summary, our findings support that early-stage patients may be better suited for immunotherapy and appropriate chemotherapy.
Conclusion
We identified 11 immune-related lncRNAs in cervical squamous cell carcinoma that were used to construct a prognostic signature. Then we compared the differences in immune checkpoints, clinicopathological characteristics, and drug treatments between the high-risk and low-risk groups. The results provide suggestions for the clinical treatment of cervical cancer, but the application of these results must be verified by a multicenter, large-sample clinical study.
Data Availability
The datasets supporting the conclusions of this article are available in the Genotype-Tissue Expression (GTEx) database and the cancer genome atlas (TCGA) database.
References
Sawaya GF, Smith-McCune K, Kuppermann M. Cervical cancer screening: more choices in 2019. JAMA. 2019;321(20):2018–9.
Wen X, Liu S, Cui M. Effect of BRCA1 on the concurrent chemoradiotherapy resistance of cervical squamous cell carcinoma based on transcriptome sequencing analysis. Biomed Res Int. 2020;2020:3598417.
Kumar L, Harish P, Malik PS, Khurana S. Chemotherapy and targeted therapy in the management of cervical cancer. Curr Probl Cancer. 2018;42(2):120–8.
Sol ES, Lee TS, Koh SB, Oh HK, Ye GW, Choi YS. Comparison of concurrent chemoradiotherapy with cisplatin plus 5-fluorouracil versus cisplatin plus paclitaxel in patients with locally advanced cervical carcinoma. J Gynecol Oncol. 2009;20(1):28–34.
Marquina G, Manzano A, Casado A. Targeted agents in cervical cancer: beyond bevacizumab. Curr Oncol Rep. 2018;20(5):40.
Long J, Lin J, Wang A, Wu L, Zheng Y, Yang X, Wan X, Xu H, Chen S, Zhao H. PD-1/PD-L blockade in gastrointestinal cancers: lessons learned and the road toward precision immunotherapy. J Hematol Oncol. 2017;10(1):146.
Chung HC, Ros W, Delord JP, Perets R, Italiano A, Shapira-Frommer R, Manzuk L, Piha-Paul SA, Xu L, Zeigenfuss S, Pruitt SK, Leary A. Efficacy and safety of pembrolizumab in previously treated advanced cervical cancer: results from the phase II KEYNOTE-158 Study. J Clin Oncol. 2019;37(17):1470–8.
Kung JT, Colognori D, Lee JT. Long noncoding RNAs: past, present, and future. Genetics. 2013;193(3):651–69.
Statello L, Guo CJ, Chen LL, Huarte M. Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol. 2021;22(2):96–118.
Denaro N, Merlano MC, Lo Nigro C. Long noncoding RNAs as regulators of cancer immunity. Mol Oncol. 2019;13(1):61–73.
Zhang L, Xu X, Su X. Noncoding RNAs in cancer immunity: functions, regulatory mechanisms, and clinical application. Mol Cancer. 2020;19(1):48.
Sang LJ, Ju HQ, Liu GP, Tian T, Ma GL, Lu YX, Liu ZX, Pan RL, Li RH, Piao HL, Marks JR, Yang LJ, Yan Q, Wang W, Shao J, Zhou Y, Zhou T, Lin A. LncRNA CamK-A regulates Ca(2+)-signaling-mediated tumor microenvironment remodeling. Mol Cell. 2018;72(1):71-83.e7.
Zhang J, Li S, Zhang X, Li C, Zhang J, Zhou W LncRNA HLA-F-AS1 promotes colorectal cancer metastasis by inducing PFN1 in colorectal cancer-derived extracellular vesicles and mediating macrophage polarization. Cancer Gene Ther 2021.
Jiang R, Tang J, Chen Y, Deng L, Ji J, Xie Y, Wang K, Jia W, Chu WM, Sun B. The long noncoding RNA lnc-EGFR stimulates T-regulatory cells differentiation thus promoting hepatocellular carcinoma immune evasion. Nat Commun. 2017;8:15129.
Zhang Y, Liu Q, Liao Q. Long noncoding RNA: a dazzling dancer in tumor immune microenvironment. J Exp Clin Cancer Res. 2020;39(1):231.
Sauerbrei W, Royston P, Binder H. Selection of important variables and determination of functional form for continuous predictors in multivariable model building. Stat Med. 2007;26(30):5512–28.
Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–73.
Wang W, Zhao Z, Yang F, Wang H, Wu F, Liang T, Yan X, Li J, Lan Q, Wang J, Zhao J. An immune-related lncRNA signature for patients with anaplastic gliomas. J Neurooncol. 2018;136(2):263–71.
Li Y, Jiang T, Zhou W, Li J, Li X, Wang Q, Jin X, Yin J, Chen L, Zhang Y, Xu J, Li X. Pan-cancer characterization of immune-related lncRNAs identifies potential oncogenic biomarkers. Nat Commun. 2020;11(1):1000.
Zhang YY, Li M, Xu YD, Shang J. LncRNA SNHG14 promotes the development of cervical cancer and predicts poor prognosis. Eur Rev Med Pharmacol Sci. 2019;23(9):3664–71.
Hao Y, Yan Z, Zhang A, Hu S, Wang N, Luo XG, Ma W, Zhang TC, He H. IL-6/STAT3 mediates the HPV18 E6/E7 stimulated upregulation of MALAT1 gene in cervical cancer HeLa cells. Virus Res. 2020;281:197907.
Shao S, Wang C, Wang S, Zhang H, Zhang Y. LncRNA STXBP5-AS1 suppressed cervical cancer progression via targeting miR-96–5p/PTEN axis. Biomed Pharmacother. 2019;117:109082.
Liu C, Tian X, Zhang J, Jiang L. Long non-coding RNA DLEU1 promotes proliferation and invasion by interacting with miR-381 and enhancing HOXA13 expression in cervical cancer. Front Genet. 2018;9:629.
Gasser S, Lim LHK, Cheung FSG. The role of the tumour microenvironment in immunotherapy. Endocr Relat Cancer. 2017;24(12):T283-t295.
Chen Q, Li T, Yue W. Drug response to PD-1/PD-L1 blockade: based on biomarkers. Onco Targets Ther. 2018;11:4673–83.
Garon EB, Hellmann MD, Rizvi NA, Carcereny E, Leighl NB, Ahn MJ, Eder JP, Balmanoukian AS, Aggarwal C, Horn L, Patnaik A, Gubens M, Ramalingam SS, Felip E, Goldman JW, Scalzo C, Jensen E, Kush DA, Hui R. Five-year overall survival for patients with advanced non-small-cell lung cancer treated with pembrolizumab: results from the phase I KEYNOTE-001 Study. J Clin Oncol. 2019;37(28):2518–27.
Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51.
Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18(1):220.
Plattner C, Finotello F, Rieder D. Deconvoluting tumor-infiltrating immune cells from RNA-seq data using quanTIseq. Methods Enzymol. 2020;636:261–85.
Dienstmann R, Villacampa G, Sveen A, Mason MJ, Niedzwiecki D, Nesbakken A, Moreno V, Warren RS, Lothe RA, Guinney J. Relative contribution of clinicopathological variables, genomic markers, transcriptomic subtyping and microenvironment features for outcome prediction in stage II/III colorectal cancer. Ann Oncol. 2019;30(10):1622–9.
Racle J, de Jonge K, Baumgaertner P, Speiser DE, Gfeller D Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife 2017, 6.
Li T, Fan J, Wang B, Traugh N, Chen Q, Liu JS, Li B, Liu XS. TIMER: a web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017;77(21):e108–10.
Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q, Li B, Liu XS. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res 2020, 48 (W1), W509-w514.
Zhang H, Li R, Cao Y, Gu Y, Lin C, Liu X, Lv K, He X, Fang H, Jin K, Fei Y, Chen Y, Wang J, Liu H, Li H, Zhang H, He H, Zhang W. Poor clinical outcomes and immunoevasive contexture in intratumoral IL-10-producing macrophages enriched gastric cancer patients. Ann Surg 2020.
Tamminga M, Hiltermann TJN, Schuuring E, Timens W, Fehrmann RS, Groen HJ. Immune microenvironment composition in non-small cell lung cancer and its association with survival. Clin Transl Immunology. 2020;9(6):e1142.
Finn OJ. Immuno-oncology: understanding the function and dysfunction of the immune system in cancer. Ann Oncol 2012, 23 Suppl 8 (Suppl 8), viii6–9.
Eskander RN, Tewari KS. Immunotherapy: an evolving paradigm in the treatment of advanced cervical cancer. Clin Ther. 2015;37(1):20–38.
Enwere EK, Kornaga EN, Dean M, Koulis TA, Phan T, Kalantarian M, Köbel M, Ghatage P, Magliocco AM, Lees-Miller SP, Doll CM. Expression of PD-L1 and presence of CD8-positive T cells in pre-treatment specimens of locally advanced cervical cancer. Mod Pathol. 2017;30(4):577–86.
Dyer BA, Zamarin D, Eskandar RN, Mayadev JM. Role of immunotherapy in the management of locally advanced and recurrent/metastatic cervical cancer. J Natl Compr Canc Netw. 2019;17(1):91–7.
Heeren AM, Punt S, Bleeker MC, Gaarenstroom KN, van der Velden J, Kenter GG, de Gruijl TD, Jordanova ES. Prognostic effect of different PD-L1 expression patterns in squamous cell carcinoma and adenocarcinoma of the cervix. Mod Pathol. 2016;29(7):753–63.
Karim R, Jordanova E, Piersma S. Tumor-expressed B7–H1 and B7-DC in relation to PD-1+ T-cell infiltration and survival of patients with cervical carcinoma. Clin Cancer Res. 2009;15:6341–7.
Lorusso D, Petrelli F, Coinu A, Raspagliesi F, Barni S. A systematic review comparing cisplatin and carboplatin plus paclitaxel-based chemotherapy for recurrent or metastatic cervical cancer. Gynecol Oncol. 2014;133(1):117–23.
Acknowledgements
We thank the researchers who gave their data for this analysis. And we also would like to acknowledge the GTEx and TCGA databases.
Funding
This study was funded by grants from the Scientific and Technological Project of Henan Province (172102310077 and funds for Creative Research Team of Henan Province.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Lv, X., liu, L., Li, P. et al. Constructing a Novel Signature Based on Immune-Related lncRNA to Improve Prognosis Prediction of Cervical Squamous Cell Carcinoma Patients. Reprod. Sci. 29, 800–815 (2022). https://doi.org/10.1007/s43032-022-00851-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43032-022-00851-z