
Abstract
Anti-Müllerian hormone (AMH) downregulates the level of stem cell factor (SCF) via the cAMP/PKA signaling pathway in human granulosa cells (GCs). Little information is available on the molecular mechanism underlying the interaction. This study is aimed at determining whether AMH regulates expression of SCF via the cAMP-PKA-CREB signaling pathway in human GCs. In the present study, we verified the binding of cAMP-response element-binding protein (CREB) to promoter of SCF in human GCs. Furthermore, the effect of CREB was tested on the SCF promoter, and the site of CREB binding to SCF promoter was identified using truncations as well as assays of SCF-promoted mutation and CREB mutation. To investigate the correlation among AMH, SCF promoter, and CREB, pGL-Basic-SCF+CREB was transfected into overexpressed AMH GCs (AMH-high GCs), low expressed AMH GCs (AMH-low GCs), and normal GCs (GCs), respectively. Finally, immunofluorescence, double immunostaining, and Western blot were carried out in AMH-high and AMH-low GCs to confirm the AMH-mediated regulation of SCF expression by inhibiting the phosphorylation of CREB (pCREB) in GCs. Results indicated CREB interacted with SCF promoter and significantly enhanced the transcription level of SCF. The CREB binding site was localized at 318–321 bp of SCF gene promote. AMH inhibits the expression of SCF by phosphorylation of CREB via the PKA signaling pathway in GCs. These findings provide an in-depth understanding of the molecular mechanism underlying AMH suppressing the follicle growth, which would aid in the development of a novel therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Anti-Müllerian hormone (AMH) is a member of the transforming growth factor-beta (TGF-β) superfamily [1], which acts on tissue differentiation and growth. AMH was originally identified due to its function in male sex differentiation. AMH is expressed in the Sertoli cells of the fetal testis, and it suppresses the Müllerian ducts [2]. AMH expression is initiated in primary follicles, which is maximal in small antral follicles and preantral structures, subsequently wanes up to ovulation [3, 4], and is thereby undetectable in human follicles > 8 mm in size and those that disappear completely in luteal bodies and atretic follicles [5]. To date, several studies have assessed the level of serum AMH as a potential clinical marker of response to gonadotrophins and ovarian reserve [6, 7]. Nilsson et al. [8] found that AMH regulates key transcription factors by Smad protein via signaling pathways in GCs, followed by gene transcription of other cytokines. A strong correlation was verified between AMH expression and the resting pool of follicles, indicating that AMH levels might predict the occurrence of menopause [9, 10].
Stem cell factor (SCF), a granulosa-derived growth factor, activates the phosphoinositide 3-kinase (PI3K) pathway in the oocyte via the oocyte surface c-Kit receptor, which might serve as an intra-oocyte network that regulates both the early development of ovarian follicles and oocyte growth [11]. Thus, AMH and SCF were proved as the key factors in follicle recruitment and development. The balance between AMH and SCF is essential for follicle development and maturation [12, 13].
Despite increasing clinical importance of AMH and SCF in fertility, little information is available on the molecular mechanism underlying the interaction. Our previous study demonstrated the negative correlation between AMH and SCF in human GCs, and SCF was shown to be downregulated by AMH of human GCs via the cAMP/PKA pathway [13]. Interestingly, AMH inhibited the phosphorylation of CREB (pCREB) through the cAMP pathway and resulted in reducing the FSH-stimulated transcription of estrogen synthase [14]. A recent study proposed that cAMP/PKA-responsive element (CRE) exists on the promoter of the human SCF gene [15]. Thus, we speculated that AMH inhibits the phosphorylation of CREB in the cAMP/PKA pathway, thereby downregulating the SCF transcription in human GCs. In the present study, we assessed the combination of CREB and SCF promoter using chromatin immunoprecipitation (ChIP) and electrophoretic mobility shift assay (EMSA) in GCs. Furthermore, we tested the effect of CREB on SCF promoter and identified the site of CREB binding to SCF promoter using truncations as well as assays of SCF-promoted mutation and CREB mutation. Strikingly, the luciferase activity was analyzed when CREB-overexpressing plasmid was transfected into the GC group, AMH-high GC group, and AMH-low GC group, respectively, to verify the effect of AMH on SCF and CREB. Finally, the SCF and pCREB expressions were observed in the GCs, AMH-high GCs, and AMH-low GCs.
Materials and Methods
Patients
A total of 35 female patients who underwent IVF therapy from March to September 2017 at the Reproductive Medicine Center of General Hospital of Ningxia Medicine University were enrolled in this study. Ovarian stimulation was performed after downregulation using a gonadotropin-releasing hormone agonist (GnRH-a, leuprolide acetate; Abbott), followed by gonadotropins [16]. These patients aged from 23 to 35 years experienced infertility due to tubal obstruction or male factors.
GC Collection and Cell Culture
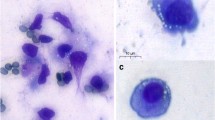
Follicular fluid (FF) samples were collected during oocyte retrieval as described previously [17]. The patients’ follicles were categorized into two groups according to follicular size at the time of retrieval: the small follicles group (diameter ≤ 12 mm) and the large follicles group (diameter > 12 mm). In each ovary, the large follicles were aspirated, following by that of the small follicles (individually aspirated). Subsequently, the line was washed using 2.0 mL phosphate-buffered saline (PBS). FF samples and the purified GCs of the small follicles were pooled for each patient, and the follicular aspirate contaminated with blood was discarded. After oocyte confirmation, FF from each patient’s follicles with diameter ≤ 12 mm was collected in sterile tubes and centrifuged at 2500 rpm for 10 min and the supernatant was aspirated. Then, the pellets were resuspended, transferred to 50% Percoll gradient (Pharmacia, American) and centrifuged at 2500 rpm for 22 min to purify human GCs. After washing two times and recentrifugation, sheets of hGC cells were digested by hyaluronidase (2:1) for 30 min to separate them. The GCs were removed using a pipette and washed with PBS. Subsequently, the cells were washed two times with PBS and centrifuged at 1000 rpm for 5 min twice. The cells were resuspended and cultured at 37 °C under 5% CO2 in 1640 medium (Gibco, 31800-014, USA) containing 10% fetal calf serum (FBS; HyClone, SH30084.03, USA), 100 U/mL penicillin, and 100 g/mL streptomycin (Invitrogen, USA). GCs were stained for presence of FSH-R (Santa Cruz Biotechnology) to estimate the purity of the cell cultures. In all experiments, over 85% of the GC staining were positive for the FSH-R (Fig. 1).

Human GC verification. Granular cell culture. Observe the expression of FSH receptor (FSHR) protein by immunofluorescence
Truncation and Mutation of SCF Promoter and Plasmid Construction
The SCF promoter was analyzed by UCSC (http://genome.ucsc.edu/) genome browser, intercepting the promoter regions of SCF genes of different lengths and mutations in the specific sequences based on the predicted sites (Fig. 2a). The different SCF promoters, 2000 bp, 1700 bp, 1500 bp, and 1300 bp, Mutant1, Mutant2, and Mutant3 of SCF promoter, and CREB-overexpressing plasmid were inserted upstream of the firefly luciferase gene in pGL3-Basic and pRL-TK (Promega, Madison, WI, USA) vectors following manufacturer’s protocols.

CREB interacts with the transcription factor SCF. The mutations of SCF promoter (a): the mutant 1 site was on 91–94 bp of SCF promoter, the mutant 2 site was on 270–273 bp of SCF promoter, and the mutant 3 site was on 318–321 bp of SCF promoter. Human GC lysates were subjected to ChIP using an anti-CREB antibody (b). PCR analysis of SCF DNA expression; group I: SCF DNA was pulled down from the cell by ChIP, while no band can be found in the IgG group III; (II) RT-Input1: the template is the entire genomic DNA; (IV) ddH2O as a template to test the specificity of the ChIP Kit; (V) anti-RNA polymerase II group; (VI) RT-Input2: the primer is for GAPDH. EMSA analysis (c). Bands were detected in the test group (II) and the mutation probe cold competition group (IV); (I) negative control group; (III) detection probe cold competition group. The relative luciferase activity analysis when different cutoffs of SCF promoter and CREB were transfected into GCs with plasmids (d). **P < 0.01 vs. III. (E) The relative luciferase activity analysis when different mutations of SCF promoter and CREB were transfected into GCs. *P < 0.05, **P < 0.01 vs. III, #P < 0.01 vs. IV. Data represent means ± SD (n = 3, each)
Overexpression and Gene Silencing of AMH in GCs
AMH was amplified by PCR and inserted into pcDNA3.1 vector (Invitrogen). GCs were cultured for 24 h up to 55–70% confluency, following which the cells were transduced using 6 μg/mL polybrene (Santa, USA) in DMEM. At a multiplicity of infection (MOI), stable clones were selected after 2 weeks using 400 g/mL geneticin (G418). The GCs were seeded overnight and transfected on the following day (at 50% confluency) with AMH using Lipofectamine 2000 (Invitrogen). AMH-specific and scrambled siRNA sequences were synthesized by Wanleibio (Shenyang, China) and transfected into cells using Lipofectamine RNAiMAX Reagent (Invitrogen) according to the manufacturer’s instructions. The transfected cells were cultured for 48 h before analysis.
Chromatin Immunoprecipitation-PCR
ChIP-PCR was performed as described previously [18]. GCs were seeded on coverslips and washed with PBS. ChIP was carried out using an anti-CREB antibody (Cell Signaling Technology, USA) and the Pierce Agarose ChIP Kit (Wanleibio, WLA106a, China) according to the protocol described previously. DNA was analyzed using a LightCycler 480 Probes Master Kit (Roche, Germany) according to the manufacturer’s instructions. The expression of SCF promoters and GAPDH was assessed by quantitative RT-PCR (qRT-PCR) using the primers listed in Table 1.
Electrophoretic Mobility Shift Assay
GC protein was extracted using the BAC kit (Wanleibio, WLA004, China), and the protein concentration was detected. Next, a variety of reagents were added (Table 2) and incubated at room temperature for 10 min. Then, the probe (Sangon, Shanghai, China) was added into mixture and placed in the dark for 20–30 min at room temperature. The product was resolved on 6% polyacrylamide gel and transferred to a membrane. Next, the membrane was cross-linked with ultraviolet for 10 min and blocked in 15-mL blocking solution for 30 min at room temperature. Then, the membrane was probed with streptavidin-HRP (1:5000), followed by streptavidin-HRP reaction solution for 20 min at room temperature. Finally, the immunoreactive bands were developed by ECL luminescent solution (WLA003, Wanleibio).
Luciferase Reporter Assay
The plasmids and the internal control Renilla luciferase plasmid were transiently transfected using Lipofectamine 2000 (Invitrogen). The luciferase assays were performed using a dual-luciferase reporter assay system (Promega). The luciferase activity was tested by a multifunctional microplate reader SpectraMax M5 (Sunnyvale, USA). Firefly luciferase activity was normalized to Renilla luciferase activity for each sample.
Western Blot Analyses
After 48-h post-transfection, an equivalent amount of protein from the AMH-high cell group, AMH-low cell group, and normal GC group was resolved by 12% SDS-PAGE and transferred to PVDF membranes. The protein concentrations of the supernatants were determined using a BCA protein assay (Wanleibio, China) using BSA as the standard. The membranes were blocked with non-fat milk for 1 h at room temperature and then probed overnight with the primary antibodies at 4 °C: anti-AMH (1:2000, Wuhan Mitaka, China), anti-pCREB (1:3000, Abcam, USA), anti-CREB (1:3500, Cell Signaling Technology, USA), anti-β-actin (1:1000, Abcam), or anti-GAPDH (1:1000, Abcam). Subsequently, suitable secondary antibodies were applied. The bands were visualized with the enhanced chemiluminescence detection reagent (Wanleibio).
Quantitative Real-Time PCR
To assess the mRNA concentrations, total RNA was extracted from cultured cells using TRIzol Reagent (Invitrogen). Quantitative RT-PCR (qRT-PCR) was performed to detect the expression of AMH and GAPDH using LightCycler 480 Probes Master Kit (Roche Diagnostics). All real-time PCR reactions were conducted in quadruplicate on an Exicycler 96 fluorescence quantifier (Bioneer, Korea). The primers are listed in Table 1.
Immunofluorescence
After 48-h post-transfection, the GCs were blocked in 3–4% paraformaldehyde for 15 min at room temperature, followed by PBS washes, permeabilization with 0.25% Triton X-100 for 10 min, and incubation with PBST for 35 min at room temperature. Then, the cells were incubated with SCF antibody (1:1500; Abcam, USA) overnight at 4 °C and subsequently with goat anti-rabbit IgG-TRITC (1:5000 dilution, Santa Cruz, USA) for 1 h at room temperature in the dark. Finally, the cells were incubated with 0.5 μg/mL DAPI for 1 min. The images were acquired using a Nikon microscope (Nikon TE2000, Japan). Flow cytometry was employed to detect the fluorescence intensity of SCF.
Immunofluorescence Double Immunostaining
GCs were fixed with 1.8% paraformaldehyde in 0.1 M phosphate buffer for 10 min and washed with 1% TBST. The GC cells were permeabilized using 1% Tween 20 in TBS for 30 min, followed by blocking with 3% hydrogen peroxide for 10 min. Next, the cells were immunostained using a monoclonal rabbit anti-pCREB antibody (1:100; Cell Signaling Technology) for 48 h at 4 °C, followed by incubation with FITC-labeled donkey anti-rabbit IgG antibody (Santa Cruz), peroxidase-labeled sheep anti-fluorescein antibody (Santa Cruz), and FITC-labeled tyramide (PerkinElmer, USA). In addition, monoclonal mouse anti-rat SCF antibody (1:10,000, Abcam) and Alexa Fluor 594–labeled anti-mouse IgG antibody (1:100, Santa Cruz) were applied for 1 h. The fluorescence images of the immunostained GCs were captured using a digital charge-coupled device camera (DP50, Olympus, Japan).
Statistical Analysis
Statistical analyses were performed using SPSS v.17.0 and GraphPad v.6.0 software. Data were presented as the mean ± SE (standard error) of the mean from at least three independent experiments for each group. ANOVA analysis and the post-least significant difference test were used to assess the differences between groups for all in vitro. P value < 0.05 was considered statistically significant.
Results
SCF Promoter Is a Direct Target of CREB
Bioinformatic analysis [19] demonstrated that the SCF transcript contains a CREB-binding site. In order to confirm the interaction between SCF and CREB, ChIP assays were carried out using an anti-CREB antibody. The resulting bands were detected in groups I, II, V, and VI. The SCF DNA was pulled down from the GCs by ChIP in the test group (group I), while no band was detected in the IgG group (group III) indicating that CREB interacted with SCF promoter (Fig. 2b). The result of ChIP-PCR was further verified using EMSA. Similar bands were detected in the test group and the mutation probe cold competition group (Fig. 2c). Taken together, CREB was found to interact with SCF promoter.
Deletion Studies Identified the Site of CREB Binding to SCF Promoter
To determine whether CREB modulated SCF expression, the luciferase activity was measured when CREB and different cutoffs of SCF promoter were transfected into GCs using pGL3-Basic and pRL-TK plasmids. Consequently, the luciferase activity was significantly elevated in group IV treated with CREB+2000-bp SCF promoter compared with group III (Fig. 2d, Table 3). In addition, the SCF expression was improved by increasing the CREB levels. Next, the binding sites of CREB on SCF promoter were predicted by bioinformatics [19]. The mutations of these binding sites on SCF promoters and CREB were constructed as pGL3-Basic and pRL-TK plasmids and transfected into GCs. The maximal luciferase activity was detected in group IV (P < 0.05), indicating that CREB could improve the SCF expression (Fig. 2e, Table 4). The minimal luciferase activity was detected in group VII as compared with groups V and VI (P < 0.01). Interestingly, no statistically significant differences were observed between groups III and VII. Thus, the binding site of CREB on SCF promoter was found be located at 318–321 bp.
AMH Downregulated the SCF Expression in Granulosa Cells
To demonstrate the functional link between AMH, SCF, and CREB, we transfected the GCs and found enhanced and decreased AMH levels in human GCs. As a result, AMH-high GCs, AMH-low GCS, and AMH normal expression GCs were established successfully. AMH protein (Fig. 3a, b) and mRNA expressions (Fig. 3c) were tested using Western blot and RT-PCR, respectively. Furthermore, the expression of SCF in AMH-transfected cells was observed by immunofluorescence. The expression of SCF was significantly decreased in the AMH-high GC group (P < 0.01 vs. control and AMH-low cells). In addition, statistically significant differences were not observed between human GCs treated with AMH-low and the normal GC group (Fig. 3d, e).

The ability of CREB to increase SCF expression could be blocked by increasing AMH levels. AMH protein expression were analyzed with Western blot (a, b). Expression of AMH mRNA was assessed by RT-PCR in the GC group, AMH-high GC group, and AMH-low GC group; error bars represent mean ± SEM (c). SCF expression in human GCs using TRITC fluorescence-labeled antibody immunofluorescence system (d, e). a–i Immunofluorescence analysis on GCs with anti-SCF (blue) antibodies identified the effect of the GCs (a–c), AMH-high group (d–f), and AMH-low group (g–i), respectively. Data were the average of three independent experiments. The luciferase activity analysis (f) when pGL-Basic-SCF+CREB overexpression and pRL-TK were transfected into the GCs, AMH-lower, and AMH-high groups, respectively (*P < 0.05)
AMH Mediates the Binding of CREB to SCF Promoter
AMH downregulated the SCF via cAMP/PKA signaling in human GCs [13]. CREB interacts with SCF promoter to increase the SCF expression. In order to further affirm the correlation between AMH, SCF, and CREB, the luciferase activity was measured when CREB-overexpressing plasmid was transfected into normal GCs, AMH-high GCs, and AMH-low GCs. However, the SCF expression was minimal in the AMH-high GC group (P < 0.05) despite CREB was overexpressed (Fig. 3f). Thus, the ability of CREB to increase SCF expression could be blocked by increasing AMH levels. Nevertheless, AMH-mediated downregulation of SCF was not effectuated via the expression of CREB.
AMH Inhibits the Phosphorylation of CREB
Phosphorylated CREB traversed into the nucleus, which reduced the FSH-stimulated transcription of estrogen synthase through autocrine and paracrine mechanisms [14]. Moreover, the expressions of pCREB and total CREB were confirmed using Western blot in GCs, AMH-high GCs, and AMH-low GCs. The pCREB levels were significantly diminished in GCs treated with overexpressed AMH (P < 0.05). However, the CREB expression did not differ in GCs, AMH-high GCs, and AMH-low GCs (Fig. 4a, b). In addition, whether AMH inhibits the phosphorylation of CREB by reducing the transcription of SCF was further explored. Also, double immunocytochemical staining confirmed the pCREB and SCF expression in GCs and AMH-high GCs. Subsequently, immunostaining showed that the percentage of pCREB and SCF was significantly decreased in the AMH-high GC group as compared with the control GCs (P < 0.05) (Fig. 4c).

AMH decreased SCF transcription by inhibiting the pCREB expression. a, b Western blotting indicated that pCREB expression was significant decreased in AMH-high GCs (*P < 0.05). There was no difference in total CREB levels. c Photomicrographs of pCREB-immunoreactive, SCF expression in the AMH-high GC group compared with the normal GC group. The GCs were fixed with paraformaldehyde and subjected to double immunostaining for pCREB (green) and SCF (red), which were labeled with FITC and Alexa Fluor 594, respectively
Discussion
Our previous study demonstrated that AMH downregulates SCF via the cAMP/PKA pathway [13]. However, the molecular mechanism underlying the AMH-mediated regulation of SCF in GCs is yet to be elucidated. This study is aimed at determining the function and molecular mechanism of AMH on SCF in female GCs. In the current study, the most significant finding was that the increasing concentrations of AMH decreased the expression of SCF by inhibiting the phosphorylation of CREB in GCs. Similar studies showed that AMH is involved in both autocrine and paracrine signaling pathways through AMHRII. This AHMRII receptor is also used to affect the SCF expression via the cAMP/PKA signaling pathway [20]. Both oocytes and somatic cells (granulosa, theca cells, and cumulus cells) secrete key cytokines into FF, which provide an essential microenvironment for the growth and development of oocytes [21]. The balance of cytokines is crucial for the follicular development in the microenvironment. AMH signaling is mediated through the downstream signaling molecules: Smad1, Smad5, and Smad8 [22]. Moreover, AMH prevents the early depletion of the follicle pool in the ovary by inhibiting the initial recruitment [23].
In the present study, The CREB-overexpressing plasmid was transfected into the normal GCs, AMH-high GC group, and AMH-low GC group, and results showed that the SCF expression was minimal in the AMH-high GC group. In addition, the CREB-promoted SCF transcription disappeared when AMH was overexpressed in human GCs, although the expression of CREB was sufficient. Some papers revealed that AMH inhibited the phosphorylation of CREB via the cAMP pathway, resulting in reduced FSH-stimulated transcription of estrogen synthase [14]. Western blot and double immunostaining results indicated that the expression of pCREB in AMH-high GCs was lower than that in normal GCs. In this study, we found that CREB enhances the SCF transcription using luciferase reporter assay. When 2000-bp SCF promoter+CREB was transfected into GCs, the SCF transcription was increased distinctly.
SCF belongs to the colony-stimulating factor (CSF) family and is a critical cytokine in the ovary that can promote the maturation and growth of oocytes. cAMP-mediated transcription regulates several physiological processes in eukaryotes, including circadian rhythm, gametogenesis, and neuroendocrine functions. The stimulation of this pathway is mediated via phosphorylation by protein kinase A (PKA) of a single serine in the structurally similar transcription factors CREB [24]. To identify the site of CREB binding to SCF promoter, ChIP, EMSA, truncations, SCF-promoted mutation, and CREB mutation assays were employed. The current study indicated that CREB was localized on SCF promoter and the binding site was on 318–321 bp of SCF promoter in female GCs. Moreover, we demonstrated that AMH decreased the level of pCREB in the cAMP pathway, which in return, prevented the entry of CREB into the nucleus and affected the transcription of SCF (Fig. 5). Thus, the regulatory signaling weakened the ovular growth-promoted action of SCF and inhibited the development of follicles. The current findings on the regulation between the AMH and SCF may provide a novel treatment direction for clinical patients with follicular growth retardation due to high AMH level. However, the isolated GCs are different from cells proliferating within a follicle. Thus, the molecular mechanisms underlying the regulation between AMH and SCF in vivo need to be further clarified.

Potential mechanisms of AMH regulate SCF via the cAMP/PKA signaling pathway in human granular cells
Data Availability
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Cate RL, Mattaliano RJ, Hession C. Isolation of the bovine and human genes for Mullerian inhibiting substance and expression of the human gene in animal cells. Cell. 1986;45(5):685–9834.
Blanchard MG, Josso N. Source of the anti-Müllerian hormone synthesized by the fetal testis: Müllerian-inhibiting activity of fetal bovine Sertoli cells in tissue culture. Pediatr Res. 1974;8(12):968–71.
Modi D, Bhartiya D, Puri C. Developmental expression and cellular distribution of Mullerian inhibiting substance in the primate ovary. Reproduction. 2006;132(3):443–53.
Be’zard J, Vigier B, Tran D. Immunocytochemical study of anti-Mu¨llerian hormone in sheep ovarian follicles during fetal and postnatal development. J Reprod Fertil. 1987;80:509–16.
Weenen C, Laven JS, Von Bergh AR. Antimullerian hormone expression pattern in the human ovary: potential implications for initial and cyclic follicle recruitment. Mol Hum Reprod. 2004;10(2):77–83.
Fiçicioglu C, Kutlu T, Baglam E, Bakacak Z. Early follicular antimullerianhormone as an indicator of ovarian reserve. Fertil Steril. 2006;85:592–6.
La Marca A, Giulini S, Tirelli A, Bertucci E, Marsella T, Xella S, et al. Antimullerian hormone measurement on any day of the menstrual cycle strongly predicts ovarian response in assisted reproductive technology. Hum Reprod. 2007;22:766–71.
Nilsson E, Rogers N, Skinner MK. Actions of antimullerian hormone on the ovarian transcriptome to inhibit primordial to primary follicle transition. Reproduction. 2007;134:209–21.
Sowers MR, Eyvazzadeh AD, McConnell D. Antimullerian hormone and inhibin B in the definition of ovarian aging and the menopause transition. J Clin Endocrinol Metab. 2008;93:3478–83.
Lie Fong S, Baart EB, Martini E. Anti-Mu ¨llerian hormone: a marker for oocyte quantity, oocyte quality and embryo quality? Reprod BioMed Online. 2008;16:664–70.
Reddy P, Shen L, Ren C, et al. Activation of Akt (PKB) and suppression of FKHRL1 in mouse and rat oocytes by stem cell factor during follicular activation and development. Dev Biol. 2005;281:160–70.
McLaughlin EA, McIver SC. Awakening theoocyte: controlling primordial follicle development. Reproduction. 2009;137:1–11.
Hu R, Wang FM, Yu L, et al. Antimullerian hormone regulates stem cell factor expression in human granulosa cells. Fertil Steril. 2014;102:1742–50 e1741.
Kanakkaparambil R, Singh R, Li D, Webb R, Sinclair KD. B-vitamin and homocysteine status determines ovarian response to gonadotropin treatment in sheep. Biol Reprod. 2009;80:743–52.
Gruijters MJG, Visser JA, Durlinger ALL, Themmen AP. Antimullerian hormone and its role in ovarian function. Mol Cell Endocrinol. 2003;211:85–90.
Liu B, Zhang L, Guo RW, Wang WJ, Duan XQ, Liu YW. The serum level of C-reactive protein in patients undergoing GnRH agonist protocols for in vitro fertilization cycle. Clin Exp Obstet Gynecol. 2014;41:190–4.
Yalcinkaya E, Cakiroglu Y, Do ger E, et al. Effect of follicular fluid NO, MDA and GSH levels on in vitro fertilization outcomes. J Turk Ger Gynecol Assoc. 2013;14:136–41.
Hu YW, Zhao JY, Li SF. RP5833A20.1/miR-382-5p/NFIA-dependent signal transduction pathway contributes to the regulation of cholesterol homeostasis and inflammatory reaction. Arterioscler Thromb Vasc Biol. 2015;35:87–101.
Rapicavoli NA, Qu K, Zhang J, et al. A mammalian pseudogene lncRNA at the interface of inflammation and anti-inflammatory therapeutics. Elife. 2013;2:e00762.
Hu R, Lou Y, Wang F-M. Effects of recombinant human AMH on SCF expression in human granulosa cells. Cell Biochem Biophys. 2013;67(3):1481–5.
Macklon NS, Fauser BC. Ovarian reserve. Semin Reprod Med. 2005;23:248–56.
Visser JA. AMH signaling: from receptor to target gene. Mol Cell Endocrinol. 2003;211:65–7.
Hirobe S, He WW, Gustafson ML. Müllerian inhibiting substance gene expression in the cycling rat ovary correlates with recruited or graafian follicle selection. Biol Reprod. 1994;50(6):1238–43.
Lassot I, Ségéral E, Berlioz-Torrent C. ATF4 degradation relies on a phosphorylation-dependent interaction with the SCF ubiquitin ligase. Mol Cell Biol. 2001;21(6):2192–202.
Acknowledgments
We thank the Laboratory of Fertility Maintenance of Ningxia Medical University and the Clinical Pathogen Microbiology Laboratory of the General Hospital of Ningxia Medical University for providing an experimental platform. And we thank professor Hai Wang for modifying the article.
Funding
This work was supported by the Natural Science Foundation of China (No. 81660257), Ningxia Natural Science Foundation (NZ16125, NZ16143), Personnel Agency Overseas talent Project, 2017 Autonomous Region Leader in Science and Technology Innovation, First-Class Discipline Construction Founded Project of Ningxia Medical University and the School of Clinical Medicine (NXYLXK2017A05), and 2018 Advantageous Subjects Project of Ningxia Medical University (XY201807). Key Research and Development program of Ningxia Hui Autonomous Region (2019BFG02005).
Author information
Authors and Affiliations
Contributions
YXF and FMW conceived and designed the study with inputs from RH. YXF and FMW have the same contribution to this article. FMW and RH were responsible for the supervision and coordination of the project. YXF and XE performed most of the in vitro experiments. HMY collected the FF. TH and HW led the data analysis with inputs from YXF, Yanfei-Wang, XE, Yafei-Wang, and HMY. The first draft of the manuscript was written by YXF and FMW; then, RH, XE, Yafei-Wang, and Yanfei-Wang contributed to revise and review the manuscript. All authors read and approved the final manuscript before submission.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
The use of human follicular fluid was reviewed and approved by the Ethics Committee of Ningxia Medical University and was performed in accordance with the approved guidelines. Written informed consent was obtained from each participating patient.
Competing Interests
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Fu, YX., Wang, FM., Ou-yang, XE. et al. Anti-Müllerian Hormone Regulates Stem Cell Factor via cAMP/PKA Signaling Pathway in Human Granulosa Cells by Inhibiting the Phosphorylation of CREB. Reprod. Sci. 27, 325–333 (2020). https://doi.org/10.1007/s43032-019-00033-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43032-019-00033-4