
Abstract
Epidermophyton floccosum is one of the most common agents of human superficial fungal infections, compared with genus Trichophyton and Microsporum, it possesses uniqueness in ecology traits and rarely causing hair infections. E. floccosum is so far the only representative species of genera Epidermophyton, and it is known as anthropophilic dermatophytes. To further reveal the genome sequences and clues of virulence factors, thus in this study, we sequenced the genome of E. floccosum (CGMCC (F) E1d), and performed comparative genomic analysis with other dermatophytes. It is revealed that E. floccosum owns the largest genome size and similar GC content compared with other dermatophytes. A total of 7565 genes are predicted. By comparing with the closest species N. gypseum, our study reveals that number and structure of adhesion factors, secreted proteases and LysM domain might contribute to the pathogenic and ecological traits of E. floccosum. Mating genes is also detected in genome data. Furthermore, we performed AFLP analysis trying to discuss intraspecific differences of E. floccosum, but no significant relationship is found between genotype and geographical distribution. Upon above, our study provides a deeper understanding and strong foundation for future researches about E. floccosum.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dermatophytes cause the most common fungal infections in humans. Among the seven genera (Trichophyton, Microsporum, Epidermophyton, Lophophyton, Paraphyton, Nannizzia and Arthroderma) [1], Trichophyton, Microsporum and Epidermophyton species are the three most common causative agents in clinical trials. The genus Epidermophyton was first reported in 1870 and named as Acrothecium floccosum [2]. In earlier studies, genus Epidermophyton was composed of two species, E. floccosum and E. stockdaleae, for the latter was geophilic and rarely caused human infections, and they also differed in antifungal susceptibilities and temperature tests [3, 4]. While based on the newly taxonomy of dermatophytes, E. floccosum is now the only representative species in the genus Epidermophyton [1]. E. floccosum, consistent with other dermatophytes, can cause superficial infections like tinea corporis and onychomycosis which is more common in tropical and subtropical areas like Iran [5] and Africa [6]. Severe disseminated infections were also reported as well [7, 8]. But it is widely acknowledged that E. floccosum rarely causes hair infections, the reason behind is still unknown.
Recently, with widely applications of next and third generation sequencing platforms, many pathogenic fungi have been sequenced as a consequence. While for dermatophytes, these tools have not been fully applied yet, and might be one of the reason why the pathogenic pattern still not so clear [9]. Whole genome sequences were first obtained in Arthroderma benhamiae and Trichophyton verrucosum [10], and some clinical important dermatophytes like Trichophyton rubrum, Trichophyton tonsurans, Trichophyton equinum, Microsporum canis, Nannizzia gypseum, Arthroderma vanbreuseghemii [11], Trichophyton violaceum [12] were sequenced later. Based on available data, researchers have found that unlike other pathogenic fungi, four gene classes are enriched in dermatophyte genomes, namely proteases secreted to degrade keratin, kinases involved in adaptation to skin, important secondary metabolites for interaction with hosts and LysM domain acting on host immune response. Although gene contents are conserved across dermatophyte genomes, it is widely accepted that dermatophytes differ in morphology, ecology and invasion process [11]. Genome study provides a potential way for exploring reasons behind these divergences. Here we sequenced genome of E. floccosum (CGMCC (F) E1d), and did phylogenetic analysis of related dermatophytes. As reported before by molecular method [1], E. floccosum shared the closest relationship with N. gypseum. However, E. floccosum is anthropophilic mainly causing nail and glabrous skin infections, while N. gypseum is zoophilic and geophilic causing hair and skin infections, and the two species also differ in their morphological characters. Therefore, genomic comparison was performed between these two species, trying to reveal reasons accounting for pathogenic and ecology traits of E. floccosum.
Sexual reproduction is quite common among eukaryotes. For fungi, sexual reproduction not only is related to host fitness, but also related to influences virulence in some species [13]. Mating type (MAT) locus, harboring two different transcription factors genes (MAT1-1 and MAT1-2). Based on previous studies, geophilic and zoophilic dermatophytes are confirmed to reproduce sexually, while anthropophilic dermatophytes are considered to lose their ability for sexual reproduction when adapting to human host [14]. Genomic comparative analysis of T. rubrum and related dermatophytes revealed that genes concerning mating and meiosis are conserved across dermatophytes [11]. E. floccosum is known as anthropophilic dermatophyte, thus we suppose that it also tends to adopt asexual way to reproduce like T. rubrum, and we try to testify it with both molecular marker and genomic data acquired. Amplified fragment length polymorphism (AFLP) method has been extensively applied in population genetic, epidemiological studies, and it is suited for intraspecific differentiation of closely related groups [15]. So far, there is still limited molecular epidemiology study of E. floccosum, hence in this study, we try to discussion genetic variation within E. floccosum with AFLP method as well.
Materials and Methods
Fungal Strain and Genomic DNA Extraction
The strain E. Floccosum (CGMCC (F) E1d) isolated from a Chinese patient was grown on potato dextrose agar (PDA) medium at 28 °C for 14 days. Genomic DNA extraction was performed by using the Fungi DNA Kit (OMEGA) according to the manufacturer’s instructions. The quality of genomic DNA was then quantified by using TBS-380 fluorometer (Turner BioSystems Inc., Sunnyvale, CA). High qualified DNA sample (OD260/280 = 1.8–2.0, > 6 μg) was utilized to construct fragment library.
Sequencing, Assembly and Annotation
The sequencing procedure was performed by Illumina Hiseq combined with Pacific Biosciences sequencing platforms. At least 1 μg qualified genomic DNA was used for Illumina sequencing library construction. Paired-end libraries with insert sizes of ~ 400 bp were prepared following Illumina’s standard genomic DNA library preparation procedure. The qualified Illumina pair-end library would be used for Illumina Hiseq sequencing at Shanghai Biozeron Biotechnology Co., Ltd (PE150 mode). For Pacific Biosciences sequencing, 20 k insert whole genome shotgun libraries were generated and sequenced on a Pacific Biosciences RS instrument using standard methods. An aliquot of 8 μg DNA was spun in a Covaris g-TUBE (Covaris, MA) at 6000 RPM for 60 s using an Eppendorf 5424 centrifuge (Eppendorf, NY). DNA fragments were then purified, end-repaired and ligated with SMRT bell sequencing adapters following manufacturer’s recommendations (Pacific Biosciences, CA). Resulting sequencing libraries were purified three times using 0.45 × volumes of Agencourt AMPure XPbeads (Beckman Coulter Genomics, MA) following the manufacture’s recommendations.
We used ab initio prediction method to get gene models for strain (CGMCC (F) E1d). Gene models were identified using Augustus (http://bioinf.uni-greifswald.de/augustus/binaries/). Then all gene models were blast against non-redundant (NR in NCBI) database, SwissProt (http://uniprot.org), KEGG (http://www.genome.jp/kegg/), and COG (http://www.ncbi.nlm.nih.gov/COG) to do functional annotation by blastp module. In addition, tRNA were identified using the tRNAscan-SE (v1.23, http://lowelab.ucsc.edu/tRNAscan-SE) and rRNA were determined using the RNAmmer (v1.2, http://www.cbs.dtu.dk/services/RNAmmer/).
Phylogenetic Analysis
We identified clustering of single-copy genes of acquired dermatophytes genomes with OrthoMCL [16]. Individual amino acid sequences were aligned with MUSCLE v3.8.31 [17]. The phylogenetic tree was inferred by maximum likelihood method with PhyML v3.0 program [18] and 100 bootstraps were used to infer branch support. Beyond E. floccosum, genomic sequences of other dermatophytes including M. canis, N. gypseum, T. benhamiae, T. equinum, T. interdigitale, T. mentagrophytes, T. rubrum, T. soudanense, T. tonsurans, T. verrucosum and T. violaceum were downloaded from NCBI genome database (https://www.ncbi.nlm.nih.gov/genome/).
Comparative Genomics
Comparative genomic analysis was conducted between E. floccosum (CMCC (F) E1d) and N. gypseum (MS_CBS118893). Protein sequences were aligned using CD-HIT v4.6.1 [19] (identity > 40%, coverage > 50%).
Adhesion Analysis
For adhesion prediction, three software products were tested, SignalP [20] was used to identify signal peptide, TMHMM [21] was used to predict transmembrane protein domains with default parameters, and Big-PI Predictor [22] was used to identify GPI-anchor sites. Potential adhesions were identified with the following parameters: SignalP positive, TMHMM negative and Big-PI positive.
Proteases Analysis
Local database related to keratin degradation of dermatophytes [23] including endoproteases and exoproteases were downloaded from Uniprot database (https://www.uniprot.org/). Orthologs were selected when E values < 1e-3 and similarities > 90%.
LysM Domain Analysis
Proteins with LysM domains were identified with Pfam database (http://pfam.xfam.org/) based on multiple sequence alignments and hidden Markov models (HMMs) as previously reported [11].
AFLP Analysis
A total of 19 E. floccosum strains were collected from the Institute of Dermatology, Chinese Academy of Medical Sciences and CBS (Table 1). All strains were identified by both morphological methods and sequencing of internal transcribed spacer (ITS) regions [24]. AFLP genotyping was performed according to previously described methods [25, 26]. Briefly, first step was usage of two restriction endonucleases (HpyCH4 IV and Msel) to obtain restriction fragments. HpyCH4 IV adapter, Msel adapter and T4 DNA ligase were adopted to perform a combined restriction-ligation procedure. Fluorescently labeled primer (HpyCH4IV) was used for sample PCR reaction. The products were 10 × diluted and combined with GeneScan LIZ500 internal size standard (Appied Biosystems, Foster City, CA, USA). After denaturation and cool down process, the samples were subjected to 96 capillary 3730xl DNA Analyzer platform (Applied Biosystem). Raw data were imported into Genemapper v4.1 software (Applied Biosystem) and analyzed by using UPGMA clustering and Sorensen's Coefficient. DNA fragments in the range of 50–500 bp were submitted for analysis.
Mating Genes
Mating type analysis was performed for 19 isolates with primers TR-α and TR-HMG separately as described previously [27]. PCR amplification was carried out at the following condition: initial denaturation at 94 °C for 5 min, 35 cycles of denaturation at 95 °C for 45 s, annealing at 55 °C for 90 min, and extension at 72 °C for 60 s, with final extension at 72 °C for 10 min. Amplified products were analyzed visually by 1% (w/v) agarose gel electrophoresis in TBE buffer at 90 V for 30 min. The products of TR-α primers are indicative of the MAT1-1 mating type, or the products of TR-HMG primers are indicative of the MAT1-2 mating type. Since mating genes of MAT1-1 and MAT1-2 had been revealed in genome data of related dermatophytes, blast was performed of genome sequences of CGMCC (F) E1d accordingly.
Results and Discussion
Genome Sequencing and Annotation
The genome of E. floccosum (CGMCC (F) E1d) was successfully sequenced using Illumina Hiseq combined with Pacific Biosciences platforms and the sequence was submitted to NCBI (ID: PRINA528555). The estimated genome size of 24.4 Mb. Compared with genome size of other clinical important dermatophytes [11], T. rubrum (22.5 Mb), T. interdigitale (23.0 Mb), M. canis (23.1 Mb) and N. gypseum (23.2 Mb), E. floccosum owns the largest genome size. The GC content is 48.5%, which is in similar with other dermatophytes. The number of assembled scaffolds is 46 with N50 of 1.44 Mb, and the length of largest one is 4.72 Mb. 7565 protein coding genes are predicted accounting for 49.39% of genome. When refers to the genome contents, they are relatively conservative within dermatophytes, which further confirms the conclusions of previous study [11].
Phylogeny Analysis
A phylogenetic tree was done between 12 dermatophytes (Fig. 1). Genomic data of genus Trichophyton are more sufficient than that of Microsporum. Considering the similarity of gene sequences, we can conclude that genomes are highly similar between the dermatophytes. N. gypseum contains the largest number of unique genes, while T. violaceum contains the least ones. Among all selected dermatophytes, E. floccosum shares the closest relationship with N. gypseum which consistent with previous study with gene markers [1]. It is widely known that E. floccosum is anthropophilic mainly causing infections of glabrous skin and nails. While for N. gypseum, which belongs to geophilic species, it is a pathogen of hair and skin of human and animals. Thus comparative genomic analysis was performed between these two species trying to reveal the reason of the specificity in ecology and pathogenicity of E. floccosum.

Phylogenetic relationship and gene conservation of dermatophytes, blue color means genes conserved in all included dermatophytes, green color means genes conserved in more than one dermatophytes, and yellow color means those are species specific genes
Comparative Genomic Analysis
Adhesion Analysis
Adherence to host surface is first and indispensable step for invasion. Table 2 shows probable adhesions of E. floccosum and N. gypseum (19 and 22 separately), most of the genes (29/41) are annotated as hypothetical protein for limited database. Several genes have definite function which are common within these two species like cu-Zn superoxide dismutase (E1dA0486 and MGYG-04560), carboxypeptidase S1 (E1dA4066 and MGYG-03675), UTR2 protein (E1dA6473 and MGYG-06064). E1dA2196 of E. floccosum is 1, 3-beta-glucanosyltransferase gel3 which is specific to E. floccosum. The 1, 3-beta-glucanosyltransferase GEL family remains in Aspergillus fumigatus and Neurospora crassa, and it is reported that gel3 is an important factor for mycelial growth [28, 29]. The importance of gel3 in E. floccosum needs more fundamental studies.
Protease Analysis
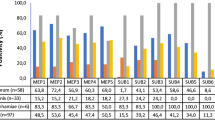
Secretion of various proteases plays a vital role for keratin degradation and is considered to be important virulence factors. Enrichment of proteases of dermatophytes has been validated with previous genomic analysis [11, 30]. Among all secreted proteases, subtilisin (S8A family) and fungalysin (M36) of extracellular endoproteases are prevalent in dermatophytes [12]. Our study (Fig 2) shows that highly similarity is found between E. floccosum and N. gypseum in M36 (Mep3-Mep5). For S8A family, which has been proven involving in mutualisms with hosts of many fungi [31]. In T. rubrum, the most prevalent dermatophytes in China, sub7 expresses highest on nail-keratin [32]. In our study, 5 genes encode sub7 for E. floccosum while 1 for N. gypseum, which may be a partial reason for more nail infections for E. floccosum. In addition, it has been proved that sub1 and sub3 expression, sub3 especially, are required for adherence in guinea pig model for M. canis [33, 34]. In our analysis, no gene encodes sub1 or sub3 annotated with local database in E. floccosum, thus we hypothesized that sub1 and sub3 may partly explain the anthropophilic character of E. floccosum. However, gene expression analysis is needed to testify this hypothesis.

Proteases genes predicted of E. floccosum and N. gypseum. Including endoproteases (M35, M36 and S8) and exoproteases (S9, M28, M24, S33 and S10)
LysM domain analysis
The LysM was originally considered as an enzyme degrading bacterial cell walls and first reported in fungus Cladosporium fulvum, acting as a virulence factor [35]. Hitherto, LysM is found among fungi with various lifestyles [36]. It is supposed that LysM effectors in dermatophytes may take part in breaking down certain products of fungal cell walls which can serve as triggers of host immunity during invasion process, resulting in chitin mask, therefore avoiding host immunostimulation [11, 36]. LysM domain can also facilitate adhesion of pathogens to human skin [37]. Figure 3 shows the LysM structure of E. floccosum and N. gypseum. The number of LysM genes within dermatophytes varies (10–31) [38]. E. floccosum, in this study, only possesses 7 related genes. The number of LysM genes varies greatly within fungi, the small number of LysM genes can serve as proof that chitin protection is not a necessary method for this fungus. Secretion signals, similar with N. gypseum, are also detected in E. floccosum. Based on the structure of LysM proteins, they can be classified into 5 types (A–E), type A and type B constitute structure of both E. floccosum and N. gypseum, with number varies. Type A is considered to play a role in invasion process, and type B is thought to take part in hydrolytic activity [36]. The number of LysM genes and type A, as we suggest, might play a role in their different reaction to host immunity thus resulting in different pathogenic process. What’s more, opposite direction in LysM domain is revealed between the two species, while the reason and meaning of this need further researches.

Structure of LysM gene cluster of E. floccosum (E1d) and N. gypseum (CBS118893)
Mating Genes
The results of gel electrophoresis are shown in Figs. 4 and 5. All isolates show positive amplification result with TR-HMG primers (MAT1-2) and the fragments are about 700 bp. In genome sequence, N. gypseum (CBS118893) was reported to be type MAT1-1, comparing with it, E. floccosum (CGMCC (F) E1d) is revealed to be type MAT1-2. Reproduction is known as a powerful way adapting to environment alteration, E. floccosum, anthropophilic fungus, also lose its ability to reproduce sexually when colonize or infect humans as predicted. What’s more, opposite direction is also found between the two species, further studies needed as well.

Gel electrophoresis result with TR-HMG primers of E. floccosum which is indicative of MAT1-2, fragments about 700 bp

Gel electrophoresis result with TR-α primers of E. floccosum which is indicative of MAT1-1
AFLP Analysis
AFLP method has been successfully done in several Trichophyton and Microsporum species [15] but not in E. floccosum. In this study, The AFLP analysis was efficiently performed in E. floccosum and two outgroup strains (30,078 and 30,007), the phylogenetic analysis obtained with UPGMA is shown in Fig. 6. Four clusters (group1-4) could be distinguished at about a cutoff value of 90% similarity. Group 2 contain only one isolate from Pakistan. Group 4 is the most common group contained isolates from different countries (China, the Netherlands, Germany, Iran and India). Isolates from the Netherlands (n = 5) were all in group 4. Consistent with previous studies [39,40,41], existence of genetic diversity in E. floccosum also validated with AFLP method. Unlike random amplified polymorphic DNA PCR (RAPD) method [40], all isolates can be divided into four rather than three main gene types. In geographical distributions, isolates from the Netherlands (5/16) are located in the main gene type, while isolates from China, Germany and Iran spread scattered otherwise. Limited by number and distribution of tested strains, the significance of this genotype needs further studies. In earlier studies, E. floccosum and E. stockdaleae were two distinct species of the genus Epidermophyton, for the latter behaves differently in antifungal susceptibilities, temperature tests and seldom causes human infections [21, 22]. E.floccosum is now recognized to be the only representative species by new taxonomy [1]. In this study, CBS100148 isolated from India (formerly classified as E. stockdaledae), it belongs to the main gene type and shares the closest relationship with CGMCC (F) E1g (E. floccosum) from China, it does not form a distinct gene type, this phenomenon further confirms the reliability of newly taxonomy. In addition, considering the classification varies with methods alteration, we propose that standard criteria need to be made in the near future.

AFLP analysis of 19 E. floccosum and 2 outgroups. The scar bar indicates the percentage similarity
Conclusion
To our best knowledge, this is the first report of the genome sequences of E. floccosum, and we did phylogenetic analysis based on genome data of other clinical important dermatophytes. It is verified that dermatophytes share similar gene content. The number and structure of protease like sub3 and sub7, adhesion factors and LysM genes may contribute to the specificity of E. floccosum. In addition, E.floccosum also lose its ability to reproduce sexually for anthropophilic feature. Four genotypes acquired with AFLP method, seems not correlated with geographical distribution, the significance needs larger samples and further study.
References
de Hoog GS, Dukik K, Monod M, Packeu A, Stubbe D, Hendrickx M, et al. Toward a novel multilocus phylogenetic taxonomy for the dermatophytes. Mycopathologia. 2017;182(1–2):5–31. https://doi.org/10.1007/s11046-016-0073-9.
McCormack P, Benham RW. An unusual finding in Epidermophyton floccosum. J Investig Dermatol. 1952;19(5):315–7. https://doi.org/10.1038/jid.1952.104.
Cabañes FJ, Bragulat MR, Bruguera T, Abarca L. The growth of Epidermophyton floccosum and E. stockdaleae at different temperatures. Mycopathologia. 1990;112(3):157–63. https://doi.org/10.1007/bf00436647.
Cabañes FJ, Abarca L, Bragulat M, Bruguera T. Sensitivity of some strains of the genus Epidermophyton to different antifungal agents. Mycopathologia. 1989;105(3):153–6. https://doi.org/10.1007/bf00437247.
Zamani S, Sadeghi G, Yazdinia F, Moosa H, Pazooki A, Ghafarinia Z, et al. Epidemiological trends of dermatophytosis in Tehran, Iran: a five-year retrospective study. J Mycol Med. 2016;26(4):351–8. https://doi.org/10.1016/j.mycmed.2016.06.007.
Nweze EI, Eke IE. Dermatophytes and dermatophytosis in the eastern and southern parts of Africa. Med Mycol. 2018;56(1):13–28. https://doi.org/10.1093/mmy/myx025.
Qiangqiang Z, Limo Q, Qixian Q. Case report. Disseminated tinea of the verrucous type due to epidermophyton floccosum. Mycoses. 2001;44(7–8):326–9.
Mary ES, Mark GT. Invasive disease due to Epidermophyton floccosum in an immunocompromised patient with Behçet’s syndrome. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 1997;25(1):153–4.
Rivera ZS, Losada L, Nierman WC. Back to the future for dermatophyte genomics. MBio. 2012. https://doi.org/10.1128/mBio.00381-12.
Burmester A, Shelest E, Glöckner G, Heddergott C, Schindler S, Staib P, et al. Comparative and functional genomics provide insights into the pathogenicity of dermatophytic fungi. Genome Biol. 2011;12(1):R7. https://doi.org/10.1186/gb-2011-12-1-r7.
Martinez DA, Oliver BG, Graser Y, Goldberg JM, Li W, Martinez-Rossi NM, et al. Comparative genome analysis of Trichophyton rubrum and related dermatophytes reveals candidate genes involved in infection. MBio. 2012;3(5):e00259-12. https://doi.org/10.1128/mBio.00259-12.
Zhan P, Dukik K, Li D, Sun J, Stielow JB, Gerrits van den Ende B, et al. Phylogeny of dermatophytes with genomic character evaluation of clinically distinct Trichophyton rubrum and T. violaceum. Stud Mycol. 2018;89:153–75. https://doi.org/10.1016/j.simyco.2018.02.004.
Hsueh YP, Heitman J. Orchestration of sexual reproduction and virulence by the fungal mating-type locus. Curr Opin Microbiol. 2008;11(6):517–24. https://doi.org/10.1016/j.mib.2008.09.014.
Metin B, Heitman J. Sexual reproduction in dermatophytes. Mycopathologia. 2017;182(1–2):45–55. https://doi.org/10.1007/s11046-016-0072-x.
Restrepo CM, Llanes A, Lleonart R. Use of AFLP for the study of eukaryotic pathogens affecting humans. Infect Genet Evol J Mol Epidemiol Evol Genet Infect Dis. 2018;63:360–9. https://doi.org/10.1016/j.meegid.2017.09.017.
Li L, Stoeckert CJ Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13(9):2178–89. https://doi.org/10.1101/gr.1224503.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7. https://doi.org/10.1093/nar/gkh340.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–21. https://doi.org/10.1093/sysbio/syq010.
Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics (Oxford, England). 2012;28(23):3150–2. https://doi.org/10.1093/bioinformatics/bts565.
Nielsen H. Predicting Secretory Proteins With SignalP. Methods Mol Biol (Clifton, NJ). 2017;1611:59–73. https://doi.org/10.1007/978-1-4939-7015-5_6.
Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305(3):567–80. https://doi.org/10.1006/jmbi.2000.4315.
Eisenhaber B, Schneider G, Wildpaner M, Eisenhaber F. A sensitive predictor for potential GPI lipid modification sites in fungal protein sequences and its application to genome-wide studies for Aspergillus nidulans, Candida albicans, Neurospora crassa, Saccharomyces cerevisiae and Schizosaccharomyces pombe. J Mol Biol. 2004;337(2):243–53. https://doi.org/10.1016/j.jmb.2004.01.025.
Mercer DK, Stewart CS. Keratin hydrolysis by dermatophytes. Med Mycol. 2019;57(1):13–22. https://doi.org/10.1093/mmy/myx160.
Makimura K, Murayama SY, Yamaguchi H. Detection of a wide range of medically important fungi by the polymerase chain reaction. J Med Microbiol. 1994;40(5):358–64. https://doi.org/10.1099/00222615-40-5-358.
Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M, et al. AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res. 1995;23(21):4407–14. https://doi.org/10.1093/nar/23.21.4407.
Gao Y, Zhan P, Hagen F, Menken SBJ, Sun J, Rezaei-Matehkolaei A, et al. Molecular epidemiology and in vitro antifungal susceptibility of Trichophyton schoenleinii, agent of tinea capitis favosa. Mycoses. 2019;62(5):466–74. https://doi.org/10.1111/myc.12889.
Kosanke S, Hamann L, Kupsch C, Moreno Garcia S, Chopra A, Gräser Y. Unequal distribution of the mating type (MAT) locus idiomorphs in dermatophyte species. Fungal Genet Biol FG B. 2018;118:45–53. https://doi.org/10.1016/j.fgb.2018.07.003.
Gastebois A, Fontaine T, Latgé JP, Mouyna I. beta(1–3)Glucanosyltransferase Gel4p is essential for Aspergillus fumigatus. Eukaryot Cell. 2010;9(8):1294–8. https://doi.org/10.1128/ec.00107-10.
Kamei M, Yamashita K, Takahashi M, Fukumori F, Ichiishi A, Fujimura M. Deletion and expression analysis of beta-(1,3)-glucanosyltransferase genes in Neurospora crassa. Fungal Genet Biol FG & B. 2013;52:65–72. https://doi.org/10.1016/j.fgb.2012.12.001.
Alshahni MM, Yamada T, Yo A, Murayama SY, Kuroda M, Hoshino Y, et al. Insight into the draft whole-genome sequence of the dermatophyte Arthroderma vanbreuseghemii. Sci Rep. 2018;8(1):15127. https://doi.org/10.1038/s41598-018-33505-9.
Muszewska A, Taylor JW, Szczesny P, Grynberg M. Independent subtilases expansions in fungi associated with animals. Mol Biol Evol. 2011;28(12):3395–404. https://doi.org/10.1093/molbev/msr176.
Chen J, Yi J, Liu L, Yin S, Chen R, Li M, et al. Substrate adaptation of Trichophyton rubrum secreted endoproteases. Microb Pathog. 2010;48(2):57–61. https://doi.org/10.1016/j.micpath.2009.12.001.
Descamps F, Brouta F, Monod M, Zaugg C, Baar D, Losson B, et al. Isolation of a Microsporum canis gene family encoding three subtilisin-like proteases expressed in vivo. J Investig Dermatol. 2002;119(4):830–5. https://doi.org/10.1046/j.1523-1747.2002.01784.x.
Baldo A, Mathy A, Tabart J, Camponova P, Vermout S, Massart L, et al. Secreted subtilisin Sub3 from Microsporum canis is required for adherence to but not for invasion of the epidermis. Br J Dermatol. 2010;162(5):990–7. https://doi.org/10.1111/j.1365-2133.2009.09608.x.
Bolton MD, van Esse HP, Vossen JH, de Jonge R, Stergiopoulos I, Stulemeijer IJ, et al. The novel Cladosporium fulvum lysin motif effector Ecp6 is a virulence factor with orthologues in other fungal species. Mol Microbiol. 2008;69(1):119–36. https://doi.org/10.1111/j.1365-2958.2008.06270.x.
de Jonge R, Thomma BP. Fungal LysM effectors: extinguishers of host immunity? Trends Microbiol. 2009;17(4):151–7. https://doi.org/10.1016/j.tim.2009.01.002.
Kar B, Patel P, Free SJ. Trichophyton rubrum LysM proteins bind to fungal cell wall chitin and to the N-linked oligosaccharides present on human skin glycoproteins. PLoS ONE. 2019;14(4):e0215034. https://doi.org/10.1371/journal.pone.0215034.
Persinoti GF, Martinez DA, Li W, Döğen A, Billmyre RB, Averette A, et al. Whole-genome analysis illustrates global clonal population structure of the ubiquitous dermatophyte pathogen Trichophyton rubrum. Genetics. 2018;208(4):1657–69. https://doi.org/10.1534/genetics.117.300573.
Ansari S, Ahmadi B, Norouzi M, Ansari Z, Afsarian MH, Lotfali E, et al. Epidermophyton floccosum: nucleotide sequence analysis and antifungal susceptibility testing of 40 clinical isolates. J Med Microbiol. 2019;68(11):1655–63. https://doi.org/10.1099/jmm.0.001074.
Khosravi A, Behzad F, Sabokbar A, Shokri H, Haddadi S, Masoudi-Nejad A. Molecular typing of Epidermophyton floccosum isolated from patients with dermatophytosis by RAPD-PCR. J Basic Microbiol. 2010;50(Suppl 1):S68-73. https://doi.org/10.1002/jobm.201000089.
De Baere T, Summerbell R, Theelen B, Boekhout T, Vaneechoutte M. Evaluation of internal transcribed spacer 2-RFLP analysis for the identification of dermatophytes. J Med Microbiol. 2010;59(Pt 1):48–54. https://doi.org/10.1099/jmm.0.013870-0.
Acknowledgements
This study was funded by The National Natural Science Foundation of China (No. 81972949), the Scientific and Technological Innovation Projects of Medicine and Health of Chinese Academy of Medical Sciences (No. 2016-I2M-3-021), the Nanjing Incubation Program for National Clinical Research Center (No. 2019060001) and the Basical Scientific Research Fund Projects of Chinese Academy of Medical Sciences (No. 2018PT31013).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have on conflict of interest.
Additional information
Handling Editor: Vishnu Chaturvedi
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Liu, J., Ge, L., Mei, H. et al. Comparative Genomics and Molecular Analysis of Epidermophyton floccosum. Mycopathologia 186, 487–497 (2021). https://doi.org/10.1007/s11046-021-00567-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11046-021-00567-9