
Abstract
Neoscytalidium dimidiatum and Bipolaris species are fungal plant pathogens that have been reported to cause human diseases. Recently, we have isolated numerous N. dimidiatum and Bipolaris species from the skin scrapings and nails of different patients. In this work, we have sequenced the genome of one strain of N. dimidiatum. The sequenced genome was compared to that of a previously reported Bipolaris papendorfii genome for a better understanding of their complex lifestyle and broad host-range pathogenicity. Both N. dimidiatum UM 880 (~ 43 Mb) and B. papendorfii UM 226 (~ 33 Mb) genomes include 11,015–12,320 putative coding DNA sequences, of which 0.51–2.49% are predicted transposable elements. Analysis of secondary metabolism gene clusters revealed several genes involved in melanin biosynthesis and iron uptake. The arsenal of CAZymes related to plants pathogenicity is comparable between the species, including genes involved in hemicellulose and pectin decomposition. Several important gene encoding keratinolytic peptidases were identified in N. dimidiatum and B. papendorfii, reflecting their potential pathogenic role in causing skin and nail infections. In this study, additional information on the metabolic features of these two species, such as nutritional profiling, pH tolerance, and osmotolerant, are revealed. The genomic characterization of N. dimidiatum and B. papendorfii provides the basis for the future functional studies to gain further insights as to what makes these fungi persist in plants and why they are pathogenic to humans.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neoscytalidium dimidiatum is a dematiaceous hyphomycetes belonging to the order Botryosphaeriales in the class Dothideomycetes. The nomenclature for Neoscytalidium as Neoscytalidium dimidiatum isolate has been a controversial issue due to the production of both arthroconidial (Scytalidium dimidiatum) and pycnidial (Natrassia mangiferae) synanamorphs [1]. Based on the DNA sequence data of 28S rDNA, Crous et al. (2006) reclassified several lineages in the family Botryosphaeriaceae [2]. According to the taxonomic revision, a new genus Neoscytalidium with type species Neoscytalidium dimidiatum to include all isolates that produce arthroconidia typical of S. dimidiatum, including Hendersonula toruloidea and S. hyalinum was proposed [1,2,3]. This revision separated Neoscytalidium dimidiatum from a closely related group (Nattrassia mangiferae) that lacked this typical characteristic [2]. To date, the genus Neoscytalidium contains only two species: N. dimidiatum and N. novaehollandiae.
N. dimidiatum is a thermotolerant fungus, which causes sooty cankers on various fruit plants, bark trees, and ornamentals, such as physic nut, dragon fruit, orange, and common fig in tropical and subtropical regions [4,5,6,7,8,9]. Neoscytalidium canker causes wilting and dieback of tree branches when temperature reaches a hot regime and in dry weather condition [10, 11]. Heat stress turns N. dimidiatum into an opportunistic plant pathogen which infects fruit plants and trees [10, 11]. Neoscytalidium dimidiatum is not only known as phytopathogen but is also notorious for causing onychomycosis [12, 13] and dermatophytosis-like skin infection [14]. The diagnosis of such infections is challenging, since N. dimidiatum infection is frequently misdiagnosed as dermatophytosis based on direct microscopic examination [15]. Evidence has emerged that this fungus also causes subcutaneous phaeohypomycosis [16], mycetoma [17], cerebral phaeohyphomycosis [18], sinusitis [19], endophthalmitis [20], and fungemia [21], mainly affecting immunocompromised patients.
For the past 10 years, the isolation of many dematiaceous fungus, mainly N. dimidiatum and Bipolaris species (teleomorph of Cochliobolus) from the superficial skin and nail of patients of University Malaya Medical Centre (UMMC), a tertiary hospital in Malaysia, has been reported. Bipolaris is a large genus of dematiaceous hyphomycetes that fall under the order Pleosporales in the class Dothideomycetes. Like N. dimidiatum, Bipolaris species and its asexual state Cochliobolus are well-known plant pathogens and have been reported to cause devastating disease epidemics of crop plants, including maize, oat, rice, sorghum, and wheat [21]. Bipolaris species has been reported to cause fruit and stem end rot in dragon fruit in Malaysia [22]. In another independent study, a serious stem canker caused by N. dimidiatum was found to be infecting most red-fleshed dragon fruit plantation in Malaysia [23]. These suggest an increasing trend of N. dimidiatum and Bipolaris species isolation from both plants and humans in Malaysia. Although Neoscytalidium sp. and Bipolaris sp. are morphologically different and not closely related phylogenetically [24], they might share similar strategies to colonize and adapt to plant and human hosts.
In this work, we identified and generated a draft genome sequence of N. dimidiatum UM 880 that was recovered from the skin scraping of a patient suffering from dermatophytosis. The published B. papendorfii UM 226 genome [25] enables us to compare the genome sequence assemblies and annotations for N. dimidiatum and B. papendorfii. The in-depth comparative analysis of these two pathogens relative to one another and to other related fungal genomes is the first step in understanding their lifestyle and fungal nutritional strategy as well as gain insight into genes that form unique features of their pathogenic success. Knowledge of the basic biology and phenotype of these pathogen is useful in the development of a more suitable strategies for disease management.
Methods
Ethics statement
The isolates of N. dimidiatum UM 880 and B. papendorfii UM 226 used in this study were obtained from an archived fungal collection in a mycology laboratory of a teaching hospital, University of Malaya Medical Centre (UMMC), Kuala Lumpur, Malaysia [25].
Fungal isolates
Both N. dimidiatum UM 880 and B. papendorfii UM 226 were isolated from the skin scraping of patients in the UMMC. The isolates were processed according to the laboratory’s standard operating procedures (SOP) [24, 25].
For scanning electron microscopy (SEM) examination, samples were fixed in 4% (v/v) glutaraldehyde for 6 h at 4 °C, followed by three washing with 0.1 M sodium cacodylate buffer, pH 7.2 (10 min each). The samples were post-fixed in 1% osmium tetroxide (w/v) for 2 h at 4 °C, washed three times with 0.1 M sodium cacodylate buffer, and dehydrated with increasing concentration of acetone, started with 35% (v/v) for 10 min, 50% (v/v) for 10 min, 75% (v/v) for 10 min, 95% (v/v) for 10 min, and finally three times with 100% (v/v). The samples were air dried with critical point dryer (Leica, Wetzlar, Germany), mounted onto aluminum stubs, sputter coated with gold with sputter coater (Leica, Germany), and visualized with LEO 1455 VP SEM (Carl-Zeiss, Germany).
Antifungal susceptibility
The in vitro antifungal susceptibilities of N. dimidiatum UM 880 and B. papendorfii UM 226 were examined using the Epsilometer Test (Etest, Biomerieux, France) to determine the minimum inhibitory concentration (MIC) of antifungal drugs as previously described [24]. To prepare the fungal inocula, N. dimidiatum UM 880 and B. papendorfii UM 226 cell suspensions were adjusted to 0.5 McFarland standard. The isolate was tested against eight common antifungal agents (amphotericin B, five azoles, viz., fluconazole, itraconazole, posaconazole, voriconazole, ketoconazole, and two echinocandins, viz., anidulafungin and caspofungin), and the minimum inhibitory concentrations (MICs) were examined after 24-h incubation.
DNA sequencing and multilocus phylogenetic analysis
Three different unlinked nuclear loci, the internal transcribed spacer region (ITS), small subunit of the ribosomal RNA gene (SSU), and large subunit of the ribosomal RNA gene (LSU) were used to identify the fungal isolate N. dimidiatum UM 880. Total DNA extraction, PCR amplification, DNA sequencing, and BLASTn search were performed as described previously [24, 25]. Unique ITS, SSU, and LSU sequences from the isolate N. dimidiatum UM 880, together with an additional species of N. novaehollandiae and an outgroup strain of Macrophomina phaseolina (Table S1), were subjected to phylogenetic analysis. Multiple sequence alignments of collected ITS, LSU, and SSU sequences were generated using M-Coffee [26].
Genomic DNA extraction, genome sequencing, and assembly
Genomic DNA extraction was carried out as previously described [25]. Whole genome sequencing of N. dimidiatum UM 880 was carried out using Illumina HiSeq 2000 Sequencer (Illumina Inc., San Diego, CA, USA) in a 2 × 90 bp paired-end mode on 500-bp and 5-kb library sizes. Illumina library was prepared using TruSeq v3 Reagent Kits (Illumina Inc., San Diego, CA, USA). The selected library fragments were purified through gel electrophoresis, which were then selectively enriched and amplified by PCR. The 500-bp Illumina sequenced read was then combined with the 5-kb Illumina sequenced read for further processing. Both sets of sequenced reads were first pre-processed using FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) trimming bases with a Phred quality below Qv20 from the 3′-end of the reads. The trimmed reads shorter than 30 bp and reads with 40% bases having Qv ≤ 20 were filtered out, retaining small-insert reads ≥ 80 bp and large-insert reads ≥ 30 bp. Two bases were trimmed from the 5′-terminal of all reads. Pre-processed reads from both libraries were assembled with Velvet version 1.2.07 [27] with k-mer setting = 63, insert length = 503, -ins_length_sd = 103, -min_pair_count = 15, insert_length2 = 5000, ins_length2_sd = 500, and min_contig_lgth = 200. Additional parameter of -shortMatePaired = yes was set for large insert library. The generated contigs assembled from Velvet were further scaffolded using SSPACE Basic v2.0 [28] with more stringent parameters than software default to achieve higher accuracy assembly (parameters: -z 100, -k 15, -a 0.3, -n 30, and -T 10). GapFiller v1.10 (-m = 60, -o = 15, -r = 0.8, –n = 30, -t = 30, and -T = 10) was used to perform gap filling by utilizing paired-end sequencing data from both libraries [29, 30].
Genome annotation
Interspersed repetitive elements and low-complexity DNA sequences were masked using RepeatMasker version open-3.3.0 with the Repbase fungal library version rm-20120418. Individual rRNA and tRNA were predicted using RNAmmer v1.2 [31] and tRNAscan-SE v1.3.1 [32], respectively. Putative transposable elements were identified using Transposon-PSI (http://transposonpsi.sourceforge.net) by PSI-TBLASTN searches with a collection of (retro-) transposon open reading frame (ORF) homology profiles. Protein coding sequences were predicted using GeneMark-ES version 2.3e [33]. Gene annotation was completed using BLAST (Basic Local Alignment Search Tool) searches against the NCBI nr and SwissProt databases.
Functional gene and pathway analysis
GO and KEGG metabolic pathway matches were performed using local BLAST2GO tools [34]. Classifications of predicted proteins were carried out using KOG [35]. Protein domain families were matched to Pfam database using InterProScan 5 [36]. The best protein models of the N. dimidiatum UM 880 and B. papendorfii UM 226 genomes were submitted to database of automated carbohydrate-active enzyme annotation (dbCAN) to functionally annotate CAZymes. Comparative analysis was performed against other fungi with different lifestyles, including saprophytic fungi (Neurospora crassa OR74A and Trichoderma reesei QM6a), facultative parasitic fungi (Aspergillus nidulans FGSC A4), biotrophic fungi (Cladosporium flavum, Ustilago maydis, and Blumeria graminis), necrotrophic fungi (Cochliobolus heterostrophus C4 and C. heterostrophus C5), hemi-biotrophic fungi (Cochliobolus sativus ND90Pr, Magnaporthe oryzae 70–15 version 8, and Fusarium graminearum PH-1), and symbiotic fungus (Laccaria bicolor) [37, 38]. Peptidases were functionally annotated by submitting the predicted protein models to the MEROPS database [39]. SignalP version 4.1 [40] was used to predict cleavage sites and signal peptide/non-signal peptide to identify small secreted proteins after discarding proteins with transmembrane domains that were determined by TMHMM version 2.0 [41]. One transmembrane domain located at the N-terminal 40 amino acids is acceptable as the domain is responsible to the secretion signal. Genomic mapping of fungal secondary metabolite backbone genes and associated genes was carried out using web-based SMURF (Secondary Metabolite Unknown Regions Finder) (www.jcvi.org/smurf/) [42]. Amino acid sequences with e-value threshold ≤ 1e − 5 and alignment length over 70% of its own length and over 50% match identity were selected and assigned as the annotation of predicted genes.
Phenotype microarrays
Biolog PM analysis (Biolog Inc., Hayward, CA, USA) was employed to examine the metabolic profile of Bipolaris papendorfii and Neosyntalidium dimidiatum. Ten MicroPlate panels (PM1 to PM10), which included carbon, nitrogen, phosphorus, sulfur, nutrient supplements, peptide, osmolytes, and pH sources, were used in this study as summarized in Table S14. Bipolaris papendorfii and Neosyntalidium dimidiatum were grown on SDA at 30 °C for 7 days. Colonies on SDA were extracted with sterile, wetted cotton swab, and inoculated into 15 mL of yeast solution (Biolog Inc.). The turbidity of the cell suspension was adjusted to 62% transmittance at 590 nm. PM1-10 inoculating fluids were prepared according to the manufacturer’s protocol. The cell suspension was inoculated to PM1-10 inoculating fluid as indicated in Table S15. The prepared inoculum suspension (100 µL) was inoculated into each well of microplate panel PM1-10 and incubated at 30 °C for 120 h. Samplings were done in duplicates and readings were taken using Epoch Microplate Spectrophotometer (BioTek Instruments, Inc., Winooski, VT, USA). The utilization of substrate was monitored at 490 nm in PM 1–8 microplate, while the growth or biomass reaction to osmolytes and pH were monitored at 750 nm in PM 9–10 microplate. Water was used as the negative control for each microplate. Data for each assay from duplicate runs were averaged to obtain the mean value.
Statistical analyses
Data were evaluated using SPSS Inc. software (version 20) (SPSS Inc., Chicago, USA). All data are presented as mean from two separate runs (n = 2), unless stated otherwise. The statistical difference between sample means for analyses involving supplementation of nitrogenous source at pH 4.5 and 9.5 as well as supplementation of osmolyte at 6% sodium chloride was analyzed with one-way ANOVA. The multiple comparison of means was analyzed by Tukey’s test. An independent T-test was used to determine the significant difference between means for pH, salt, and urea tolerance test. The statistical level of significance was preset at α = 0.05.
Results and discussion
Identification of N. dimidiatum UM 880
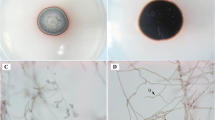
The N. dimidiatum UM 880 colonies on sabouraud dextrose agar (SDA) were fast growing with dense, deeply floccose, and cottony aerial mycelium (Fig. 1A and 1B). The colony reached diameters of 90 mm and filling the Petri within 4 to 6 days at 30 °C (Fig. 1A). The surface of the colonies was initially grayish white, becoming dark gray, and produced blackish brown diffusible pigment within 6-day incubation (Fig. 1A). It had a black coloration on the reverse side (Fig. 1C).

Macroscopic and microscopic morphology of N. dimidiatum UM 880. The surface (A), close-up (B), and reverse (C) view of colony morphology of N. dimidiatum UM 880 after being cultured for 7 days on SDA. Lactophenol cotton blue mount showing D typical arthroconidia of N. dimidiatum (× 400 magnification, bars 20 µm). Two-celled conidia are also shown (arrows). Scanning electron micrograph showing E the arthroconidia were cylindrical with smooth to slightly verruculose walled and arranged in chains
Microscopic examination of slide culture preparations demonstrated broad branched, septate, subhyaline to dark brown hyphae (6–10 µm wide) with no conidiophores (Fig. 1D). They are constricted at their prominent thick septations, giving the pseudohyphae appearance. Contiguous of one- to two-celled, pigmented arthroconidia (3–6 × 5–12 µm) with rectangular, oval, and barrel-shaped were produced (Fig. 1D). The arthroconidia were flattened on the ends; two-celled conidia were separated by a thick septum (Fig. 1D). No pycnidium was produced by N. dimidiatum UM 880. The SEM microphotograph of the N. dimidiatum UM 880 revealed that the arthroconidia were cylindrical with smooth to slightly verruculose walled and arranged in chains (Fig. 1E). The end of each conidium was central convex dome in shape and was connected to other conidium. Based on the typical morphological features as described previously [43], the N. dimidiatum UM 880 clinical isolate was identified as N. dimidiatum.
Molecular identification was performed to confirm that the N. dimidiatum UM 880 isolate is truly N. dimidiatum as suggested by its morphological features. The internal transcribed spacer region (ITS), small subunit of the ribosomal RNA gene (SSU), and large subunit of the ribosomal RNA gene (LSU) sequences were obtained by amplification and bidirectional sequencing. PCR amplification of the ITS, LSU and SSU gene regions yielded specific amplicons of approximately 560 bp (ITS), 1200 bp (SSU), and 2000 bp (LSU), respectively (Fig. S1). By querying ITS, LSU, and SSU nucleotide sequences against those deposited in the NCBI-nucleotide database, the sequenced ITS gave 100% query coverage and 100% (579/579) identity with N. dimidiatum strain CBS 499.66; the sequenced LSU gave 72% query coverage and 100% (1159/1159) identity with N. dimidiatum strain CBS 145.78; the sequenced SSU gave 100% query coverage and 100% (1024/1024) identity with N. dimidiatum strain CMW 26170. The sequenced ITS, LSU, and SSU gene regions were used to construct a phylogram using combined gene analysis with the additional 16 ex-type strains of Neoscytalidium species (Fig. 2 and Table S1). Multilocus phylogenetic analysis revealed that the N. dimidiatum UM 880 isolate was clustered together with the group of N. dimidiatum instead of being grouped together into N. novaehollandiae cluster (Fig. S2). Taken together, the morphological and molecular characterization of N. dimidiatum UM 880 isolate revealed that it is N. dimidiatum.

KOG and KEGG classifications of proteins in N. dimidiatum UM 880 and B. papendorfii UM 226. A KOG class annotation distribution of N. dimidiatum UM 880 and B. papendorfii UM 226 genomes. A, RNA processing and modification; B, chromatin structure and dynamics; C, energy production and conversion; D, cell cycle control, cell division, chromosome partitioning; E, amino acid transport and metabolism; F, nucleotide transport and metabolism; G, carbohydrate transport and metabolism; H, coenzyme transport and metabolism; I, lipid transport and metabolism; J, translation, ribosomal structure, and biogenesis; K, transcription; L, replication, recombination, and repair; M, cell wall/membrane/envelope biogenesis; N, cell motility; O, post-translational modification, protein turnover, chaperones; P, inorganic ion transport and metabolism; Q, secondary metabolites biosynthesis, transport, and catabolism; R, general function prediction only; S, function unknown; T, signal transduction mechanisms; U, intracellular trafficking, secretion, and vesicular transport; V, defense mechanisms; W, extracellular structures; X, unnamed protein; and Z, cytoskeleton. B Distribution of predicted proteins from N. dimidiatum UM 880 and B. papendorfii UM 226 genomes that are involved in metabolic pathways by KEGG database
Properties of the N. dimidiatum and B. papendorfii genomes
The N. dimidiatum UM 880 genome was sequenced to ~ 93-fold depth of genome sequence coverage. In this study, a total of 33,078,316 paired reads (3.3 Gb) of a 500-bp insert-size library and 10,699,352 paired reads (1.1 Gb) of a 5-kb insert-size library were generated by Illumina HiSeq 2000 Sequencing system. The assembly yielded a combined total length of 42,687,746 bases in 2230 contigs (≥ 200 bases) which were then placed into 110 scaffolds (≥ 1000 bases) with paired-end sequencing data from both libraries. The B. papendorfii UM 226 genome was generated by a hybrid assembly of Illumina short reads and PacBio long reads with a lower number of contigs and a higher N50 value (Table 1). The larger genome for N. dimidiatum UM 880 genome (~ 43 Mb) as compared to the B. papendorfii UM 226 genome (~ 33 Mb) [25] is most likely due to the lower sequence coverage, as small scaffolds may fall into larger scaffold gaps. The G + C content of N. dimidiatum UM 880 (54.16%) was comparable to B. papendorfii UM 226 (50.65). Overall, the summary of the sequence statistics is shown in Table 1.
Transposable elements
Transposable elements (TEs) are ubiquitous components in fungal genomes. Transposable elements play an important role in the genetic variation and actively affect gene structure to allow them to adapt in adverse environments, especially for fungus that lacks sexual stages [44]. Dematiaceous fungal genomes contain different amounts of TEs. For example, the assembled genome of B. papendorfii UM 226, Cochliobolus heterostrophus race O genome, Sporothrix schenckii, and Sporothrix brasiliensis contain 2.49%, 5.90%, 0.34%, and 0.62% of TEs, respectively [25, 45, 46]. Transposable elements identified in N. dimidiatum UM 880 and B. papendorfii UM 226 genomes were categorized into classes and annotated as class I (LTR and LINE) and class II transposons. The assembled genome of N. dimidiatum UM 880 contains low TEs content (0.51%), whereas these “jumping genes” occupy 2.49% of the B. papendorfii UM 226 genome (Table 2). The high TE content in B. papendorfii UM 226 might impact on its genome organization, evolution, and genomic functions, including gene silencing, regulation of gene expression, or gene mutation. Despite the smaller genome size of the B. papendorfii UM 226, it has a fourfold higher TE content and has either class I (retrotransposons) or class II DNA transposons as compared to N. dimidiatum UM 880. LINE (long interspersed nuclear element) was mostly identified in the N. dimidiatum UM 880 genome (Table 2), which is common for fungal genomes [46, 47]. Bipolaris papendorfii UM 226, however, contains high copy number of DDE_1 transposases, which contain functional domains to mediate DNA-binding, protein–protein interaction, and “cleavage-and -joining” activities [47]. Gypsy element belonging to the LTR superfamily is widely distributed in the genome. A total of 83 and 279 copies of Gypsy elements were identified in N. dimidiatum UM 880 and B. papendorfii UM 226 genomes, respectively. Santana et al. (2014) reported that an estimated 50% of TEs identified in the C. heterostrophus race O genome was Gypsy elements [46]. Gypsy elements were found to be located in the non-coding regions adjacent to the PKS1, PKS2, OXI1, LAM1, RED2, and RED3 genes, but their exact function remains unknown [46]. No SINE (short interspersed nuclear elements) retrotransposon was identified in neither N. dimidiatum UM 880 nor B. papendorfii UM 226 genomes.
Gene content
A total of 137 tRNAs and 23 rRNAs (20 8S/5.8S rRNA, one 18S rRNA, and two 28S rRNA) were identified in the N. dimidiatum UM 880 genome, whereas B. papendorfii UM 226 genome contains 128 tRNAs, 12 5S rRNA, three 18S rRNA, and three 28S rRNA. The N. dimidiatum UM 880 genome revealed a total of 12,320 putative coding DNA sequences (CDS) with gene density of 2.88 genes/10 kb. The assembly size and the total number of predicted genes in the N. dimidiatum UM 880 genome were compared to other dematiaceous fungi. Neoscytalidium dimidiatum UM 880 has a relatively larger genome but lower gene density compared to other dematiaceous fungi (Table S2), reflecting larger amounts of non-coding DNA in the N. dimidiatum UM 880 genome. On average, there were 3.09 exons per gene and average size of protein coding genes was 1451 bp in N. dimidiatum UM 880 genome. In addition, N. dimidiatum UM 880 genome revealed that a total of 11,577, 7656, and 9155 protein-coding sequences were homologous to known proteins deposited in the NCBI nr, SwissProt, and InterPro databases, and are comparable to that of B. papendorfii UM 226 (Tables 3 and S3).
Hypothetical proteins are predicted proteins, which are characterized by low identity to annotated proteins and have not been functionally characterized [48]. Surprisingly, both N. dimidiatum UM 880 and B. papendorfii UM 226 genomes harbor large number of hypothetical proteins or unannotated proteins. The N. dimidiatum UM 880 and B. papendorfii UM 226 genomes contain a total of 6041 and 10,330 hypothetical proteins/unannotated proteins but lacked significant homologies to known proteins from NCBI nr database (Table S4). InterPro protein sequence analysis revealed at least one domain was identified in 3098 and 7614 hypothetical proteins/unannotated proteins in N. dimidiatum UM 880 and B. papendorfii UM 226 genomes, respectively (Table S4). Most of the hypothetical proteins and unannotated proteins contain protein kinase domain (169 and 290 in N. dimidiatum UM 880 and B. papendorfii UM 226 genomes, respectively), suggesting the importance of these unknown proteins involved in phosphorylation to govern various cellular processes, such as metabolism, transcription, cell movement, apoptosis, and differentiation by changing cellular location, enzyme activity, or association with other proteins.
The N. dimidiatum UM 880 and B. papendorfii UM 226 genomes were also mapped to Eukaryotic Clusters of Orthologs (KOG), Gene Ontology (GO), and the Kyoto Encyclopedia of Genes and Genomes (KEGG) databases to characterize the predicted proteins. In N. dimidiatum UM 880 genome, a total of 6182 proteins were ascribed to 26 different functional groups based on KOG classification (Fig. 2A and Table S5). The category [R] general function prediction contains the greatest number of genes (1234 genes) and shows that many proteins were not classified to a distinct group (Fig. 2A). The categories [O] post-translational modification, protein turnover, chaperones (542 genes); [Q] secondary metabolites biosynthesis, transport, and catabolism (539 genes); [I] lipid transport and metabolism (467 genes); [C] energy production and conversion (438 genes); and [T] signal transduction mechanisms (423 genes) were the top five most abundant KOG groups (Fig. 2A). Neoscytalidium dimidiatum UM 880 and B. papendorfii UM 226 genomes contain a large fraction of putative genes that are associated with post-translational modifications, protein turnover, and chaperones (Fig. 2A). Post-translational modifications of fungal cell surface proteins, such as reversible peptide modifications ubiquitination, neddylation, and sumoylation, are vital in fungal pathogenicity [49]. The putative genes that are related to ubiquitination, neddylation, and sumoylation are listed in Table S6.
Gene ontology (GO) analysis revealed that the N. dimidiatum UM 880 and B. papendorfii UM 226 have 7202 and 7155 genes with at least one GO assignment, respectively. All genes that matched to “cellular component,” “molecular function,” and “biological process” categories of GO (Table 4) were further examined. A 0.05 p-value of Pearson chi-square test was used as the cutoff for the significance of the differences between the two datasets. The differences between the two species are considered remarkable when the p-value is below this significant level. In the “cellular component” ontology, we found that genes that mapped to the cell (GO:0005623) and organelle (GO:0043226) were significant higher in N. dimidiatum UM 880 as compared to the B. papendorfii UM 226. In the “molecular function” ontology, N. dimidiatum UM 880 contains higher number of genes involved in transporter activity but lower number of genes involved in binding activity. In the “biological process” ontology, N. dimidiatum UM 880 contains more genes assigned to the “response to stimulus” (GO:0050896) category but lesser genes in the “metabolic process” (GO:0008152) category than B. papendorfii UM 226. In the “metabolic process” category, the “nitrogen compound metabolic process” (GO:0006807), “macromolecule metabolic process” (GO:0043170), “primary metabolic process” (GO:0044238), and “oxidation reduction” (GO:0055114) are categories which showed highly significant relationship. In addition, there was a significant relationship observed in the category of “response to chemical stimulus” (GO:0042221), under the subset of “response to stimulus.” Of the 1110 genes assigned to this category in N. dimidiatum UM 880, 201 genes were further assigned to the “response to drug” (GO:0042493) category (168 genes in B. papendorfii UM 226), 274 genes into “response to xenobiotic stimulus” (GO:0009410) category (237 genes in B. papendorfii UM 226), and 246 genes to “response to inorganic substance” (GO:0010035) category (207 genes in B. papendorfii UM 226). Particularly, the numbers of genes assigned to “drug transport” (GO:0015893) and “multidrug transport” (GO:0006855) are higher in N. dimidiatum UM 880 (78 and 26 genes, respectively) than in B. papendorfii UM 226 (39 and 28 genes, respectively). This might contribute to the higher tolerance of drugs in N. dimidiatum UM 880 as compared to B. papendorfii UM 226 as will be discussed in the “Antifungal susceptibility profile and drug resistance genes” section.
To further gain insight into the gene functions in N. dimidiatum UM 880, we successfully assigned 1812 predicted proteins to their orthologous genes in metabolic pathways in the KEGG database (Table S7). The top five categories in KEGG metabolic pathway in both fungi are carbohydrate metabolism, amino acid metabolism, lipid metabolism, nucleotide metabolism, and xenobiotics biodegradation and metabolism (Fig. 2B). Overall, B. papendorfii UM 226 has noticeably more KEGG annotated genes involved in the metabolic pathway as compared to the N. dimidiatum UM 880 (Fig. 2B). However, it should be noted that the numbers of genes encoding enzymes for secondary metabolite biosynthesis are more abundant in N. dimidiatum UM 880 than in B. papendorfii UM 226 (Fig. 2B), which is in agreement with the KOG annotation (Fig. 2A). The metabolism and biosynthesis of secondary metabolites in N. dimidiatum UM 880 and B. papendorfii UM 226 are further discussed in the “Secondary metabolism” section.
Secondary metabolism
Dothideomycetes fungi harbor large number of gene-encoding enzymes for secondary metabolite production, including polyketide synthase (PKS) and non-ribosomal peptide synthetase (NRPS) that are vital in niche exploitation as well as pathogenicity [50, 51]. Many of these secondary metabolites often serve as virulence factors, which are required for these necrotrophs [52, 53]. With this in mind, we screened the N. dimidiatum UM 880 and B. papendorfii UM 226 genomes to identify genes related to secondary metabolism that are important for virulence and survival such as host specific toxins (HST), non-host specific toxin (NHST), and melanin. Secondary Metabolite Unique Regions Finder (SMURF) analysis predicted 39 and 32 gene clusters encoding backbone enzymes in N. dimidiatum UM 880 and B. papendorfii UM 226 genomes, respectively (Table 5).
Dematiaceous fungi are organisms that are known to produce melanin during formation of fungal spore for deposition in the cell wall. Fungal melanins are essential for survival, pathogenicity, and increase in their competitive ability to occupy plethora of niches. Jahn et al. (2000) indicated that loss of melanin pigment is associated with enhanced susceptibility to reactive oxygen species (ROS) [54]. Most fungal melanins are biosynthesized via 1,8-dihydroxynaphthalene (DHN) pathway using polyketide synthases (PKs) to form melanin precursors. The PKSs are encoded by the most widely distributed fungal PKS genes, such as PKS1 in Colletotrichum lagenarium [51] and PKS18 in Cochliobolus heterostrophus [55]. In this study, PKS1 (UM880_8812, 70.2% identical), PKS15 (UM880_2257, 51% identical), alternapyrone synthase, PKSN (UM880_2826, 56% identical), and PKS13 (UM880_9993, 53% identical) were identified in N. dimidiatum UM 880 genome; PKS1 (UM226_1411, 93% identical), PKS2 (UM226_3388, 84% identical), PKS4 (UM226_8230, 79% identical), PKS7 (UM226_6241, 65% identical), PKS9 (UM226_7130, 77% identical), and PKS11 (UM226_7830, 85% identical) were found in B. papendorfii UM 226 genome. Like B. papendorfii UM 226 [25], N. dimidiatum UM 880 may produce 1,8-dihydroxynaphthalene (DHN) melanin by a set of DHN melanin-synthesis-related proteins, including PKS1, 1,3,6,8-tetrahydroxynaphthalene reductase (BRN2) (UM880_8808, 90%), transcription factor CMR1 (UM880_8809, 94.1%), and three scytalone dehydratases (UM880_2773, 85%; UM880_3280, 98.9%; and UM880_7245, 50.3%). A small gene cluster comprising PKS1 BRN2, and CMR1 was identified in N. dimidiatum UM 880 genome (Fig. 3A). The mutual orientation of PKS1, BRN2, and transcription factor CMR1 is the same as found in B. papendorfii UM 226 (Fig. 3A). Amino acid alignment revealed that N. dimidiatum UM 880 PKS1 (NDPKS1) is 70.7% identical to B. papendorfii UM 226 PKS1 (BPPKS1). Both the NDPKS1 and BPPKS1 contain features of a non-reducing fungal type I PKS with a starter unit of ACP transacylase (SAT), β-ketoacyl synthase (KS), acyltransferase (AT), product template (PT), acyl carrier protein (ACP), and thioesterase (TE) [56]. NDPKS1 and BPPKS1 contain domain order of KS-AT-ACP-ACP-TE similar to Bipolaris oryzae PKS1 that is involved in melanin biosynthesis [57] (Fig. 3B). Neoscytalidium dimidiatum UM 880 contains two mitogen-activated protein kinases, Chk1 (UM880_5115, 91.7%) and Mps1 (UM880_607, 86%), that play important role to regulate transcription factor CMR1 at the transcriptional level [55].

Melanin biosynthetic genes identified in N. dimidiatum UM 880 and B. papendorfii UM 226 genomes. A Schematic representation of the biosynthetic gene cluster for the PKS in N. dimidiatum UM 880 and B. papendorfii UM 226 genomes. Numbers are in kilobases. B Domain analysis of NDPKS1, BPPKS1, and Bipolaris oryzae PKS1
The 6-methylsalicylic-acid synthase belongs to a class of PKSs that is involved in building of an aryl moiety in many bioactive secondary metabolites, such as avilamycin, calicheamicin cholorothricin, maduropeptin, pactmycin, and polyketomycin [58]. In this work, we found a gene encoding 6-methylsalicylic-acid synthase (UM880_11018, 85.2%) in N. dimidiatum UM 880. As described in Ding et al. (2010), UM880_11018 harbors multiple functional domains in an order of KS-AT-dehydratase (DH)-ketoreductase (KR)-ACP [58]. The presence of this encoding gene in N. dimidiatum UM 880 suggests that this isolate may produce antibiotic substances.
Iron uptake and homeostasis
Fungal pathogens can acquire iron from vertebrate hosts to survive and proliferate in the hosts. Additionally, iron acquisition is important for the infection process in fungi [59]. Fungi commonly use two high-affinity iron uptake mechanisms: (i) non-ribosomally synthesized secreted iron chelators (siderophores) and (ii) non-siderophore reductive iron assimilation (RIA) [60,61,62,63]. Fungal siderophores are synthesized by multi-domain NRPSs which are encoded by NRPS2 (its homolog SidC in Aspergillus fumigatus) and NRPS6 (its homolog SidD in Aspergillus fumigatus) [61]. On the other hand, the RIA involves three sequential processes, starting with the reduction of Fe3+ to Fe2+ by ferric reductase, followed by re-oxidation of Fe3+ by the ferroxidase, and finally the transport of the insoluble Fe3+ across the membrane by the high-affinity iron permease Ftr1 [62]. In this study, we identified ferrichrome-type NRPS, NRPS2 (UM880_724) and fusarinine-type NRPS, NRPS6 (UM226_3398) in N. dimidiatum UM 880 and B. papendorfii UM 226, respectively. This finding revealed that N. dimidiatum UM 880 employs the intracellular siderophore, ferricrocin, but B. papendorfii UM 226 uses the extracellular siderophore, fusarinine C for iron acquisition. NRPS6 is a virulence factor for Fusarium graminearum, Cochliobolus heterostrophus, C. miyabeanus, and A. brassicicola [61]. Knockdown of the gene in these fungi caused a significant reduction in virulence and hypersensitive to oxidative stress [61, 64]. Here, we showed that UM226_3398 is a pathogenicity-associated gene predicted by the pathogen-host interaction database (PHI-base). However, ferricrocin plays no role in virulence of C. heterostrophus, but is required for sexual development [64]. In addition, two genes encoding L-ornithine-N5-monooxygeneses were found in N. dimidiatum UM 880 (UM880_818 and UM880_8078) and B. papendorfii UM 226 (UM226_2340 and UM226_3401). The L-ornithine-N5-monooxygenase catalyzes the committed step for the biosynthesis fusarinine C and ferricrocin. In this study, a total of seven putative siderophore transporter-encoding gene orthologs to SIT1, MIRB, and MIRC were also discovered in N. dimidiatum UM 880, while one putative siderophore transporter similar to MIRC was identified in B. papendorfii UM 226 (Table S3). In A. fumigatus, utilization of ferricrocin required siderophore transporters Sit1 and Sit2, and both MirB and MirC have been implicated to be important for the transportation of siderophores [65].
Most fungi employ more than one iron-uptake mechanism in iron homoeostasis. Aspergillus fumigatus employs siderophore and RIA mechanism to acquire iron [63]. Our findings showed that both N. dimidiatum UM 880 and B. papendorfii UM 226 harbor siderophore and siderophore transporters to acquire iron, and they have all essential gene-encoding enzymes involved in the three-step RIA process. In N. dimidiatum UM 880, we found a metalloreductase Fre1p (UM880_12166), four ferroxidases Fet3 (UM880_431, UM880_937, UM880_2901, and UM880_11949), and two iron permease Ftr1 (UM880_2900 and UM880_432), whereas a metalloreductase Fre8 (UM226_5148), two ferroxidases Fet3 (UM226_875 and UM226_5495), and an iron permease Ftr1 (UM226_876) were identified in B. papendorfii UM 226. As previously reported [66, 67], ferroxidase and iron permease encoding gene clusters were determined in both N. dimidiatum UM 880 and B. papendorfii UM 226. In N. dimidiatum UM 880 and B. papendorfii UM 226, the iron permeases Ftr1 were located adjacent to ferroxidases Fet3.
Genomes of both fungi contain genes involved in hemicellulose and pectin decomposition
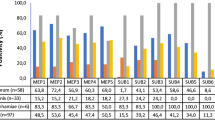
The plant cell wall is composed of polysaccharides cellulose, hemicellulose (composed of xyloglucans, xylans, mannans, glucomannans, and beta-glucan), and pectin (composed of galacturonan and rhamnogalacturonan). The capabilities to breakdown plant cell wall carbohydrates for nutrient sources or facilitate infection via host–pathogen interactions are important features of fungal lifestyle. Fungi can produce various carbohydrate-active enzymes (CAZymes) to degrade complex plant polysaccharide materials to simple monomers for carbon sources [38]. The abundance of fungal CAZymes demonstrates a remarkable functional diversity and fungal lifestyle due to the vast structural and functional diversity of the complex network of different plant cell wall polysaccharides. The N. dimidiatum UM 880 genome was mapped to the CAZy database to identify the presence of CAZyme-coding homologous genes. The CAZyme content in the genome of N. dimidiatum UM 880 was compared to B. papendorfii UM 226 and other fungi from different nutritional modes or infection mechanisms to determine their digestive potential against various plant cell wall polysaccharides. The lifestyle of N. dimidiatum and B. papendorfii was determined by comparing their CAZyme content and distribution to saprophytic fungi (Neurospora crassa OR74A and Trichoderma reesei QM6a), facultative parasitic fungi (Aspergillus nidulans FGSC A4), biotrophic fungi (Cladosporium flavum, Ustilago maydis, and Blumeria graminis), necrotrophic fungi (Cochliobolus heterostrophus C4 and C. heterostrophus C5), hemi-biotrophic fungi (Cochliobolus sativus ND90Pr, Magnaporthe oryzae 70–15 version 8, and Fusarium graminearum PH-1), and symbiotic fungus (Laccaria bicolor). The N. dimidiatum UM 880 genome contains 669 gene-encoding putative CAZymes, comprising 68 carbohydrate binding module (CBM), 162 carbohydrate esterases (CE), 305 glycoside hydrolases (GH), 104 glycosyl transferases (GT), and 30 polysaccharide lyases (PL) (Table S8). At the first glance, the CAZyme contents in N. dimidiatum UM 880 and B. papendorfii UM 226 genomes are larger than saprotrophs, biotrophs, and symbiotic fungi but comparable to other plant pathogens (necrotrophs and hemibiotrophs) (Fig. 4A and Table S8). In general, plant pathogenic fungi contain more CAZymes than saprotrophic, biotrophic, and symbiotic fungi [37]. The high number of CAZymes in N. dimidiatum UM 880 and B. papendorfii UM 226 genomes suggests their capability in degrading plant carbohydrates to access internal plant tissues and causing infection in plants.

CAZyme class annotation distribution of N. dimidiatum UM 880 and B. papendorfii UM 226 genomes. A Comparison of the distribution of CAZyme catalytic domains between N. dimidiatum UM 880, B. papendorfii UM 226, and fungi from various lifestyles. CBM, carbohydrate binding module; CE, carbohydrate esterase; GH, glycoside hydrolase; GT, glycosyltransferase; PL, polysaccharide lyase. B Comparison of the plant cell wall degrading potential from CAZyme analysis between N. dimidiatum UM 880, B. papendorfii UM 226 and fungi from various lifestyles
CAZyme catalytic domains of CE, GH, and PL are well known as cell wall degrading enzymes [68]. The genomes of N. dimidiatum UM 880 and B. papendorfii UM 226 contain 196 and 186 CAZymes unambiguously associated with the degradation of plant cell wall polysaccharides cellulose, hemicellulose, pectin, as well as hemicellulose or pectin side chains (Table S9). The number of plant cell wall–degrading enzymes in N. dimidiatum UM 880 and B. papendorfii UM 226 genomes was not far off to plant pathogenic fungi but higher than in the other non-pathogenic fungi analyzed except for C. fulvum (Fig. 4B). As previously noted [38], C. fulvum differs from other biotrophs, which contains more plant cell wall–degrading enzyme families of GH28, GH31, GH35, GH43, CE5, and CE16 (Table S9). Besides the quantitative similarity, the N. dimidiatum UM 880 shows qualitative similarity to the B. papendorfii UM 226, especially, the cellulolytic and hemicellulolytic capacities as well as the cleavage activity of hemicellulolytic and pectin side chains (Fig. 4B and Table S5). The number of putative cellulose–degrading enzymes encoded in N. dimidiatum UM 880 and B. papendorfii UM 226 is smaller than in plant cell wall–degrading pathogens (necrotrophs and hemibiotrophs), indicating that they do not extensively digest cellulose. However, their potential cellulose-degrading activity is comparable to that of saprotrophic, symbiotic, and facultative parasitic fungi (Table S9). In particular, biotrophic fungi have a low cellulose-degrading capacity as compared to that of N. dimidiatum UM 880 and B. papendorfii UM 226. The members of GH61 family are copper-dependent lytic polysaccharide monooxygenases that play important role to enhance lignocellulose degradation by catalyzing the oxidative cleavage of cellulose [69]. However, the GH61 was not identified in both N. dimidiatum UM 880 and B. papendorfii UM 226, whereas necrotrophic and hemibiotrophic fungi have an average of 22 genes (minimum 15, maximum 25). Biotrophic, saprophytic, and facultative parasitic fungi contain lower number of GH61 CAZymes. The hemicellulose degrading capacity is significantly larger for N. dimidiatum UM 880 and B. papendorfii UM 226 than in other plant cell wall–degrading fungi (Fig. 4B and Table S9). The unique feature of the N. dimidiatum UM 880 and B. papendorfii UM 226 is the presence of many CE1 proteins (31 and 42, respectively), whereas other analyzed fungi have an average of four genes (minimum 0, maximum 8) (Table S9). Both N. dimidiatum UM 880 and B. papendorfii UM 226 contain, respectively, two and one CE1 acetyl xylan esterase that directed against the plant cell wall xylan. The two xylanase families GH10 and GH11, however, are equivalent in size to other fungi, except for L. bicolor and B. graminis, which lack of any families GH10 and GH11. Neoscytalidium dimidiatum UM 880 contain a noticeably higher number of CAZymes putatively involved in pectin degradation as compared to the B. papendorfii UM 226 (64 vs. 38 putative pectin-specific CAZymes, respectively) and other fungi (Table S9). The pectin-degrading capacity for to B. papendorfii UM 226 is comparable to that of necrotrophic and hemibiotrophic fungi in the number of enzymes in all pectin-related CAZY families as well as the ratio of pectin-degrading lyases versus hydrolases, except for M. oryzae 70–15 version 8 (Table S9). Neoscytalidium dimidiatum UM 880 has higher number of polygalacturonases (family GH28), pectin methylesterases (family CE8), and pectate lyases (families PL1 and PL3) than in all the other plant cell wall–degrading fungi (Table S9), suggesting that the fungus has high capacity of pectin degradation. The observations also propose a common preference for soft plant tissues, such as fruits or flowers that are rich in pectin. Different plant cell wall–degrading fungi likely employ different methods to degrade plant tissues. In this work, the results suggest a preference of N. dimidiatum UM 880 and B. papendorfii UM 226 for hemicellulose and pectin rather than for cellulose. Although the cellulose-degrading capacity is similar between them, their ability to degrade soft tissue components, especially pectin, is much different. Even if it lacks experimental supports, the lower number of cellulose-degrading enzymes in both N. dimidiatum UM 880 and B. papendorfii UM 226 suggests that they employ another strategy to digest cellulose.
Secreted peptidases
Dermatophytes harbor the ability to secrete broad specturm of peptidases to degrade compact hard keratin to be used as nutritional sources, and the ability to degrade keratin is one of the most important pathogenic attributes of dermatophytes like Trichophyton rubrum and Microsporum canis [70]. The degradation process releases free amino acids from keratin (as source of nutrients) and ultimately leads to production of ammonia, which alters the host tissue to an alkaline pH, producing a host environment that is optimal for these proteolytic enzymes [70]. At this stage of knowledge, N. dimidiatum and B. papendorfii are not known as dermatophytes. However, the isolation of N. dimidiatum UM 880 and B. papendorfii UM 226 from the human skin lesions had raised our interest to search for genes encoding keratin-degradation proteases in these genomes. In line with this information, peptidase-encoding genes were classified in the N. dimidiatum UM 880 and B. papendorfii UM 226 genomes using the MEROPS database, with emphasis on the secreted peptidases (Table S10). Neoscytalidium dimidiatum UM 880 and B. papendorfii UM 226 genomes contain 199 and 153 peptidases, respectively. Of these, a total of 58 and 35 of the deduced peptidases contain a secretion signal in N. dimidiatum UM 880 and B. papendorfii UM 226 genomes, respectively. This preliminary data showed that N. dimidiatum UM 880 possesses greater metabolic capability for protein degradation as compared to B. papendorfii UM 226. Burmester et al. (2011) revealed that keratinolytic peptidases that belong to the families M14 (metallocarboxypeptidases), M28 (aminopeptidases), M35 (deuterolysin), M36 (fungalysin), S8 (subtilisin), and S10 (carboxypeptidases) have expanded in dermatophytes Arthroderma benhamiae and Trichophyton verrucossum [71]. In this study, collections of predicted secreted peptidases in M14, M28, M35, M36, S8, and S10 families of N. dimidiatum UM 880 and B. papendorfii UM 226 were compared to those A. benhamiae and T. verrucossum (Table 6). Neoscytalidium dimidiatum UM 880 contains lower number of peptidases families S8, S14, and M28, while B. papendorfii UM 226 contains lesser peptidases families S8 and S10 as compared to A. benhamiae and T. verrucossum. No deuterolysin was found in B. papendorfii UM 226. Additionally, fungalysin was not identified in both N. dimidiatum UM 880 and B. papendorfii UM 226.
The data was further analyzed by cross-checking with known keratinolytic peptidases in N. dimidiatum UM 880 and B. papendorfii UM 226 genomes based on literature review. Here, we managed to identify a total of potential 13 and 18 keratinolytic peptidases in N. dimidiatum UM 880 and B. papendorfii UM 226 genomes, respectively (Table 7). Of the secreted peptidases, one gene (UM880_9235) in N. dimidiatum UM 880 and two genes (UM226_975 and UM226_2139) in B. papendorfii UM 226 showed 46%, 51%, and 45% identity to T. rubrum metallopeptidase TruMCPA (ABW79919), respectively. Multiple sequence alignment was constructed and the conserved catalytic sites, zinc binding, substrate binding, and cysteine residues were identified (Fig. S3). Two genes in N. dimidiatum, UM880_7307 and UM880_4501, were identified as putative leucine aminopeptidase LAP1 and LAP2 with 49% and 46% identities to T. rubrum LAP1 and LAP2, respectively, whereas in B. papendorfii, a total of six putative aminopeptidases were determined, comprising two putative LAP1 (UM226_9084 and UM226_9684) and four putative LAP2 (UM226_5777, UM226_3516, UM226_7154, and UM226_10463). The hydrolytic activities of both LAP1 and LAP2 peptidases are the same [72]. The consensus active residues and binding sites of these predicted peptidases are the same as reported [72] (Figs. S4 and S5).
Additionally, the S9 family dipeptidyl peptidases (DPPs) were identified in both genomes. Both genomes contain a gene encoding DPPIV with 61% similarity to T. rubrum and A. fumigatus DPPIV in N. dimidiatum UM 880 (UM880_10172) and 56% and 57% identities similarity to T. rubrum and A. fumigatus DPPIV in B. papendorfii UM 226 (UM226_84). However, B. papendorfii UM 226 has two additional genes compared to N. dimidiatum UM 880 that encode DPPV. The consensus motif catalytic triad of the putative DPPIVs and DPPVs were identified in the multiple sequence alignments (Figs. S6 and S7).
We also identified three secreted peptidases each from N. dimidiatum UM 880 and B. papendorfii UM 226 that shared similarity to serine carboxypeptidases (S10) associated with keratin degradation in T. rubrum (TruSCPs) and its orthologs in A. fumigatus (Table 7). Most of the peptidases shared higher identity to the A. fumigatus cp1 and cp2 genes, except the gene UM226_5845 shared highest identity to TruSCPB (60% identity, AAS76666). In addition, a gene each from N. dimidiatum UM 880 (UM880_11056) and B. papendorfii UM 226 (UM226_4504) shared 67.67% and 67.11% identities, respectively, to T. rubrum SCPC [GenBank: AAS76668]. Neoscytalidium dimidiatum UM 880 and B. papendorfii UM 226 possess four and two putative genes, respectively with > 40% identity to dermatophyte subtilases and the ortholog of A. fumigatus ALP2. Of these, three N. dimidiatum UM 880 and two B. papendorfii UM 226 genes were shown to be similar to M. canis subtilisin 3 (McaSub3). McaSub3 is required for adherence of the fungus to the epidermal layer of host [73]. Although both fungi do not contain the full set of genes encoding subtilases, the potential of their keratinolytic activity shall not be neglected as it is suggested that the multiple keratinases work in concert to ensure successful pathogenesis [74].
Apart from causing superficial skin and nails infections, N. dimidiatum was reported to cause invasive infections [75, 76]. In N. dimidiatum UM 880, six secreted metallopeptidases similar to the Pleurotus ostreatus peptidase PoFE (M43.008) were identified. The conserved active residue and metal ligands were identified (Fig. S8). The PoFE carries fibrinolytic activity and were shown able to dissolve blood clots by degrading the fibrin polymers [77]. As the coagulation and deposition of fibrin resulted from inflammation are essential host defense mechanism against infectious agents [78], the fibrinolysis reaction by these peptidases might enable the invasion of the fungus in human.
Antifungal susceptibility profile and drug resistance genes
Given that the lack of established guidelines on MIC breakpoints for dematiaceous fungi, MIC of ≤ 1 µg/mL was used as an indicator of potential susceptibility for the tested drugs as previously noted [24]. James et al. (2016) showed that clinically isolated N. dimidiatum from Malaysia were susceptible to miconazole, clotrimazole, voriconazole, and amphotericin B [79]. In this study, N. dimidiatum UM 880 and B. papendorfii UM 226 were susceptible to posaconazole, voriconazole, fluconazole, ketoconazole, anidulafungin, caspofungin, and amphotericin B. Neoscytalidium dimidiatum UM 880 was resistant to itraconazole (MIC = 4 µg/mL), whereas B. papendorfii UM 226 was susceptible to itraconazole (MIC = 0.012 µg/mL) (Table 8). The molecular mechanisms of azole resistance have been well described in yeast and mold isolates. Potential resistance mechanisms include alterations in the target enzyme (lanosterol 14α demethylase) or overexpression of various efflux pumps [80,81,82]. Lanosterol 14α demethylase is encoded by the ERG11/CYP51 gene, that is targeted by azoles drugs to inhibit fungal ergosterol biosynthesis. Putative Cyp51A (UM880_6807, 69% identical to Aspergillus flavus) and ERG11 (UM880_1345, 66% identical to Leptosphaeria maculans) were identified in N. dimidiatum UM 880; a putative ERG11 (UM226_4997, 88% identical to Leptosphaeria maculans) weas identified in B. papendorfii UM 226. These three protein sequences were compared to the previously published ERG11 protein sequences from azole-susceptible Candida albicans isolates (GenBank accession no. XM_711668 and AIX03623). Common ERG11 mutations (A114S, Y132F, K143R, F145L, S405F, D446E, G448E, G450E, and G464S) that are associated with azole resistance [80, 81] were not observed in UM880_6807, UM880_1345, and UM226_4997. A nonsynonymous CYP51A mutation (M172V) that was observed in Aspergillus fumigatus [82] was identified in N. dimidiatum UM880_6807 and UM880_1345 but not in B. papendorfii UM226_4997 (Fig. S9). Furthermore, the UM880_6807 and UM880_1345 contain a mutation at the codon 255 (D255K) which was not identified in UM226_4997 as well as in azole-susceptible C. albicans [83, 84] and A. fumigatus [82].
Overexpression of major facilitator superfamily (MFS) transporter encoded by MDR1 (multidrug resistance) and ATP-binding cassette (ABC) transporters encoded by CDR1 and CDR2 (Candida drug resistance) are responsible for azoles resistance development [85, 86]. Neoscytalidium dimidiatum UM 880 harbors three MDR1 (UM880_2808, UM880_8079, and UM880_9846) and a CDR1 (UM880_4210). As previously noted [87, 88], UM880_2808, UM880_8079, and UM880_9846 were predicted to contain 12 putative transmembrane domains by TMpred (Fig. S10), suggesting they harbor transmembrane transporter activity. The Ndt80p transcription factor plays a key role in modulating azole tolerance by regulating ERG11 gene expression [88]. The binding of Ndt80p to the promoters of ERG biosynthetic genes is essential to activate their transcription in response to azole drugs [89]. Neoscytalidium dimidiatum UM 880 contains a putative Ndt80 transcription factor (UM880_7612, 87% identical to Neofusicoccum parvum UCRNP2). The 500 bp upstream regions of the UM880_6807, UM880_1345, and UM880_4210 genes were analyzed in silico. The putative promoter sequence was analyzed for potential NDT80 transcription factor binding sites using JASPAR core fungi database [90]. Among them, only UM880_1345 gene contains a putative Ndt80 binding consensus located between − 70 and − 90 (data not shown). Neither the MDR1, CDR1, nor NDT80 transcription factor was found in the B. papendorfii UM 226 genome.
Apart from ABC transporters CDR1 and CDR2, CDR4 has been reported to be involved in azole resistance in filamentous fungi [91]. Knockout of the CDR4 gene increased the susceptibility of Neurospora crassa to azole drugs, including ketoconazole, fluconazole, and itraconazole [91]. We identified a putative CDR4 (UM880_8630, 85% identical to N. parvum UCRNP2) in N. dimidiatum UM 880 but not in B. papendorfii UM 226 genome. Taken together, the ERG11, MDR1, CDR1, CDR4, and the NDT80 transcription factors may function in concert to confer high itraconazole resistance in the N. dimidiatum UM 880.
Comparative nutritional profiling of N. dimidiatum UM 880 and B. papendorfii UM 226
Phenotype microarray (PM) analysis was used to obtain a global understanding of the nutritional metabolism of N. dimidiatum UM 880 and B. papendorfii UM 226. A total of 379 different compounds were tested using PM plates 1 to 8, including 190 different carbon sources, 95 nitrogen sources, 59 phosphorus sources, 35 sulfur sources, 94 nutrient supplements, and 282 peptide nitrogen sources. Phenotypic differences based on their ability to metabolize carbon, nitrogen, phosphorous, sulfur, nutrient supplements, as well as peptide nitrogen sources were noted.
By KEGG annotation analysis, the genes predicted to be involved in central metabolic pathways for carbohydrates utilization were more abundant in B. papendorfii UM 226 compared to N. dimidiatum UM 880 (Fig. 2B). In agreement with the KEGG data (Fig. 2B), B. papendorfii UM 226 (62 carbon sources) was metabolically active on carbon source as compared to N. dimidiatum UM 880 (19 carbon sources) (Table S11). PM analysis showed that B. papendorfii UM 226 is capable of using simple sugars and complex carbohydrates as carbon sources for growth. Bipolaris papendorfii UM 226 can utilize monosaccharides (α-D-glucose, D-fructose, D-Psicose, D-ribose, L-rhamnose, D-xylose), disaccharides (maltose, sucrose, D-trehalose), trisaccharide (raffinose), tetrasaccharide (stachyose), synthetic sugar (lactulose), as well as polysaccharides (mannan, pectin). In addition, B. papendorfii UM 226 could utilize a wide range of sugar alcohol (adonitol, dulcitol, i-erythritol, D-arabitol, D-mannitol, maltitol, xylitol) and carboxylic acids (D-tartaric acid, δ-amino valeric acid, γ-hydroxybutyric acid, β-hydroxybutyric acid, α-keto valeric acid, γ-amino butyric acid), which could not be utilized by N. dimidiatum UM 880. Based on KEGG analysis, genes involved in glycolysis, gluconeogenesis, pentose phosphate pathway, fructose, mannose, sucrose, galactose, and starch metabolism were annotated in both N. dimidiatum UM 880 and B. papendorfii UM 226 although no evidence from PM analysis to support N. dimidiatum UM 880 activities on these carbon substrates (Table S12). Overall, B. papendorfii UM 226 has a greater ability to utilize a wide array of carbon substrates.
PM data revealed markedly differences in nitrogen metabolism when N. dimidiatum UM 880 and B. papendorfii UM 226 were compared (Table S11) although a comparable number of genes predicted for nitrogen metabolism was annotated in N. dimidiatum UM 880 and B. papendorfii UM 226 genomes (29 and 31 genes, respectively) (Table S13). Neoscytalidium dimidiatum UM 880 was able to utilize 56 out of 95 nitrogen sources, while B. papendorfii UM 226 could utilize only eight of the tested nitrogen substrates. The nitrogen sources selective for B. papendorfii UM 226 growth were organic nitrogen (ammonia), uric acid, L-amino acid (L-proline, L-alanine, and L-glutamine), D-amino acid (D-valine), and dipeptides (Ala-Asp and Gly-Gln). Interestingly, we found that N. dimidiatum UM 880 genome contains a nitrate transport protein (UM880_1228) and a nitrite reductase protein (UM880_5592), which are found to be absent in B. papendorfii UM 226. Furthermore, it should be noted that no phosphorus, sulfur, and nutritional supplements was utilized by B. papendorfii UM 226 (Table S11). On the other hand, N. dimidiatum UM 880 was found able to utilize 14 phosphorus, sulfur, as well as a nutritional supplement. In addition, N. dimidiatum UM 880 showed the greater ability to metabolize peptide nitrogen substrates (183 peptide nitrogen substrates) as compared to the B. papendorfii UM 226 (117 peptide nitrogen substrates). Taken together, we speculated that the reduced utilization of carbon sources could be compensated by the utilization of nitrogen, phosphorus, sulfur, nutritional supplement, and peptide nitrogen. Although N. dimidiatum UM 880 and B. papendorfii UM 226 contain a comparable number and distribution of CAZymes, their nutrient utilization profile varies significantly. PM data revealed that N. dimidiatum UM 880 could utilize a diverse array of substrates, which further demonstrate its metabolic versatility. Thus, it is not surprising that N. dimidiatum has a broad geographical and host range [7]. In contrast, B. papendorfii UM 226 utilizes limited sources of nutrients as compared to that of N. dimidiatum UM 880.
Growth of N. dimidiatum UM 880 and B. papendorfii UM 226 in response to environmental factors
Fungi are commonly exposed to broadly fluctuating environmental conditions where adaptation is crucial for their survival. In particular, an ability to respond to a wide pH range allows them to cope with rapid changes in their extracellular settings. In fact, pH is also one of the major environmental factors affecting pathogenicity. Therefore, the growths of B. papendorfii UM 226 and N. dimidiatum UM 880 at various pH were analyzed in this study. Our results revealed that B. papendorfii UM 226 grew better compared to N. dimidiatum UM 880 at all pH range. B. papendorfii UM 226 grew well at a wide range of pH ranging from strongly acidic to alkaline (pH 3.5 to 10), with an optimum growth between pH 4 and 8. On the other hand, the maximum growth of N. dimidiatum UM 880 was at pH 4.5–5.5. Lambers et al. (2006) has demonstrated that the pH of natural skin surface is at an average of 4.7 [92]. The ability of both B. papendorfii UM 226 and N. dimidiatum UM 880 to grow at this pH (~ pH 4.7) may account for their high occurrence in dermal surface and hence causing dermatitis disorder. In addition, these fungal especially B. papendorfii UM 226 can most likely continue to grow on the skin surface even after application of cosmetic product or soap since they can adapt to adverse alkaline pH condition (Fig. 5).

Mycelial growth of B. papendorfii UM 226 and N. dimidiatum UM 880 at pH 3.5–10. Results are expressed as mean ± standard error; n = 2. The means of B. papendorfii UM 226 and N. dimidiatum UM 880 were significantly different (p < 0.05; independent T-test) at each pH
Noting the versatility of these strains at wide pH range, we also evaluated the effect of various supplementations on the fungal growth at both acidic and alkaline conditions. Our results indicated that the supplementation of nitrogen sources did not affect the growth of N. dimidiatum UM 880 at alkaline condition (Fig. 6). Interestingly, supplementations with L-histidine, L-phenylalanine, and L-tryptophan are shown to exhibit additional inhibitory effect for B. papendorfii UM 226 at alkaline condition (pH 9.5). On the other hand, supplementation of anthranilic acid is shown to inhibit the growth of B. papendorfii UM 226 and N. dimidiatum UM 880 at pH 4.5 (a pH close to natural skin surface condition) as their growths in media supplemented with anthranilic acid were significantly (P < 0.05) lower (60–92% lower) compared to the control (Fig. 7). Other nitrogenous sources evaluated in this study had very limited effect on the growth of both strains. This may suggest that supplementation of anthranilic acid L-histidine and L-tryptophan on cosmetic agents such as alkaline soap or topical creams may likely inhibit the growth of B. papendorfii UM 226 and N. dimidiatum UM 880 on skin surface, hence, potentially reducing the risk for dermatitis disease due to these fungi.

Mycelial growth of B. papendorfii UM 226 and N. dimidiatum UM 880 in media (pH 9.5) containing various nitrogenous sources. Results are expressed as mean from two independent runs. Control represents media (pH 9.5) without supplementation. ab means for B. papendorfii UM 226 with different lowercase letters are significantly different (p < 0.05; ANOVA with Tukey’s post hoc test). A means for N. dimidiatum UM 880 are not significantly different (p < 0.05; ANOVA with Tukey’s post hoc test)

Mycelial growth of B. papendorfii UM 226 and N. dimidiatum UM 880 in media (pH 4.5) containing various nitrogenous sources. Results are expressed as mean from two independent runs. Control represent media (pH 4.5) without supplementation. abcdefgh means for B. papendorfii UM 226 with different lowercase letters are significantly different (p < 0.05; ANOVA with Tukey’s post hoc test). AB means for N. dimidiatum UM 880 with different uppercase letters are significantly different (p < 0.05; ANOVA with Tukey’s post hoc test)
The growth of B. papendorfii UM 226 in saline condition was concentration dependent and generally showed a better salt tolerance as compared to N. dimidiatum UM 880 (Fig. 8). Although there was a gradual decrease in the growth of B. papendorfii UM 226 as the concentration of NaCl increases, the mycelial growth of B. papendorfii UM 226 remained high (above OD = 1) even at 9% NaCl. Unlike B. papendorfii UM 226, low concentration of NaCl (2–3% NaCl) promoted the growth of N. dimidiatum UM 880 and the growth starts to decline as the concentration exceed 4% NaCl. In order to increase the ability to grow at high-salinity condition, microorganism would require a special adaptation to maintain the osmotic equilibrium across the cell membrane. Cells of almost all organisms commonly utilize organic-osmolyte mechanism when exposed to hyperosmolality and high salt or solute condition [91]. Therefore, we also screened for the potential osmolytes by including a range of compounds in the solution containing 6% NaCl. Unlike most microorganism which required betaine or glycerol to restore their osmotolerance properties [93], our data showed that both glutamic acid and proline provided a better osmoprotectant effect for both strains of fungal especially B. papendorfii UM 226 as compared to betaine (Fig. 9). On the other hand, dimethyl sulfonyl propionate was shown to exert additional inhibitory effect for both strains when cultured in 6% NaCl condition (Fig. 9). This data would be important to provide a fundamental knowledge in developing potential drug targets for antifungal strategies for both these strains.

Mycelial growth of B. papendorfii UM 226 and N. dimidiatum UM 880 in solution containing various concentration of NaCl. Results are expressed as mean ± standard error; n = 2. The means of B. papendorfii UM 226 and N. dimidiatum UM 880 were significantly different (p < 0.05; independent T-test) at each concentration of NaCl

Mycelial growth of B. papendorfii UM 226 () and N. dimidiatum UM 880 () in media containing 6% NaCl and supplemented with various osmolytes. Results are expressed as mean from two independent runs. Control represent media containing 6% NaCl only. abcd means for B. papendorfii UM 226 with different lowercase letters are significantly different (p < 0.05; ANOVA with Tukey’s post hoc test). A means for N. dimidiatum UM 880 are not significantly different (p > 0.05; ANOVA with Tukey’s post hoc test)
In addition to NaCl, this study also evaluated the effect of various other salt including sodium lactate, sodium formate, sodium nitrate, and sodium nitrite. Bipolaris papendorfii UM 226 and N. dimidiatum UM 880 showed a concentration-dependent growth in all the salt media (Table S14). Similar observation was demonstrated in media containing urea where a gradual decrease of mycelial was observed in both strain as the concentration of urea increases. It is critical to understand the growth pattern of the fungi in urea because urea has been commonly used in treatment of dermatological diseases and as potent dermal emollient [94]. In general, B. papendorfii UM 226 was shown to exhibit better (p < 0.05) mycelial growth compared to N. dimidiatum UM 880 in media containing urea. Our results suggest that increasing the concentration of urea may decrease the mycelial growth. The mycelial growth for both strains drastically decreased by 88–91% in media containing 7% urea compared to that of containing 2%. The OD750 for both strains were almost negligible in media containing 7% urea (Fig. 10). Previous study has demonstrated that Neoscytalidium infections, even always superficial, are often difficult to treat [95] while information for treatment of B. papendorfii in skin remains limited. Therefore, our data is important to provide a fundamental information for clinical management of these fungi.

Mycelial growth of B. papendorfii UM 226 and N. dimidiatum UM 880 in solution containing various concentration of urea. Results are expressed as mean ± standard error; n = 2. The means of B. papendorfii UM 226 and N. dimidiatum UM 880 were significantly different (p < 0.05; independent T-test) at 2–6% of urea. The means for both strains were not significantly different (p > 0.05) at 7% of urea
Conclusion
In this study, we take an organized approach to expand our knowledge of the genomic, genetic, as well as metabolic profiles of two plant pathogens that were isolated from humans. Comparative genomics of N. dimidiatum and B. papendorfii revealed several striking differences in genome size, GC content, gene content, and transposons. The genome sequences further demonstrated a remarkable difference in the amount and types of putative secondary metabolites. Unlike N. dimidiatum, B. papendorfii contain a gene encoding dimethylallyl tryptophan synthase (DMATS). The putative 6-methylsalicylic-acid synthase gene was unique to N. dimidiatum. The biological function of most of this predicted gene remains to be determined to gain insight whether it contributes to the biosynthesize of various antibiotics. A survey of the CAZYmes in the N. dimidiatum and B. papendorfii with other fungi from different lifestyles showed that they have similar mode of infection in plants. The distribution of CAZyme-encoding genes demonstrated a preference of N. dimidiatum and B. papendorfii for hemicellulose and pectin as nutrient sources. Additionally, rare human diseases caused by these fungi may be associated with the presence of several putative keratinolytic peptidases. In particular, we also identified several genes associated with azole resistance of N. dimidiatum, which were missed in the B. papendorfii genome. Our PM analysis proved that N. dimidiatum could utilize a diverse array of nutrient sources particularly noticeable in the case of nitrogen, phosphorus, and sulfur sources as compared to B. papendorfii that mainly utilizes carbon as nutrient source. The most significant finding in the data was that B. papendorfii is a salt-tolerant mold, which can adapt to the wide pH range, suggesting it can tolerate and survive in hostile and diverse environments. However, substrate utilization observed under different growth conditions is still unknown. Therefore, future study on the substrate utilization under diverse growth conditions is vital to gain better awareness of their lifestyles and subsequently their infection model.
Data availability
The data that support the results of this study are accessible via GenBank assembly accession: GCA_900092665.1. The genome of B. papendorfii UM 226 is accessible via GenBank assembly accession: GCA_000817285.1.
References
Tan DH, Sigler L, Gibas CF, Fong IW (2008) Disseminated fungal infection in a renal transplant recipient involving Macrophomina phaseolina and Scytalidium dimidiatum: case report and review of taxonomic changes among medically important members of the Botryosphaeriaceae. Med Mycol 46:285–292
Crous PW, Slippers B, Wingfield MJ et al (2006) Phylogenetic lineages in the Botryosphaeriaceae. Stud Mycol 55:235–253
Phillips AJ, Alves A, Abdollahzadeh J et al (2013) The Botryosphaeriaceae: genera and species known from culture. Stud Mycol 76:51–167
Machado AR, Pinho DB, Dutra DC, Pereira OL (2012) First report of collar and root rot of physic nut (Jatropha curcas) caused by Neoscytalidium dimidiatum in Brazil. Plant Dis 96:1697
Lan GB, He ZF (2012) First report of brown spot disease caused by Neoscytalidium dimidiatum on Hylocereus undatus in Guangdong. Chinese Mainland Plant Dis 96:1702
Polizzi G, Aiello D, Vitale A (2009) First report of shoot blight, canker, and gummosis caused by Neoscytalidium dimidiatum on Citrus in Italy. Plant Dis 93:1215
Ray JD, Burgess T, Lanoiselet VM (2010) First record of Neoscytalidium dimidiatum and N. novaehollandiae on Mangifera indica and N. dimidiatum on Ficus carica in Australia. Australas Plant Dis Notes 5:48–50
Abo Rehab ME, Rashed MF, Ammar MI, El-Morsy SA (2014) Dieback and sooty canker of Ficus trees in Egypt and its control. Pak J Biol Sci 17:364–371
Hassan WA, Pasha AA, Mohammad MB (2009) Sooty canker on some thin bark trees caused by Nattrassia mangiferae. Egypt J Agric Res 87:443–456
Sadowsky A, Solel Z, Sztejnberg A (2007) Effect of heat-stress predisposition on the development of Scytalidium wilt of Star Ruby’ grapefruit, caused by Scytalidium lignicola. Eur J Plant Pathol 117:123–127
Hassan WA, Haleem RA, Hassan PH (2011) Effect of heat-stress predisposition on the development of sooty canker caused by Neoscytalidium dimidiatum (Penz.) Crous and Slippers. Acta Agrobot 64:207–212
Elewski BE (1996) Onychomycosis caused by Scytalidium dimidiatum. J Am Acad Dermatol 35:336–338
Lacaz CS, Pereira AD, Heins-Vaccari EM et al (1999) Onychomycosis caused by Scytalidium dimidiatum. Report of two cases. Review of the taxonomy of the synanamorph and anamorph forms of this coelomycete. Rev Inst Med Trop Sao Paulo 41:319–323
Arrese JE, Pierard-Franchimont C, Pierard GE (2001) Scytalidium dimidiatum melanonychia and scaly plantar skin in four patients from the Maghreb: imported disease or outbreak in a Belgian mosque? Dermatology 202:183–185
Hay RJ (2002) Scytalidium infections. Curr Opin Infect Dis 15:99–100
Rockett MS, Gentile SC, Zygmunt KH, Gudas CJ (1996) Subcutaneous phaeohypomycosis caused by Scytalidium dimidiatum in the foot of an immunosuppressed host. J Foot Ankle Surg 35:350–354
Drouhet E, Dupont B (1983) Laboratory and clinical assessment of ketoconazole in deep-seated mycoses. Am J Med 74:30–47
Geramishoar M, Zomorodian K, Zaini F et al (2004) First case of cerebral phaeohyphomycosis caused by Nattrassia mangiferae in Iran. Jpn J Infect Dis 57:285–286
Dunn JJ, Wolfe MJ, Trachtenberg J, Kriesel JD, Orlandi RR, Carroll KC (2003) Invasive fungal sinusitis caused by Scytalidium dimidiatum in a lung transplant recipient. J Clin Microbiol 41:5817–5819
Al-Rajhi AA, Awad AH, Al-Hedaithy SS, Forster RK, Caldwell KC (1993) Scytalidium dimidiatum fungal endophthalmitis. Br J Ophthalmol 77:388–390
Li PP, Cao ZY (2013) First report of Bipolaris papendorfii causing corn leaf spot in China. Plant Dis 97:1506
Valencia-Botín AJ, Kokubu H, Ruiz DR (2013) A brief overview on pitahaya (Hylocereus spp.) diseases. Australas Plant Pathol 15:437–440
Masratul HM, Baharuddin S, Latiffah Z (2013) Identification and molecular characterizations of Neoscytalidium dimidiatum causing stem canker of red-fleshed dragon fruit (Hylocereus polyrhizus) in Malaysia. J Phytopathol 161:841–849
Yew SM, Chan CL, Lee KW et al (2014) A five-year survey of dematiaceous fungi in a tropical hospital reveals potential opportunistic species. PLoS ONE 9:e104352
Kuan CS, Yew SM, Toh YF et al (2015) Dissecting the fungal biology of Bipolaris papendorfii: from phylogenetic to comparative genomic analysis. DNA Res 22:219–232
Moretti S, Armougom F, Wallace IM, Higgins DG, Jongeneel CV, Notredame C (2007) The M-Coffee web server: a meta-method for computing multiple sequence alignments by combining alternative alignment methods. Nucleic Acids Res 35:W645-648
Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829
Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W (2011) Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27:578–579
Boetzer M, Pirovano W (2012) Toward almost closed genomes with GapFiller. Genome Biol 13:R56
Nadalin F, Vezzi F, Policriti A (2012) GapFiller: a de novo assembly approach to fill the gap within paired reads. BMC Bioinformatics 13:S8
Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW (2007) RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35:3100–3108
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964
Lomsadze A, Ter-Hovhannisyan V, Chernoff YO, Borodovsky M (2005) Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res 33:6494–6506
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676
Tatusov RL, Fedorova ND, Jackson JD et al (2003) The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41
Quevillon E, Silventoinen V, Pillai S et al (2005) InterProScan: protein domains identifier. Nucleic Acids Res 33:W116-120
Ohm RA, Feau N, Henrissat B et al (2012) Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes fungi. PLoS Pathog 8:e1003037
Zhao Z, Liu H, Wang C, Xu JR (2014) Correction: comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genomics 15:6
Rawlings ND, Barrett AJ, Bateman A (2012) MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res 40:D343-350
Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786
Krogh A, Larsson B, von Heijne G, Sonnhammer EL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580
Khaldi N, Seifuddin FT, Turner G et al (2010) SMURF: genomic mapping of fungal secondary metabolite clusters. Fungal Genet Biol 47:736–741
Sigler L, Summerbell RC, Poole L et al (1997) Invasive Nattrassia mangiferae infections: case report, literature review, and therapeutic and taxonomic appraisal. J Clin Microbiol 35:433–440
Daboussi MJ (1997) Fungal transposable elements and genome evolution. Genetica 100:253–260
Teixeira MM, de Almeida LG, Kubitschek-Barreira P et al (2014) Comparative genomics of the major fungal agents of human and animal Sporotrichosis: Sporothrix schenckii and Sporothrix brasiliensis. BMC Genomics 15:943
Santana MF, Silva JC, Mizubuti ES et al (2014) Characterization and potential evolutionary impact of transposable elements in the genome of Cochliobolus heterostrophus. BMC Genomics 15:536
Brillet B, Bigot Y, Auge-Gouillou C (2007) Assembly of the Tc1 and mariner transposition initiation complexes depends on the origins of their transposase DNA binding domains. Genetica 130:105–120
Galperin MY, Koonin EV (2004) ‘Conserved hypothetical’ proteins: prioritization of targets for experimental study. Nucleic Acids Res 32:5452–5463
Leach MD, Brown AJ (2012) Posttranslational modifications of proteins in the pathobiology of medically relevant fungi. Eukaryot Cell 11:98–108
Bushley KE, Turgeon BG (2010) Phylogenomics reveals subfamilies of fungal nonribosomal peptide synthetases and their evolutionary relationships. BMC Evol Biol 10:26
Kroken S, Glass NL, Taylor JW, Yoder OC, Turgeon BG (2003) Phylogenomic analysis of type I polyketide synthase genes in pathogenic and saprobic ascomycetes. Proc Natl Acad Sci U S A 100:15670–15675
Yang G, Rose MS, Turgeon BG, Yoder OC (1996) A polyketide synthase is required for fungal virulence and production of the polyketide T-toxin. Plant Cell 8:2139–2150
Panaccione DG, Scott-Craig JS, Pocard JA, Walton JD (1992) A cyclic peptide synthetase gene required for pathogenicity of the fungus Cochliobolus carbonum on maize. Proc Natl Acad Sci U S A 89:6590–6594
Jahn B, Boukhallouk F, Lotz J, Langfelder K, Wanner G, Brakhage AA (2000) Interaction of human phagocytes with pigmentless Aspergillus conidia. Infect Immun 68:3736–3739
Eliahu N, Igbaria A, Rose MS, Horwitz BA, Lev S (2007) Melanin biosynthesis in the maize pathogen Cochliobolus heterostrophus depends on two mitogen-activated protein kinases, Chk1 and Mps1, and the transcription factor Cmr1. Eukaryot Cell 6:421–429
Wang Y, Wang J, Cheong YH, Hur JS (2014) Three new non-reducing polyketide synthase genes from the lichen-forming fungus Usnea longissima. Mycobiology 42:34–40
Moriwaki A, Kihara J, Kobayashi T, Tokunaga T, Arase S, Honda Y (2004) Insertional mutagenesis and characterization of a polyketide synthase gene (PKS1) required for melanin biosynthesis in Bipolaris oryzae. FEMS Microbiol Lett 238:1–8
Ding W, Lei C, He Q, Zhang Q, Bi Y, Liu W (1999) Insights into bacterial 6-methylsalicylic acid synthase and its engineering to orsellinic acid synthase for spirotetronate generation. Chem Biol 17:495–503
Weinberg ED (1999) The role of iron in protozoan and fungal infectious diseases. J Eukaryot Microbiol 46:231–238
Schrettl M, Bignell E, Kragl C et al (2004) Siderophore biosynthesis but not reductive iron assimilation is essential for Aspergillus fumigatus virulence. J Exp Med 200:1213–1219
Oide S, Moeder W, Krasnoff S et al (2006) NPS6, encoding a nonribosomal peptide synthetase involved in siderophore-mediated iron metabolism, is a conserved virulence determinant of plant pathogenic ascomycetes. Plant Cell 18:2836–2853
Condon BJ, Oide S, Gibson DM, Krasnoff SB, Turgeon BG (2014) Reductive iron assimilation and intracellular siderophores assist extracellular siderophore-driven iron homeostasis and virulence. Mol Plant Microbe Interact 27:793–808
Haas H, Eisendle M, Turgeon BG (2008) Siderophores in fungal physiology and virulence. Annu Rev Phytopathol 46:149–187
Condon BJ, Wu D, Kraševec N, Horwitz BA, Turgeon BG (2014) Comparative genomics of Cochliobolus phytopathogens. In: Dean R, Lichens-Park A, Kole C (eds) Genomics of plant-associated fungi: monocot pathogens. Springer, Berlin, Heidelberg, pp 41–67
Aguiar M, Orasch T, Misslinger M, Dietl AM, Gsaller F, Haas H (2021) The siderophore transporters Sit1 and Sit2 are essential for utilization of ferrichrome-, ferrioxamine- and coprogen-type siderophores in Aspergillus fumigatus. Journal of Fungi 7:768
Lian T, Simmer MI, D’Souza CA et al (2005) Iron-regulated transcription and capsule formation in the fungal pathogen Cryptococcus neoformans. Mol Microbiol 55:1452–1472
Eichhorn H, Lessing F, Winterberg B et al (2006) A ferroxidation/permeation iron uptake system is required for virulence in Ustilago maydis. Plant Cell 18:3332–3345
Ospina-Giraldo MD, Griffith JG, Laird EW, Mingora C (2010) The CAZyome of Phytophthora spp.: a comprehensive analysis of the gene complement coding for carbohydrate-active enzymes in species of the genus Phytophthora. BMC Genomics 11:525
Langston JA, Shaghasi T, Abbate E, Xu F, Vlasenko E, Sweeney MD (2011) Oxidoreductive cellulose depolymerization by the enzymes cellobiose dehydrogenase and glycoside hydrolase 61. Appl Environ Microbiol 77:7007–7015
Ciesielska A, Kawa A, Kanarek K, Soboń A, Szewczyk R (2021) Metabolomic analysis of Trichophyton rubrum and Microsporum canis during keratin degradation. Sci Rep 11:3959
Burmester A, Shelest E, Glockner G et al (2011) Comparative and functional genomics provide insights into the pathogenicity of dermatophytic fungi. Genome Biol 12:R7
Monod M, Lechenne B, Jousson O et al (2005) Aminopeptidases and dipeptidyl-peptidases secreted by the dermatophyte Trichophyton rubrum. Microbiology 151:145–155
Bagut ET, Baldo A, Mathy A et al (2012) Subtilisin Sub3 is involved in adherence of Microsporum canis to human and animal epidermis. Vet Microbiol 160:413–419
Shi Y, Niu Q, Yu X et al (2016) Assessment of the function of SUB6 in the pathogenic dermatophyte Trichophyton mentagrophytes. Med Mycol 54:59–71
Elinav H, Izhar U, Benenson S et al (2009) Invasive Scytalidium dimidiatum infection in an immunocompetent adult. J Clin Microbiol 47:1259–1263
Garinet S, Tourret J, Barete S et al (2015) Invasive cutaneous Neoscytalidium infections in renal transplant recipients: a series of five cases. BMC Infect Dis 15:535
Shen MH, Kim JS, Sapkota K et al (2007) Purification, characterization, and cloning of fibrinolytic metalloprotease from Pleurotus ostreatus mycelia. J Microbiol Biotechnol 17:1271–1283
Bergmann S, Hammerschmidt S (2007) Fibrinolysis and host response in bacterial infections. Thromb Haemost 98:512–520
James JE, Santhanam J, Lee MC et al (2017) In vitro antifungal susceptibility of neoscytalidium dimidiatum clinical isolates from Malaysia. Mycopathologia 182:305–313
White TC (1997) Increased mRNA levels of ERG16, CDR, and MDR1 correlate with increases in azole resistance in Candida albicans isolates from a patient infected with human immunodeficiency virus. Antimicrob Agents Chemother 41:1482–1487
Perea S (2000) Azole resistance in Candida albicans. Rev Esp Quimioter 13:314–317
Howard SJ, Cerar D, Anderson MJ et al (2009) Frequency and evolution of azole resistance in Aspergillus fumigatus associated with treatment failure. Emerg Infect Dis 15:1068–1076
Flowers SA, Colon B, Whaley SG, Schuler MA, Rogers PD (2015) Contribution of clinically derived mutations in ERG11 to azole resistance in Candida albicans. Antimicrob Agents Chemother 59:450–460
Xiang MJ, Liu JY, Ni PH et al (2013) Erg11 mutations associated with azole resistance in clinical isolates of Candida albicans. FEMS Yeast Res 13:386–393
Tsao S, Rahkhoodaee F, Raymond M (2009) Relative contributions of the Candida albicans ABC transporters Cdr1p and Cdr2p to clinical azole resistance. Antimicrob Agents Chemother 53:1344–1352
Anderson JB (2005) Evolution of antifungal-drug resistance: mechanisms and pathogen fitness. Nat Rev Microbiol 3:547–556
Law CJ, Maloney PC, Wang DN (2008) Ins and outs of major facilitator superfamily antiporters. Annu Rev Microbiol 62:289–305
Pao SS, Paulsen IT, Saier MH Jr (1998) Major facilitator superfamily. Microbiol Mol Biol Rev 62:1–34
Sellam A, Tebbji F, Nantel A (2009) Role of Ndt80p in sterol metabolism regulation and azole resistance in Candida albicans. Eukaryot Cell 8:1174–1183
Mathelier A, Zhao X, Zhang AW et al (2014) JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res 42:D142-147
Zhang Y, Zhang Z, Zhang X et al (2012) CDR4 is the major contributor to azole resistance among four Pdr5p-like ABC transporters in Neurospora crassa. Fungal Biol 116:848–854
Lambers H, Piessens S, Bloem A, Pronk H, Finkel P (2006) Natural skin surface pH is on average below 5, which is beneficial for its resident flora. Int J Cosmet Sci 28:359–370
Burg MB, Ferraris JD (2008) Intracellular organic osmolytes: function and regulation. J Biol Chem 283:7309–7313
Pan M, Heinecke G, Bernardo S, Tsui C, Levitt J (2013) Urea: a comprehensive review of the clinical literature. Dermatol Online J 19:20392
Khan ZU, Ahmad S, Joseph L, Chandy R (2009) Cutaneous phaeohyphomycosis due to Neoscytalidium dimidiatum: first case report from Kuwait. J Mycol Med 19:138–142
Funding
This work was supported by High Impact Research MOE Grant (UM.C/625/1/HIR/MOHE/MED/31, account no: H-20001–00-E000070) from the Ministry of Education Malaysia and partly by Putra Grant—Putra Graduate Initiative (IPS) from Universiti Putra Malaysia (GP-IPS/2018/9613300).
Author information
Authors and Affiliations
Contributions
Conceptualization: CSK, KPN, and LTLT; data curation: CSK, SMY, YCT, WYY, CCH, and SYC; funding acquisition: KPN and LTLT; investigation: CSK, SMY, HUM, SKY, JMJ, and RDV; methodology: CSK, SMY, JMJ, RDV, SLN, and SYC; project administration: CSK; resources: KPN and LTLT; software: YCT, WYY, and CCH; supervision: KPN and LTLT; validation: CSK; visualization: CSK; writing—original draft: CSK and SKY; writing—review and editing: KPN, SMY, HUM, HFS, KNH, SNM, SYC, and LTLT.
Corresponding author
Ethics declarations
Conflict of interest
The authors no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kuan, C.S., Ng, K.P., Yew, S.M. et al. Comparative genomic and phenotypic analyses of pathogenic fungi Neoscytalidium dimidiatum and Bipolaris papendorfii isolated from human skin scraping. Braz J Microbiol 54, 1351–1372 (2023). https://doi.org/10.1007/s42770-023-01032-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42770-023-01032-z