
Abstract
In this study, hybrid aerogels containing carbon nanoparticles (CNP) and multi-walled carbon nanotubes (MWCNT) were synthesized via sol–gel method using resorcinol/formaldehyde precursors through a hydrolysis-condensation reaction mechanism. Porous carbonaceous structures were achieved by freeze-drying of the organic gels followed by controlled carbonization under an inert gas. The samples were characterized by various techniques such as FTIR, BJH, FESEM, CV, and EIS. The specific surface area and total pore volume of the aerogel sample were measured to be as high as 452 m2/g and 0.782 cm3/g, respectively, thus enhancing the electric double-layer formation. Electrochemical tests on the samples showed a large specific capacitance (212 F/g) and an excellent cyclic stability over 3000 cycles. Performance of the synthesized structures was subsequently assessed as electrodes in a capacitive deionization (CDI) process. At the operating conditions of 1.6 V voltage, flow rate of 20 mL/min, and NaCl concentration of 1000 mg/L a promising adsorption capacity around 42.08 mg/g was achieved.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Due to the scarcity of freshwater supply, desalination of seawater and brackish water is of crucial importance and development of sustainable desalination technologies is urging. Acceptable limit of salt concentration in water, set by World Health Organization (WHO) is 500 ppm [1]. Nowadays, water is desalinated and commercially supplied by various technologies like electrodialysis [2], reverse osmosis [3], multi-stage flash [4], and multi effect distillation [5]. These technologies are energy intensive due to evaporation process or applying osmotic pressure for salt separation. Capacitive deionization is a promising technology for desalination of brackish water due to its environmental friendliness, ion removal efficiency, and energy effectiveness since instead of extraction of huge amount of water, ions are removed [6]. Besides, CDI technology offers wide applicability for water softening [7], heavy metal [8], phosphate and nitrate removal from water [9, 10] through a combination of chemisorption, physisorption, electrodeposition, electrophoresis, and double-layer charging processes [11].
Capacitive deionization (CDI) is an electrochemical method in which ions in a solution are attracted by electrodes charged with opposite polarity where they are adsorbed in electric double layer created at the solid–liquid interface. Prolonged use of electrodes results in their saturation. The electrodes can easily be regenerated by reversing the polarity or establishing a short circuit between the electrodes to release the adsorbed ions into the solution. Thus, CDI method can save energy like a supercapacitor and simultaneously desalinate water [12]. The operation is of cyclic mode, including adsorption/desalination cycle and desorption/washing cycle sequentially [13]. Advantages of the method include low voltage (to prevent electrolysis of water), operation at ambient pressure and temperature, lack of membrane, easy regeneration, no consumption or production of toxic chemical by the system [1].
One of the challenges of CDI implementation is selection of the electrode material possessing high specific surface area, high porosity, high electrical conductivity, fast adsorption–desorption responses and chemical and electrochemical stability within a wide range of pH. Although various kinds of carbon materials like activated carbon [14], carbon cloth [15], carbon nanotubes [16], carbon aerogel [17], activated carbon fiber [18], graphene [19], various carbon allotrope hybrids [20] have previously been studied as electrode materials in CDI systems, however, the performance requires further improvement. To name a few of the recent studies, a carbon aerogel-polypyrrole (CA-Ppy) composite (with an adsorption capacity of 10.1 mg/g) from resorcinol and formaldehyde precursors was prepared by Beke et al. through sol–gel method for the desalination of NaCl solution using the hybrid CDI method. [21]. In a study by Miao et al. the CA and Ppy/CA-x anodes were coupled with CA cathodes for capacitive deionization [22]. The CA was derived from carboxymethyl cellulose (CMC). The polypyrrole (Ppy)/CA electrodes were prepared by grafting Ppy on carbon aerogel (CA) via in-situ polymerization. The desirable desalination performance was attributed to the positively charged nitrogen atoms of Ppy which could capture Cl− well through the surface redox reaction in CDI. Liu et al. reported construction of CA from leather wastes, rich in N, O and S, by a one-step activated pyrolysis process [17]. Distortional carbon nanocrystallites (DCNs) as well as the self hetero-doping provided uneven surface charge distributions, resulting in electrons and ions conductivities as well as water wettability improvement of the electrode. The enhanced adsorption capacity was also attributed to the structural defects and doping which could act as active sites [17]. Liu et al. reported the preparation of a nitrogen-doped hierarchical porous carbon aerogel (N-HPCA) for high-performance CDI [23].The CA was derived from commercial chitosan (CS). They concluded that the N-doping largely enhanced the conductivity and surface wettability of the carbon material resulting in an enhanced CDI performance. Yeh et al. prepared the coconut shell-based activated carbon electrodes by a two-stage activation method using the activation process of KOH etching plus CO2 gasification method [24]. They suggested that the controlled mesoporosity of the electrode led to higher specific capacitance and better rate capability as compared to the commercial one. They concluded that the enhanced mesoporosity could facilitate the ion transport and also could boost the accessible surface area resulting in the improved capacitive ion storage.
Since in CDI electrosorption process mesosized pores enhance the ion adsorption [6], the objective of this work was to explore the performance of carbon nanotube/carbon nanoparticle hybrid aerogel as the electrode active material due to possession of high mesoprosity. Besides, as in carbon aerogel, carbon nanoparticles are linked together by covalent bridges and the contact points between the particles and the pores introduce an internal resistance, to increase the electrical conductivity of the electrode, in this study carbon nanotubes (due to 1D scale and high aspect ratio) were embedded in the CA as charge nanocollectors or nanopathways to reduce the internal resistance [25].
2 Materials and method
2.1 Materials
All the chemicals including resorcinol (R), formaldehyde (F), and sodium carbonate © were supplied by Merck. The COOH-functionalized multi-walled carbon nanotubes (purity > 95%) were purchased from Nanosany Corporation. The MWCNT was of 10–20 nm outer diameter and 10–30 μm length with SSA of 200 m2/g. The MWCNT electrical conductivity was greater than 100 S/cm as claimed by the manufacturer.
2.2 Preparation of aerogel
A typical aerogel was synthesized by dispersing 1.3 wt% of the functionalized MWCNT in deionized water using an ultrasonic bath for 3 h. Afterwards, 4–13 wt% of resorcinol and formaldehyde with a molar ratio of R/F = 2 and sodium carbonate as a catalyst with a molar ration of R/C = 200 were added to the solution. The mixture was then resettled in a mold and aged in an oven at 85 °C for 3 days. The resulting gel was then soaked in an organic solvent to exchange the solvent with water before being freeze-dried to yield the RF aerogel. The carbonaceous aerogel was obtained by pyrolyzing the RF aerogel at 1050 °C under N2 atmosphere for 3 h [26,27,28].
2.3 Fabrication of the CDI electrodes
To fabricate the working electrode, the aerogel as the active material was first ground adequately then it was mixed with carbon black as conductive additive and PTFE as binder with 75%, 20% and 5% proportion. Again, the mixture was ground for more adhesion and was moisturized by adding few drops of ethanol. The mixture was then pressed as a 5 cm diameter circle on a nickel foam plate with diameter of 82 mm (Fig. 1) followed by drying at 80 °C for 12 h. The active area of each electrode was about 20 cm2. The total mass loading on each electrode was 1 g whereas the capacity was reported based on the active aerogel. The electrodes were finally applied in a CDI cell made of Ertalon where they were separated by a hollow center silicon gasket with 2 mm thickness.

Schematic of the set-up (a) and the fabricated electrode (b)
2.4 CDI set-up and experiments
The experiments were conducted in batch mode by circulating the solution through the system. In each test, the solution was pumped through a peristaltic pump into the electrosorption unit cell, and returned into the reservoir under ambient conditions. The reservoir was 250 mL. The salt concentration was continuously measured and monitored by a conductivity meter (PrismaTech company) using a predetermined calibration curve. The voltage was applied by RN-3005D power supply. The saturated electrodes were regenerated by shorting the electrodes and washing them with fresh water (Fig. 1).
2.5 Characterization
The morphology and microstructures of the samples were studied by a MIRA3 TESCAN-XMU field emission scanning electron microscope (FESEM). For identification of the surface functional groups, FTIR spectra were recorded using a Bruker Vector 33 spectrometer with KBr pellets. To determine the BET specific surface areas of the samples, adsorption–desorption isotherms of N2 at 77 K were measured by an adsorption instrument (Belsorp mini-II). Pore size distribution was evaluated using the BJH method. To study the crystallinity of the structures, X-ray diffraction (XRD) analyses were carried out on a Philips PW1825 diffractometer using Co Kα radiation (wavelength of 1.79 Å) under 49 kV and 30 mA with a step size of 0.002° and 0.5 s per step.
Electrochemical behavior of the sample was studied in a 1.0 M NaCl electrolyte at room temperature using either a three-electrode system or a two-electrode system. The working electrode was prepared by mixing the active materials (80%) with acetylene black (15 wt%) and polytetrafluoroethylene (PTFE) (5 wt%). The mixture was then coated on a stainless steel substrate (1 cm2) as current collector and dried at 80 °C for 4 h before being pressed under 15 MPa. The reference electrode was Ag/AgCl and the counter electrode was platinum. Electrochemical impedance (EIS) and cyclic voltammetry (CV) were then measured. Supercapacitive characteristics of the sample was evaluated by an electrochemical workstation (PGSTAT 204 Autolab, Netherland). The specific capacitance derived from the discharge curve was calculated by [24]:
where I (A) is the current, t (s) is the discharge time, m (g) is mass of the loaded material on the electrode, ΔV (V) is the potential change during the discharge process.
3 Results and discussion
3.1 Characterization of the electrodes
Structural texture of the synthesized aerogel can be seen in Fig. 2. It is shown that carbon nanotubes are covered by carbon nanoparticles which are formed during the carbonization process most likely by seeding on their walls since the CNTs diameters (> 50 nm) are larger than the widths reported by the manufacturer (10–20 nm). In addition to large individual carbon particles, strings of interconnected smaller particles are depicted acting as conductive binder in the network of CNTs. It seems that spreading the carbon nanotubes throughout the structure strengthens the framework. The porous structure is formed as a result of solvent removal during the drying process and material decomposition through pyrolysis process.

FESEM images of the aerogel
X-ray diffraction (XRD) pattern was recorded to analyze the crystallinity and structural order of the sample. Figure 3 shows that the dominant phase is amorphous and the structure is disordered. The major peaks emerged at 2θ = 30° and 51° correspond to 002 and 100 planes of graphitic carbon, respectively [29, 30].

a XRD pattern and b FTIR spectrum of the aerogel
FTIR analysis was carried out to identify the functional groups on the aerogel surface (Fig. 3). The absorption bands emerged near 1142 cm−1 and 1562 cm−1 correspond to the C–O–C vibrations and C=C stretching vibrations of the aromatic rings. The broad band at 3415 cm−1 is assigned to the stretching vibrations of O–H bond indicating the existence of water on the surface [31]. Presence of surface oxygen containing groups enhance hydrophilicity of the aerogel.
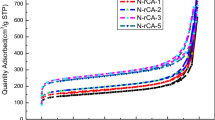
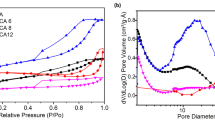
Specific surface area of the sample was measured to be 452 m2/g by standard BET (Brunauer–Emmett–Teller) analysis conducted at boiling temperature of nitrogen (77 K). The plot of adsorption/desorption of N2 at various relative pressures are depicted in Fig. 4. Nitrogen is adsorbed into the micropores at low relative pressures (beginning of the curves) and then starts filling the mesopores at higher relative pressures. According to IUPAC classification, the isotherms are of IV type whereas the hysteresis loop with closure around P/P0 ~ 0.4 is of H3 type. Presence of the hysteresis loop is due to the capillary condensation in mesoporous regions [32]. The pore size distribution determined by BJH (Barrett-Joyner-Halenda) model showed existence of micro and meso pores where t-plot measurement confirmed the dominance of meso pores (2–50 nm) in the structure (Table 1). Meso pores provide less resistance for ion transport through porous electrodes. Pores with small sizes increase the steric hindrance and restrict the mobility of ions. Hierarchical structures with meso pores enteries of micro pores facilitates fast adsorption rate as well as high capacitance [33]. According to Fig. 4 (inset), the pore sizes were not uniform and the mean pore size was assessed to be around 6.9 nm. The high specific surface area and pore volume are characteristics of aerogel structure.

Nitrogen adsorption/desorption at 77 K (inset BJH) plot of the aerogel
Cyclic voltammetry was conducted to study the electrochemical behavior of the aerogel. Figure 5a shows the CV curves at various scan rates (5–100 mV/s) representing quasi-rectangular shapes indicating desirable capacitive behavior and stability [34, 35]. As the scan rate increases the current response increases accordingly and the CV curves maintain their shapes implying that the ion transport is rapid and the electrode has a satisfactory rate performance and small internal resistance [36, 37]. Lack of recognizable redox peaks indicates that no major Faradic reaction happens and NaCl electrosorption occurs through electrical double-layer mechanism [38]. Galvanostatic charge–discharge (GCD) measurement of the sample at different current densities (1–16 A/g) is demonstrated in Fig. 5b. GCD curves show almost isosceles triangles indicating the symmetry between charge and discharge and representing an excellent reversibility and capacitive properties of the electrode material and almost the linear voltage–time dependence. The ohmic resistance of ionic transport and diffusion into the pores is reflected in the voltage drop (iR) at the turning point of charge/discharge profile which can be reduced by mesoporosity [24]. Figure 5c depicts the specific capacitance vs. scan rate (mV/s). As the scan rate increases the specific capacitance decreases which could be due to insufficient time for the electrolyte ions to diffuse into the electrode pores. At high scan rates the slope decreases indicating low resistance for ion diffusion [17]. Figure 5d shows the specific capacitance vs. current density (A/g). It can be seen that the specific capacitance at 1 A/g is as high as 293 F/g (measured by 3 electrode cell and 212 F/g measured by two-electrode cell) which can be attributed to the high surface area and desirable pore size distribution [39]. As the current density increases the specific capacitance decreases which could be due to Ohmic drop and sluggish kinetics of electrochemical activities. To examine the stability of the electrode, capacitance retention of the electrode was measured in 3000 cycles. It can be seen from Fig. 5e that the electrode retained more than 95% of its initial capacitance during the repeated charge–discharge process. To investigate the conductivity of electrode material and internal resistance of the electrode, electrolyte and ion diffusion into the electrode, electrochemical impedance spectroscopy was utilized. The Nyquist plot seen in Fig. 5f is fitted to and interpreted by an equivalent electric circuit (Fig. 5g). It shows a semicircle in the high frequency region and an oblique line corresponding to Warburg impedance at low frequency region related to the diffusion in electrolyte. The large slope suggests fast ion diffusion rate [22, 40, 41]. Charge transfer resistance (Rct) can be evaluated from the diameter of the semicircle which is in parallel with double-layer capacitance (Qdl) at the interface between the electrode and electrolyte solution. The starting point of the semicircle (Rs) represents the bulk resistance including the contact resistance.

a Cyclic voltammetry, b galvanostatic charge–discharge (GCD) measurements, c specific capacitance versus scan rate, d specific capacitance versus current, e stability test at 16 A/g, f Nyquist plot, g the equivalent circuit
3.2 CDI performance
Among several electrochemical reactions and processes involved in CDI system, like capacitive ion storage, ion kinetics, and non-Faradic chemical surface charge, and carbon redox reactions, water chemistry, the main reaction is the ion storage which is strongly influenced by surface area and pore size distribution [42]. Performance of CDI system is mainly assessed by its electrosorption capacity. For a given feed water composition and electrode geometry and material, the capacity is affected by key operating parameters like applied voltage, initial ion concentration, and flow rate of solution passing over the electrodes.
For ion electrosorption in CDI system, the applied voltage plays as driving force. Theoretically, higher applied voltage is associated with more free electric charge on the electrode surface and stronger electrostatic forces resulting in enhanced ion removal. However, since the standard potential of water electrolysis is 1.23 V, the increase in voltage should be limited. Hence, the electrode potential is a crucial factor to avoid redox reactions and minimize the power consumption. In practice, a slightly larger voltage than 1.23 V may be applied due to existence of resistance in the circuit. Deionization curves versus time at various voltages are depicted in Fig. 6a. The decrease in the solution electrical conductivity is an indication of ion adsorption. In this work, the voltage varied between 1.2 and 1.8 V and the cell voltage of 1.6 V which resulted in high capacity (Fig. 6b) was fixed for the next experiments as consistent with the previous studies [11, 43].

Variation of electrical conductivity vs. time at various voltage (a), variation of capacity vs. voltage (b)
During the experiment, the solution pH changed from 6.58 to 7.55. The increased pH might be caused by reduction of dissolved oxygen in the solution reservoir producing OH− [33]:
At higher voltage, oxidation of chloride can lower the pH [33], noting that the resistance at contact points causes a drop in the applied voltage.
Ion concentration affects the electrical conductivity of the solution so that lower concentration translates to slower kinetics. It also alters the ionic strength that determines the hydrated ion diameter. Higher ionic strength leads to smaller hydration shell and easier transport of ions into pores [33]. A balance between ion accessibility for electrosorption and co-ion electrostatic repelling within confined pores determines the optimal ion concentration [44]. Figure 7a shows variation of electrical conductivity with time at various initial salt concentrations and the affected adsorption capacity is demonstrated in Fig. 7b. Initially, as the ion concentration increases the capacity also sharply increases up to a point where further increase in concentration has small effect on the capacity which can be due to the lack of excess adsorption sites. This behavior has been observed in previous studies [40, 45].

Variation of electrical conductivity vs. time at various initial salt concentrations (a), variation of capacity vs. concentration (b)
Variation of electrical conductivity with time at various flow rates is depicted in Fig. 8a and the variation of adsorption capacity can be seen in Fig. 8b. Low flow rate is in favor of EDL formation [40] and as the flow rate increases the residence time of ions in the cell decreases; however, better mixing is achieved. Therefore, the adsorption capacity is affected by the resultant of these opposing effects up to a point where the electrode gets saturated and the capacity does not remarkably change with the flow rate.

Variation of electrical conductivity vs. time at various flow rates (a), variation of capacity vs. flow rate (b)
To study the kinetics of the electrosorption process, which is linked to the mass transport rate of ions driving from bulk solution onto the surface of electrode, the experimental data was fitted to the pseudo-second order model (Eq. 4):
where qt and qe are capacities at time (t) and at the equilibrium state, respectively, and k2 is the rate constant (g mg−1 min−1). Figure 9 shows satisfactory correlation between the experimental data and the linearized kinetic model. The parameters (qe and k2) can be obtained from slope and intercept of the curves. Table 2 shows that as the solution concentration increases, the rate constant and the equilibrium capacity also increase. The calculated qe is consistent with the experimental capacity (qe).

Adsorption data fitted to pseudo-second order kinetic model for various concentrations (V = 1.6 V, Q = 20 mL/min, Vreservoir = 250 mL)
The effective surface area (Aeff) was calculated by the following equation [44]:
where \({r}_{Na+}\) is the radius of hydrated Na+ (3.58 Å) [46], NA is Avagadro number (6.02 × 1023 mol−1), MNaCl is the molecular weight of NaCl (58.44 g mol−1). At 1000 mg g−1 salt concentration, Aeff was evaluated to be around 38.5% of the BET specific surface area. This can be attributed to the rather small mean pore diameters so that the pores (< 6.4–8 Ǻ in width depending on the electrolyte concentration) cannot be effectively used due to the EDL over- lapping effect of micropores [47]. The electrical double-layer thickness is typically of a few nanometers. In CDI, the EDL overlapping takes place within confined pores of the charged electrodes limiting accessible surface area for ions [47].
Table 3 lists the adsorption capacities of various carbonaceous electrode materials in CDI cells. Compared to these materials, the synthesized electrodes in this work is quite promising since the continuous carbon nanotubes and the interconnected carbon particles enable fast electron/ion transport. Besides, high specific surface area and high mesoporosity provide adequate ion transfer passage way.
4 Conclusion
In this investigation, carbon aerogel containing carbon nanotubes was synthesized by traditional sol–gel method. Various characterization techniques confirmed the outstanding structural properties and electrochemical behavior of the aerogel (specific capacitance of around 212 F/g and cyclic stability over 3000 cycle). The sample was subsequently utilized in a capacitive deionization system as the electrode active material. Effect of various operating parameters like applied voltage, initial salt concentration, and solution flow rate were studied in a recirculating CDI cell. It was observed that the system operated well at a voltage a bit larger than the standard potential of water electrolysis because of the circuit resistances. Increase in the flow rate initially improved the capacity until establishing a mild plateau. Furthermore, increase in the salt concentration raised the adsorption capacity. The kinetic of the electrosorption process properly followed the pseudo-second order model and the salt concentration remarkably dropped within initial 10 min. Notably, the flexible synthesis approach allows the fabrication of many other hybrid carbon aerogels to further improve the CDI performance.
Data availability
The data that support the findings of this study are available from the corresponding author, [N. Salman Tabrizi], upon reasonable request.
References
Oladunni J, Zain JH, Hai A, Banat F, Bharath G, Alhseinat E (2018) A comprehensive review on recently developed carbon based nanocomposites for capacitive deionization: from theory to practice. Sep Sci Technol 207:291–320
He W, Le Henaff A-C, Amrose S, Buonassisi T, Peters IM, Winter AG (2021) Voltage- and flow-controlled electrodialysis batch operation: flexible and optimized brackish water desalination. Desalination 500:114837. https://doi.org/10.1016/j.desal.2020.114837
Fellaou S, Ruiz-Garcia A, Gourich B (2021) Enhanced exergy analysis of a full-scale brackish water reverse osmosis desalination plant. Desalination 506:114999. https://doi.org/10.1016/j.desal.2021.114999
Khoshrou I, Jafari Nasr MR, Bakhtari K (2017) New opportunities in mass and energy consumption of the multi-stage flash distillation type of brackish water desalination process. Sol Energy 153:115–125. https://doi.org/10.1016/j.solener.2017.05.021
Liu S, Wang Z, Han M, Zhang J (2021) Embodied water consumption between typical desalination projects: reverse osmosis versus low-temperature multi-effect distillation. J Clean Prod 295:126340. https://doi.org/10.1016/j.jclepro.2021.126340
Ahmed MA, Tewari S (2018) Capacitive deionization: processes, materials and state of the technology. J Electroanal Chem 813:178–192. https://doi.org/10.1016/j.jelechem.2018.02.024
Seo S-J, Jeon H, Lee JK, Kim G-Y, Park D, Nojima H et al (2010) Investigation on removal of hardness ions by capacitive deionization (CDI) for water softening applications. Water Res 44:2267–2275. https://doi.org/10.1016/j.watres.2009.10.020
Chen R, Sheehan T, Ng JL, Brucks M, Su X (2020) Capacitive deionization and electrosorption for heavy metal removal. Environ Sci 6:258–282. https://doi.org/10.1039/C9EW00945K
Pastushok O, Zhao F, Ramasamy DL, Sillanpää M (2019) Nitrate removal and recovery by capacitive deionization (CDI). Chem Eng J 375:121943. https://doi.org/10.1016/j.cej.2019.121943
Ge Z, Chen X, Huang X, Ren ZJ (2018) Capacitive deionization for nutrient recovery from wastewater with disinfection capability. Environ Sci 4:33–39. https://doi.org/10.1039/C7EW00350A
Gabelich CJ, Tran TD, Suffet IHM (2002) Electrosorption of inorganic salts from aqueous solution using carbon aerogels. Environ Sci Technol 36(13):3010–3019. https://doi.org/10.1021/es0112745
Suss ME, Porada S, Sun X, Biesheuvel PM, Yoon J, Presser V (2015) Water desalination via capacitive deionization: what is it and what can we expect from it? Energy Environ Sci 8(8):2296–2319. https://doi.org/10.1039/C5EE00519A
Elsaid K, Elkamel A, Sayed ET, Wilberforce T, Abdelkareem MA, Olabi A-G (2022) Bio-based carbon materials for capacitive deionization CDI desalination processes. In: Olabi A-G (ed) Encyclopedia of smart materials. Elsevier, Oxford, pp 402–410
Kyaw HH, Al-Mashaikhi SM, Myint MTZ, Al-Harthi S, El-Shafey E-SI, Al-Abri M (2021) Activated carbon derived from the date palm leaflets as multifunctional electrodes in capacitive deionization system. Chem Eng Process 161:108311. https://doi.org/10.1016/j.cep.2021.108311
Laxman K, Myint MTZ, Al Abri M, Sathe P, Dobretsov S, Dutta J (2015) Desalination and disinfection of inland brackish ground water in a capacitive deionization cell using nanoporous activated carbon cloth electrodes. Desalination 362:126–132. https://doi.org/10.1016/j.desal.2015.02.010
Zhang S, Wang Y, Han X, Cai Y, Xu S (2018) Optimizing the fabrication of carbon nanotube electrode for effective capacitive deionization via electrophoretic deposition strategy. Prog Nat Sci Prog Nat Sci Mater 28:251–257. https://doi.org/10.1016/j.pnsc.2018.02.010
Liu Y, Zhang X, Gu X, Wu N, Zhang R, Shen Y et al (2020) One-step turning leather wastes into heteroatom doped carbon aerogel for performance enhanced capacitive deionization. Microporous Mesoporous Mater 303:110303. https://doi.org/10.1016/j.micromeso.2020.110303
Wang C, Chen L, Liu S, Zhu L (2018) Nitrite desorption from activated carbon fiber during capacitive deionization (CDI) and membrane capacitive deionization (MCDI). Colloids Surf A Physicochem Eng Asp 559:392–400. https://doi.org/10.1016/j.colsurfa.2018.09.072
Kalfa A, Penki TR, Cohen I, Shpigel N, Avraham E, Aurbach D et al (2020) Thermally reduced graphene oxide as an electrode for CDI processes: a compromise between performance and scalability? Desalination 492:114599. https://doi.org/10.1016/j.desal.2020.114599
Nguyen TT, Huynh LTN, Pham TN, Tran TN, Ho TTN, Nguyen TD et al (2021) Enhanced capacitive deionization performance of activated carbon derived from coconut shell electrodes with low content carbon nanotubes–graphene synergistic hybrid additive. Mater Lett 292:129652. https://doi.org/10.1016/j.matlet.2021.129652
Beke M, Velempini T, Prabakaran E, Pillay K (2023) Preparation of carbon-aerogel polypyrrole composite for desalination by hybrid capacitive desalination method. Arab J Chem 16(2):104412. https://doi.org/10.1016/j.arabjc.2022.104412
Miao L, Wang Z, Peng J, Deng W, Chen W, Dai Q, Ueyama T (2022) Pseudocapacitive deionization with polypyrrole grafted CMC carbon aerogel electrodes. Sep Purif Technol 296:121441. https://doi.org/10.1016/j.seppur.2022.121441
Liu X, Liu H, Mi M, Kong W, Ge Y, Hu J (2019) Nitrogen-doped hierarchical porous carbon aerogel for high-performance capacitive deionization. Sep Purif Technol 224:44–50. https://doi.org/10.1016/j.seppur.2019.05.010
Yeh C-L et al (2015) Improved performance in capacitive deionization of activated carbon electrodes with a tunable mesopore and micropore ratio. Desalination 367:60–68
Bordjiba T, Mohamedi M, Dao LH (2007) Synthesis and electrochemical capacitance of binderless nanocomposite electrodes formed by dispersion of carbon nanotubes and carbon aerogels. J Power Sources 172(2):991–998
Worsley MA, Pauzauskie PJ, Kucheyev SO, Zaug JM, Hamza AV, Satcher JH et al (2009) Properties of single-walled carbon nanotube-based aerogels as a function of nanotube loading. Acta Mater 57:5131–5136. https://doi.org/10.1016/j.actamat.2009.07.012
Reyhani R, Zadhoush A, Tabrizi NS, Nazockdast H, Naeimirad M (2021) Synthesis and characterization of powdered CNT-doped carbon aerogels. J Non Cryst Solids 571:121058. https://doi.org/10.1016/j.jnoncrysol
Al-Muhtaseb SA (2003) Preparation and properties of resorcinol-formaldehyde organic and carbon gels. Adv Mater 15(2):101–114
Okolo GN, Neomagus HWJP, Everson RC, Roberts MJ, Bunt JR, Sakurovs R et al (2015) Chemical–structural properties of South African bituminous coals: Insights from wide angle XRD–carbon fraction analysis, ATR–FTIR, solid state 13C NMR, and HRTEM techniques. Fuel 158:779–792. https://doi.org/10.1016/j.fuel.2015.06.027
Światowska J, Lair V, Pereira-Nabais C, Cote G, Marcus P, Chagnes A (2011) XPS, XRD and SEM characterization of a thin ceria layer deposited onto graphite electrode for application in lithium-ion batteries. Appl Surf Sci 257:9110–9119. https://doi.org/10.1016/j.apsusc.2011.05.108
Ţucureanu V, Matei A, Avram AM (2016) FTIR spectroscopy for carbon family study. Crit Rev Anal Chem 46:502–520. https://doi.org/10.1080/10408347.2016.1157013
Al-Ghouti MA, Da’ana DA (2020) Guidelines for the use and interpretation of adsorption isotherm models: a review. J Hazard Mater 393:122383. https://doi.org/10.1016/j.jhazmat.2020.122383
Jia B, Zhang W (2016) Preparation and application of electrodes in capacitive deionization (CDI): a state-of-art review. Nanoscale Res Lett 11(1):64. https://doi.org/10.1186/s11671-016-1284-1
Lv Y, Ding L, Wu X, Guo N, Guo J, Hou S et al (2020) Coal-based 3D hierarchical porous carbon aerogels for high performance and super-long life supercapacitors. Sci Rep 10:7022. https://doi.org/10.1038/s41598-020-64020-5
Samage A et al (2022) High power, long cycle life capacitive carbon from Hibiscus cannabinus, a Agri-bio-waste withsimultaneous value addition in water treatment application. Chem Eng J 435:134952
Ye S, Feng J, Wu P (2013) Deposition of three-dimensional graphene aerogel on nickel foam as a binder-free supercapacitor electrode. ACS Appl Mater Interfaces 5:7122–7129. https://doi.org/10.1021/am401458x
Xu G, Han J, Ding B, Nie P, Pan J, Dou H et al (2015) Biomass-derived porous carbon materials with sulfur and nitrogen dual-doping for energy storage. Green Chem 17:1668–1674. https://doi.org/10.1039/C4GC02185A
Divyapriya G, Vijayakumar KK, Nambi I (2019) Development of a novel graphene/Co3O4 composite for hybrid capacitive deionization system. Desalination 451(2019):102–110. https://doi.org/10.1016/j.desal.2018.03.023
Jung H-H et al (2007) Capacitive deionization characteristics of nanostructured carbon aerogel electrodes synthesized via ambient drying. Desalination 216(1):377–385
Zhu G, Wang H, Xu H, Zhang L (2018) Enhanced capacitive deionization by nitrogen-doped porous carbon nanofiber aerogel derived from bacterial-cellulose. J Electroanal Chem 822:81–88. https://doi.org/10.1016/j.jelechem.2018.05.024
Fahim M, Shah A, Bilal S (2019) Highly stable and efficient performance of binder-free symmetric supercapacitor fabricated with electroactive polymer synthesized via interfacial polymerization. Materials (Basel) 12(10):1626. https://doi.org/10.3390/ma12101626
Lee B et al (2018) Enhanced capacitive deionization by dispersion of CNTs in activated carbon electrode. ACS Sustain Chem Eng 6(2):1572–1579
Sui Z et al (2012) Green synthesis of carbon nanotube–graphene hybrid aerogels and their use as versatile agents for water purification. J Mater Chem 22(18):8767
Liu T, Serrano J, Elliott J, Yang X, Cathcart W, Wang Z, He Z, Liu G (2020) Exceptional capacitive deionization rate and capacity by block copolymer-based porous carbon fibers. Sci Adv 6(16):eaaz0906. https://doi.org/10.1126/sciadv.aaz0906
Zhang H, Liang P, Bian Y, Jiang Y, Sun X, Zhang C et al (2016) Moderately oxidized graphene–carbon nanotubes hybrid for high performance capacitive deionization. RSC Adv 6:58907–58915. https://doi.org/10.1039/C6RA10088K
Le Khac Duyen PQN, Kien LA (2016) Capacitive deionization (CDI) for desalisation using carbon aerogel electrodes. Sci Technol Dev 19(3):155–164
Hou C-H, Liu N-L, Hsu H-L, Den W (2014) Development of multi-walled carbon nanotube/poly(vinyl alcohol) composite as electrode for capacitive deionization. Sep Purif Technol 130:7–14. https://doi.org/10.1016/j.seppur.2014.04.004
Luo G, Wang Y, Gao L, Zhang D, Lin T (2018) Graphene bonded carbon nanofiber aerogels with high capacitive deionization capability. Electrochim Acta 260:656–663. https://doi.org/10.1016/j.electacta.2017.12.012
Dong Q, Wang G, Qian B, Hu C, Wang Y, Qiu J (2014) Electrospun composites made of reduced graphene oxide and activated carbon nanofibers for capacitive deionization. Electrochim Acta 137:388–394. https://doi.org/10.1016/j.electacta.2014.06.067
Chen Z, Zhang H, Wu C, Luo L, Wang C, Huang S et al (2018) A study of the effect of carbon characteristics on capacitive deionization (CDI) performance. Desalination 433:68–74. https://doi.org/10.1016/j.desal.2017.11.036
Zhang D et al (2007) Influence of diameter of carbon nanotubes mounted in flow-through capacitors on removal of NaCl from salt water. J Mater Sci 42(7):2471–2475
Ebrahimian Mashhadi M, Mozaffarian M, Mohseni M, Jafarikojour M, Dabir B (2022) Evaluation of synergistic effects of coupling capacitive deionization (CDI) and UV oxidation processes for saline water treatment. J Environ Chem Eng 10:106909. https://doi.org/10.1016/j.jece.2021.106909. (Elsaid, 2022 #1626)
Acknowledgements
This study was financed by Iran National Science Foundation (INSF, No. 97018985), and supported by Materials and Energy Research Center of Iran (No. 241119802).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tabrizi, N.S., Taleghani, M.S. Performance of CNT-CNP aerogel as electrode in capacitive deionization system. Carbon Lett. 33, 1253–1263 (2023). https://doi.org/10.1007/s42823-023-00477-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42823-023-00477-4