
Abstract
The tumor suppressor p53 plays a central role in stress responses and tumor suppression. The increasingly complex p53 network is controlled by multiple layers of mechanisms, including the genetic level, transcriptional level, and protein level. Post-translational modifications (PTMs) of p53 represent a precise and efficient form of regulation. To date, the modification of p53 by ubiquitin and ubiquitin-like proteins (UBLs) has been studied extensively, including SUMOylation, NEDDylation, FATylation, ISGylation, and the recently identified UFMylation. They affect p53 stability, conformation, localization, transcriptional activity and binding partners. Here, we review these recent discoveries and summarize our understanding of ubiquitination and UBL modifications of p53 to better comprehend the complex landscape of p53 regulation. We will discuss how the ubiquitination and UBL modifications of p53 dynamically adjust its function to respond to various stress stimuli, thereby determining cell fate.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
p53, the well-known “guardian of the genome” (Lane, 1992), has been studied for more than 40 years and is the most studied gene of all time (Dolgin, 2017). As a tumor suppressor, p53 is mutated in more than half of human tumors, which perturbs p53 function and disrupts the p53 regulatory network (Kruse & Gu, 2009a). p53 controls a broad and flexible network, including proliferation, cell cycle, apoptosis, ferroptosis, DNA repair, embryo implantation, pluripotency, angiogenesis, metabolism, inflammation, immunity, autophagy and senescence (Kastenhuber & Lowe, 2017; Vousden & Prives, 2009). Functioning as a transcriptional regulator, hundreds of target genes are regulated by p53 to execute these varied functions. For example, the canonical p53 target cyclin-dependent kinase inhibitor p21 plays a critical role in the regulation of the cell cycle and senescence (Brown et al., 1997; Marx, 1993), although several other p53 target genes such as GADD45 and PAI-1 also contribute to these responses (Vousden & Prives, 2009). PUMA and NOXA are key mediators in p53-dependent apoptosis (Chipuk et al., 2005; Oda et al., 2000). In addition, SLC7A11, GLS2, PTGS2, and SAT1 have been discovered to be directly regulated by p53 in regulation of ferroptosis (Jiang et al., 2015). p53 activates the expression of LIF, as well as numerous long non-coding RNAs (LncRNAs) such as TUNA, for the maintenance of embryonic stem cell pluripotency (Fu et al., 2020). p53 also participates in metabolism regulation, including glucose metabolism, lipid metabolism, serine metabolism and nucleotide metabolism, by direct or indirect modulation of a series of key factors or enzymes such as glucose transporters (GLUTs), TIGER, SREBP-1, carnitine palmitoyltransferase, malonyl-CoA decarboxylase, PHGDH and GMP synthetase (Liu, Zhang, et al., 2019). These examples highlight the growing complexity of our understanding of the p53 regulation network (Fig. 1), although the mechanisms underlying the ability of p53 to induce multiple biological processes remain unclear.

The p53 network. p53 controls many distinct biological processes involved in tumor suppression and progression. Regulation of p53 includes the genetic level, transcriptional level, and PTM level
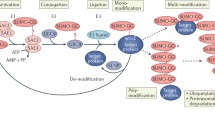
Regulation of p53 occurs at multiple levels, including the genetic level, transcriptional level, and protein level. PTMs at protein level have been extensively studied. Reported PTMs of p53 include phosphorylation, ubiquitination, acetylation, crotonylation, succinylation, methylation, SUMOylation, NEDDylation, FATylation, ISGylation, UFMylation, ADP-ribosylation, hydroxylation, β-hydroxybutyrylation and O-GlcNAcylation (Huang et al., 2014; Kruse & Gu, 2009a; Li et al., 2011; Liao et al., 2020; Liu et al., 2020; Liu, Tavana, et al., 2019; Park et al., 2016). Some of these PTMs have been well studied and shown to be essential for the activity and stability of p53, such as phosphorylation and ubiquitination (Dai & Gu, 2010). In the steady-state condition, the expression of p53 is maintained at a very low level mediated by the ubiquitin–proteasome pathway, whereas in the stressed conditions, p53 protein is accumulated and activated, a process mediated by phosphorylation, acetylation and some other PTMs like UFMylation (Kruse & Gu, 2009a; Liu et al., 2020; Rodriguez et al., 2000). Similar to ubiquitin, there are more than a dozen UBLs, including small ubiquitin-like modifiers (SUMOs), neural precursor cell expressed, developmentally down-regulated 8 (NEDD8), interferon-stimulated gene 15 (ISG15), human leukocyte antigen (HLA)-F adjacent transcript 10 (FAT10), ubiquitin-fold modifier 1 (UFM1), ubiquitin-related modifier-1 (URM1), autophagy-related protein 8 (ATG8), autophagy-related protein 12 (ATG12), fan ubiquitin-like protein 1 (FUB1), histone monoubiquitylation 1 (HUB1) and prokaryotic ubiquitin-like protein (PUP) (Pearce et al., 2008; Wang, Zhu, et al., 2017). These small proteins share a similar structure with ubiquitin and conjugate to their targets following an E1–E2–E3 enzymatic cascade (Wang, Zhu, et al., 2017). p53 has been found to be covalently modified by SUMOs, NEDD8, FAT10, ISG15 and UFM1 (Fig. 2). The functions of these UBLs are not only associated with p53 stability and conformation, but also alter its transcriptional activity and subcellular localization. There have been many excellent reviews written previously on the topic of PTMs of p53 (Gu & Zhu, 2012; Kruse & Gu, 2008, 2009a; Liu, Tavana, et al., 2019; Meek & Anderson, 2009); therefore, here we will summarize the characteristics of the ubiquitination and UBL modifications of p53, with particular focus on the biological function of UBLs of p53.

Overview of p53 PTMs. The major sites of p53 Ac acetylation, M methylation, Ub ubiquitination, S SUMOylation, N NEDDylation, I ISGylation, U UFMylation are plotted and the functions of these PTMs are indicated
Ubiquitination of p53
Ubiquitin is an 8.6-kD protein with 76-amino acids, and relies on maturation of ubiquitin precursor by de-ubiquitylating enzymes (DUBs) to expose the dipeptide glycine–glycine (Gly–Gly) for conjugation to substrates via a three-step enzymatic reaction. Thus far, 2 E1 enzymes, 38 E2 enzymes, and > 600 E3 ligases have been identified in the human genome. Ubiquitination is a dynamically reversible process, and > 100 DUBs act on ubiquitin (Komander, 2009; Zhang & Sun, 2020). The ubiquitin molecule possesses seven lysine residues (K6, K11, K27, K29, K33, K48 and K68), and all of these lysine residues together with the first methionine (M1) can be ubiquitinated to form ubiquitin chains. These chains are then transferred to different substrates leading to polyubiquitination of the targets (Swatek & Komander, 2016). K48-linked chains are the most common chains to target proteins for degradation, whereas K63-linked chains execute non-degradative functions. K11-linked chains are involved in target protein degradation by working with K48- and K63-linked chains. Other lysine-linked chains are not well understood (Kwon & Ciechanover, 2017; Swatek & Komander, 2016). Besides polyubiquitination, substrates can also be modified with only one ubiquitin, resulting in monoubiquitination, or with individual ubiquitin at several lysine residues, resulting in multi-monoubiquitination. Monoubiquitination appears to mainly regulate the substrates trafficking, activity and interactions with other proteins (Kwon & Ciechanover, 2017). Collectively, ubiquitination can target proteins for proteasomal degradation, intracellular protein trafficking, cellular signaling transduction, protein activity, and protein–protein interactions.
The protein p53 functions as a tetramer with four identical chains of 393 residues, encompassing an amino (N)-terminal transactivation domain, a proline-rich domain, a central DNA-binding domain, a tetramerization domain, and a carboxy (C)-terminal regulatory domain (Fig. 2). The C-terminal region of p53 is rich in basic amino acids and prone to be modified by various PTMs, especially by ubiquitin and UBLs (Ko & Prives, 1996; Liu et al., 2020; Liu, Tavana, et al., 2019). p53 is found to be modified by both polyubiquitination and monoubiquitination, which occurr at a series of different lysine residues in the C terminus. Mouse double minute 2 homolog (MDM2) is the major ubiquitin E3 ligase, and belongs to RING-domain E3s family that binds to and leads to p53 degradation (Haupt et al., 1997; Honda et al., 1997; Kubbutat et al., 1997). The crystal structure of the N-terminal 109-residue fragment of MDM2 interacts with the transactivation domain of p53 (Kussie et al., 1996). p53 degradation is mediated by polyubiquitination with K48-linked chains at six lysine residues (K370, K372, K373, K381, K382 and K386) of p53 (Nakamura et al., 2000; Rodriguez et al., 2000). It is reported that high levels of MDM2 promotes p53 polyubiquitination and nuclear degradation, whereas low levels of MDM2 induces monoubiquitination and nuclear export of p53 (Li et al., 2003). MDM2 is well known to be transcriptionally induced by p53 thereby forming a negative feedback loop that maintains low activity of p53 in normal conditions (Wu et al., 1993). In response to a multitude of stresses, including genotoxic stress signals, hypoxia, oncogenic activation, nutrient deprivation, ribosomal stress, chronic stress and nucleolar stress, MDM2 funnels into a common node in each stress regulatory network, which in turn regulates p53 and induces its biological responses under these adverse conditions (Hu et al., 2012; Russo & Russo, 2017; Vousden & Prives, 2009). For example, there is a sophisticated mechanism for p53 accumulation and activation following DNA damage: (i) serine (S) and tyrosine (Y) residues of MDM2 are rapidly phosphorylated by Ataxia Telangiectasia Mutated (ATM) kinase and c-Abl tyrosine kinase, which is necessary for the activity of MDM2 inhibition and p53 activation in cells and in vivo (Gannon et al., 2012; Maya et al., 2001; Saadatzadeh et al., 2017); (ii) phosphorylation of p53 at S15 and S20 also mediated by the ATM kinase family, which reduces the affinity of p53 with MDM2, resulting in p53 accumulation and activation (Chao et al., 2000; Shieh et al., 1997; Unger et al., 1999); (iii) phosphorylation of MDMX, a homologue of MDM2 that represses p53’s function (Sanford et al., 2021), leads to increased binding to MDM2 and thus be degraded by MDM2, preceding activation and accumulation of p53 (Chen et al., 2005; Pereg et al., 2005). The simultaneous phosphorylation of MDM2, MDMX and p53 ensures an effective release of p53 from the inhibitory action of MDM2 upon DNA damage, thereby inducing a well-coordinated p53 response. Furthermore, crucial evidence to support the pivotal role of MDM2 on the negative regulation of p53 activity is that the embryonic lethality in Mdm2-null mice can be rescued by the loss of p53 (Jones et al., 1995; Montes de Oca Luna et al., 1995). Contrary to the Mdm2 depletion mice, overexpressed-Mdm2 transgenic mice are predisposed to spontaneous tumorigenesis, which probably due to the increased inhibition of p53 function (Jones et al., 1998). In addition to the E3 activity of MDM2 in regulating p53, MDM2 also inhibits p53 transcriptional activity by recruiting histone deacetylase and corepressors to p53 (Chen et al., 2010), as well as controls p53 synthesis by prompting proteasomal degradation of ribosomal protein L26 (RPL26) thereby diminishing the interaction of RPL26 with p53 mRNA region (Ofir-Rosenfeld et al., 2008). Overall, MDM2 is a key regulator of the tumor suppressor p53, making it more attractive for anti-cancer drug design.
Besides MDM2, there are scores of ubiquitin E3 ligases, including constitutive photomorphogenic protein 1 (COP1), p53-induced protein with a RING-H2 domain (PIRH2), ARF-BP1, makorin ring finger protein 1 (MKRN1), MKRN2, Synoviolin, TOP1 binding arginine/serine rich protein (TOPORS), CHIP, UBE4B, ring finger protein 1 (RNF1), RNF2, RNF128, CARP1, CARP2, JFK and the TRIM-family members. These ubiquitin E3 ligases can modify p53 with K48-linked polyubiquitin chains for degradation: many of their functions in the regulation of p53 are described in Table 1 and have been summarized well previously (Pan & Blattner, 2021). The differing mechanistic pathways and physiological functions of the varied E3 ligases present a complex picture. Similar to MDM2, COP1 and PIRH2 are RING-domain containing proteins that bind to and target p53 for ubiquitination and degradation, respectively. They both can be transcriptionally upregulated by p53, and constitute an autoregulatory feedback loop that controls the function of p53 to regulate cell cycle and apoptosis (Dornan et al., 2004; Hakem et al., 2011; Ka et al., 2018; Leng et al., 2003). Importantly, the degradation of p53 mediated by COP1 is independent of MDM2 or PIRH2. COP1 can inhibit p53’s effects on cell cycle arrest and apoptosis by regulating p21 and BAX (Dornan et al., 2004). PIRH2 also targets and ubiquitinates p53 independently of MDM2, thereby suppressing p53 transactivation (Sheng et al., 2008). PIRH2 favors different lysine residues in p53, including K101, K164, K292 and K305 within the DNA-binding domain and K357, K370, K382 and K386 in the C-terminus of p53 (Shloush et al., 2011). However, the biological significance of these lysines regulated by PIRH2 remains to be determined. Furthermore, PIRH2 has been reported to coordinate with Axin, HIPK2 and TIP60 to lead to maximal activation of p53 in response to genotoxic stress through p53 phosphorylation at S46, which ultimately triggers apoptosis (Li et al., 2009). MKRN1, a member of the RING-domain E3 ligases, regulates p53 polyubiquitination at lysine residues K291 and K292, subsequently resulting in proteasomal degradation of p53 (Lee et al., 2009). The association of p53 with MKRN1 is strongly reduced upon DNA damage, thus resulting in stabilization of p53 but not p21, indicating a dual function of MKRN1 in regulating cell death under stress conditions (Lee et al., 2009). Recently, MKRN2 has been identified as a novel E3 ligase that directly interact with p53, and promotes p53 ubiquitination and degradation in vivo and in vitro (Zhang et al., 2020). Distinct from those lysines targeted by MDM2, PIRH2 and MKRN1, an atypical ubiquitin E3 ligase E4F1 mediates ubiquitination at lysine residues K319, K320 and K321 of p53 for localization on chromatin but not degradation, and stimulates a transcriptional program that specifically controls the cell cycle (Le Cam et al., 2006). MSL2, another RING-domain E3 ligase, ubiquitinates p53 at K351 and K357, and also does not affect the stability of p53, but promotes p53 cytoplasmic localization (Kruse & Gu, 2009b). Similar to MSL2, the RING-domain E3 ligase RNF38 can ubiquitinate p53 in vitro and in vivo, resulting in relocalization of p53 to discrete foci associated with promyelocytic leukemia (PML) nuclear bodies (Sheren & Kassenbrock, 2013). Other RNF proteins that can directly target p53 are RNF1, RNF2 and RNF128, which all regulate p53 stabilization and p53-dependent cell proliferation and death (Chen et al., 2013; Shen et al., 2018; Wen et al., 2014).
In contrast to the above examples regulated by the RING-domain E3 ligases, the ubiquitination of p53 also can be regulated by the HECT-domain E3 ligases, U-BOX E3 ligases, and F-BOX E3 ligases (Pan & Blattner, 2021). Human papilloma virus (HPV) E6-associated cellular protein E6-AP is the first reported HECT-domain containing E3 ligase that interacts with p53, mediating p53 ubiquitination and its subsequent degradation (Scheffner et al., 1993). WWP1 is another HECT-domain E3 ligase involved in p53 regulation. Unlike other E3 ligases, WWP1 stabilizes p53 in a ubiquitination-dependent manner with a surprisingly concomitant decrease of its transcriptional activities. The expression of WWP1 also has been found to be reduced by p53, pointing to a previously unrecognized regulatory feedback loop (Laine & Ronai, 2007). In addition to HECT-domain E3 ligase, the U-BOX E3 ligase CHIP, is able to polyubiquitinate p53 and induce its proteasomal degradation by association with the chaperones Hsc70 and Hsp90 (Esser et al., 2005). JFK, a Kelch domain-containing F-box protein, also can promote ubiquitination and the degradation of p53 by assembling with the Skp1-Cul1-F-box complex. Interestingly, JFK is also transcriptionally regulated by p53 and forms an auto-regulatory negative feedback loop with p53 (Sun et al., 2009, 2011). These findings indicate that due to the necessity of maintaining low expression levels of p53 under normal conditions, a large number of E3s form regulatory feedback loops to participate in the regulation of p53 homeostasis. In summary, these different classes of E3 ligases play a central role in p53 ubiquitination, degradation, protein stability and trafficking, and control the transcriptional activity of p53, providing a regulatory network for p53.
As we have known that ubiquitination is a reversible process, a large number of DUBs has been identified that involved in p53 management, including ubiquitin-specific proteases (USPs), ovarian tumor proteases (OTUs), and Machado–Joseph disease-like proteins (MJDs). They regulate several E3 ligases of p53 or p53 directly by removing the ubiquitin, thereby controlling p53 stabilization (Kwon et al., 2017). Herpesvirus-associated ubiquitin-specific protease (HAUSP, also known as USP7) is the first identified DUB that specifically deubiquitinates p53 both in vivo and in vitro (Li, Chen, et al., 2002). However, subsequent studies have reported that MDM2 is a preferred substrate for HAUSP rather than p53 (Cummins et al., 2004; Li et al., 2004), indicating that the regulation of p53 by HAUSP is a complex process. Similar to HAUSP, ovarian tumor domain-containing Ub aldehyde binding protein 1 (OTUB1), a member of OTUs family, can suppress MDM2-mediated p53 ubiquitination, resulting in regulation of p53 stability and activity (Sun et al., 2012). OTUB1 can also stabilize MDMX, thereby contributing to p53 phosphorylation and p53-mediated apoptosis (Chen et al., 2017). In addition, USP2a, USP2 and USP15 regulate p53 stability by deubiquitinating and stabilizing MDM2 (Stevenson et al., 2007; Wei et al., 2016; Zou et al., 2014). USP26 is also found to deubiquitinate and stabilize MDM2 (Lahav-Baratz et al., 2017), which may negatively regulate p53. USP4 can target ARF-BP1, thereby mediating ARF-BP1-associated p53 ubiquitination (Zhang et al., 2011). Furthermore, USP28 can deubiquitinate of H2A, resulting in transcriptional activation of p53, p21 and p16, thereby regulating cell proliferation (Li et al., 2019). USP47 can target ubiquitinated ribosomal protein S2 (RPS2), thereby inhibiting the interaction between RPS2 and MDM2, and alleviating RPS2-mediated suppression of MDM2, consequently inducing p53 expression under ribosomal stress (Cho et al., 2020).
Unlike the DUBs mentioned above indirectly regulating p53 stability and activity, a class of USPs, including USP3, USP5, USP9X, USP10, USP11, USP24, USP29 and USP42, has been reported to directly control p53 stability and activity, respectively (Fu et al., 2017; Kwon et al., 2017). Among them, USP3 interacts with and deubiquitinates p53 in normal fibroblast cells, and regulates normal cell transformation (Fu et al., 2017). USP10 is mainly localized in the cytoplasm where it deubiquitinates p53 at normal conditions, whereas USP10 translocates from the cytoplasm to the nucleus and subsequently influencing p53 localization in response to stress conditions (Yuan et al., 2010). In contrast to USP10, USP42 is a nuclear protein that is required for the stabilization and activation of p53 in response to various stress signals (Hock et al., 2011). Besides USPs family, OTUD5, another OTUs family member, functions to deubiquitinate and stabilize p53, thereby activating a p53 response upon DNA damage (Luo et al., 2013). Ataxin-3 (ATX-3), a member of MJDs family, has also been reported to interact with and stabilize p53, thus promoting p53-dependent apoptosis in both mammalian cells and the central nervous system of zebrafish, which providing an explanation for the pathogenic mechanism of spinocerebellar ataxia type 3 (SCA3) (Liu, Li, et al., 2016). Collectively, these DUBs together with E3 ligases of p53 dynamically regulate p53 homeostasis, thereby controlling p53 regulatory network and determining cell fate.
Crosstalk between ubiquitination, acetylation and methylation
Ubiquitination, acetylation, methylation and the UBL modifications all occur at lysine residues, especially in the C-terminal region of p53, thereby affecting the activity and stability of p53 by intersection. It has been reported that acetylated p53 can not be ubiquitinated in vitro, and the ubiquitination of p53 has been shown to be abrogated after the induction of acetylation at the C-terminal domain (Li, Luo, et al., 2002). MDM2 actively suppresses acetyltransferase CBP/p300-mediated p53 acetylation, whereas acetylated p53 is capable of inhibiting p53 ubiquitination in vivo and in vitro (Ito et al., 2001; Li, Luo, et al., 2002). To date, 10 lysine residues have been reported to be acetylated and involved in p53 transcriptional activation, stability regulation, inducing apoptosis, cell cycle arrest and ferroptosis (Tang et al., 2008). Among them, acetylation of p53 on K370, K372, K373, K381, K382 and K386 by CBP/p300 activates its transcriptional activity and enhances p53 stability by inhibiting its ubiquitination by MDM2 at these lysines (Gu & Roeder, 1997; Kruse & Gu, 2008). Notably, acetylation of p53 abrogates MDM2-mediated transcriptional repression by blocking the recruitment of MDM2 to p53-responsive promoters, which leads to p53 activation independent of its phosphorylation status (Tang et al., 2008). In addition, N-acetyltransferase 10 (NAT10) acetylates p53 at K120 and stabilizes p53 by inhibiting MDM2-mediated p53 ubiquitination. Under stress conditions, NAT10 translocates from the nucleolus to nucleoplasm, stabilizes p53 by preventing the interaction between p53 and MDM2, thereby regulating the cell cycle and apoptosis pathways dependent on p53 (Liu, Tan, et al., 2016). Interestingly, NAT10 also promotes MDM2 degradation, which synergistically stabilizes p53 (Liu, Tan, et al., 2016). K120 of p53 can be acetylated by p300/CBP-associated factor (PCAF), TIP60 and MOZ, which is important for accumulation of p53 at BAX and PUMA promoters to initiate apoptosis. K320 is the predominant acetylation site targeted by PCAF in hypoxia and the DNA damage response, and acetylated p53 at K320 is prone to recruit specific target genes that promote cell survival (Xenaki et al., 2008). Conversely, ubiquitinated p53 at K320 is preferentially located on chromatin to regulate the cell cycle, indicating an interplay between PCAF mediated acetylation and E4F1 mediated ubiquitination (Le Cam et al., 2006). PCAF also has the intrinsic ubiquitin E3 ligase activity and targets MDM2 for ubiquitination thereby regulating p53 activity (Linares et al., 2007). As critical acetyltransferases, both CBP and p300 can function as E4 ligases by enhancing polyubiquitination of p53 that is already monoubiquitinated by MDM2 in the cytoplasm (Grossman et al., 2003; Shi et al., 2009). In contrast, CBP/p300 acetylates MDM2 in the RING-domain to restrain its E3 ligase activity, in turn to inhibit p53 polyubiquitination. Therefore, CBP/p300 plays a dual role in the regulation of p53.
Acetylation is highly reversible. Acetylated lysine residues of p53 can be deacetylated by various histone deacetylases (HDACs), including HDAC1, HDAC2, HDAC6 and HDAC8, as well as Sir2-like proteins (sirtuins), sirtuin1 and sirtuin3 (Sirt1 and Sirt3) (Brandl et al., 2012; Liu, Tavana, et al., 2019). HDAC1-containing complex has been first identified to deacetylate p53 and modulate p53-dependent cell growth and apoptosis (Luo et al., 2000). Notably, MDM2 and HDAC1 can form a complex that regulates p53 ubiquitination and deacetylation in a cooperative fashion, thus controlling p53 stability and function (Ito et al., 2002). It is reported that HDAC2 can deacetylate p53 at K320 (Brandl et al., 2012), whereas HDAC6 deacetylates p53 at K120, K381 and K382 (Park et al., 2017; Ryu et al., 2017), which all these lysines also are the ubiquitination sites, indicating a regulatory network between acetylation and ubiquitination. Indeed, the HDAC6 inhibitor A452 can disrupt wild-type p53–MDM2–MDMX interaction while promote mutant p53–MDM2–MDMX interaction, thus stabilizing wild-type p53 and destabilizing mutant p53 in cancer cells (Ryu et al., 2017), which providing an encouraging evidence for the feasibility of p53-targeted anticancer therapy. In contrast to these HDACs that can directly deacetylate p53, Sirt7 has been reported to deacetylate serine/threonine kinase receptor associated protein (STRAP), and influences the interaction between STRAP and p53, thereby regulating p53 function and subsequently p53-mediated signaling pathways (Yu et al., 2020).
Besides acetylation, methylation of p53 occurs at K370, K372, K373 and K382 of p53 by different methyltransferases. The interplay between p53 methylation and acetylation is well established (Carr et al., 2012). For example, the methylation level at K382 is decreased upon DNA damage, which allows induction of K382-acetylation mediated by CBP/p300 thereby promoting p53 activity. Additionally, K372 of p53 can be methylated by a lysine-specific methyltransferase SET9, which restricts methylated p53 to the nucleus and stabilizes p53 (Chuikov et al., 2004). Although little is known regarding the crosstalk between methylation and ubiquitination in this context, it is possible that methylation directly interferes with MDM2-mediated ubiquitination or indirectly targets some important factors involved in regulating p53 stabilization. The molecular mechanism for the methylation-induced stabilization of p53 remains to be elucidated.
SUMOylation of p53
SUMOylation is the most intensively studied UBL modification. Unlike ubiquitination, there are several modifiers for SUMOylation, consisting of SUMO-1, SUMO-2, SUMO-3 and SUMO-4. Whilst the three-dimensional structure of SUMO proteins is similar to ubiquitin, they share less than 20% sequence identity with ubiquitin (Geiss-Friedlander & Melchior, 2007). The enzymatic cascade of SUMOylation is similar to ubiquitination, in that mature SUMOs with dipeptide Gly–Gly are exposed for conjunction to substrates. SUMOylation has one SUMO-E1 enzyme in the form of a heterodimer containing SAE1/Aos1 and SAE2/Uba2 subunits, and the only known SUMO-E2 enzyme UBC9. The reported SUMO-E3 ligases for SUMOylation are the protein inhibitor of activated STAT (PIAS) family as well as RanBP2. Sentrin-specific proteases (SENPs) and de-SUMOylating isopeptidase-1/2 (DESI-1/2) as well as USPL1 are responsible for de-SUMOylation. To our knowledge, SUMO-1 is distinct from the other family members and usually forms mono-SUMO for the targets, whereas SUMO-2 and SUMO-3 (referred to as SUMO-2/3) share 97% sequence identity and can form poly-SUMO chains (Geiss-Friedlander & Melchior, 2007; Saitoh & Hinchey, 2000; Ulrich, 2008). SUMO-4 only appears to be conjugated to its substrates under stressed conditions, however, the roles of SUMO-4 have yet to be uncovered (Owerbach et al., 2005; Wei et al., 2008). To date, hundreds of proteins can be SUMOylated, which involved in regulating protein–protein interaction, protein activity, stability, cellular localization, chromatin remodeling, precursor-mRNA splicing and ribosome assembly (Becker et al., 2013; Hendriks et al., 2014).
p53 is one of the SUMOylated substrates that is conjugated by SUMO-1 in osteosarcoma cells and in vitro (Rodriguez et al., 1999). The major SUMOylation site of p53 is K386, and this SUMOylation is mediated by different SUMO-E3 ligases, including the PIAS family, TOPORS, TRIM19, TRIM27, RanBP2, adenovirus E1B-55 K, and a viral protein KSHV basic-leucine-zipper (K-bZIP) (Ashikari et al., 2017; Chang et al., 2010; Chu & Yang, 2011; Kahyo et al., 2001; Pennella et al., 2010; Schmidt & Muller, 2002; Takayama et al., 2018; Weger et al., 2005). The regulation of p53’s function by SUMO-1 is still in debate. Early reports have shown that conjugation of SUMO-1 can increase the transcriptional activity of p53 (Gostissa et al., 1999; Rodriguez et al., 1999); however, PIAS1 and PIASxβ, the PIAS family members, can promote SUMOylation of p53 and strongly repress the transcriptional activity of p53 in HeLa cells (Schmidt & Muller, 2002). Interestingly, SUMO-1 modification of p53 at K386 has no effect on p53’s transcriptional activation, cellular localization, or growth regulation (Kwek et al., 2001). Recently, one study has reported that PIAS1 can promote SUMO-1 conjugation of p53 at K386 in lens epithelial cells, thus enhances p53 transcription activity by specifically upregulating BAX expression (Nie et al., 2021). One possibility is that the transactivation activity of p53 regulated by SUMOylation is preferentially mediated by different SUMO-E3 ligases, in a cell type-specific and context-dependent fashion. In addition to the PIAS family, MDM2 can increase the level of p53 SUMOylation, especially by forming a complex with ARF, an important regulator of p53 stability. However, MDM2 exhibits no SUMOylation activity in a cell-free system (Chen & Chen, 2003).
In contrast to MDM2, TOPORS and the PIAS family can directly enhance SUMOylation of p53 (Weger et al., 2005). Previous studies have reported that PIASy, another PIAS family member, binds to p53 and inhibits transactivity of p53 (Nelson et al., 2001); MDM2 can cooperate with PIASy to promote p53 SUMOylation and nuclear export (Carter et al., 2007). In addition, SUMO‑2/3 also conjugates to p53 (Chang et al., 2010; Li et al., 2006; Stindt et al., 2011). SUMO-2/3 can be modified at K386, and cells treated with H2O2 can induce p53 SUMOylation by SUMO-2/3, but not SUMO-1. Moreover, such modification can stimulate p53 transcriptional activity and play roles in premature senescence and stress response (Li et al., 2006). It is interesting to note that MDM2 can enhance the conjugation of endogenous SUMO-2/3 to p53, which is correlated with a reduction of both activation and repression of a subset of p53-target genes, including the activation genes p21, BAX and macrophage inhibitory cytokine-1 (Mic-1), and the repression genes cyclin-dependent kinase 1 (Cdk1) and cyclin A2 (Stindt et al., 2011). The addition of ARF and ribosomal protein L11 (RPL11) also strongly induced SUMOylation of p53 (Stindt et al., 2011). Mitotic arrest-deficient 1 (Mad1), a well-characterized regulator of chromosome segregation during mitosis (Rodriguez-Bravo et al., 2014; Ryan et al., 2012), interacts with the PML nuclear body scaffold, and this interaction is enhanced by SUMOylation. Upregulated Mad1 causes a reduction of p53 protein levels by displacing MDM2 from PML nuclear bodies (Wan et al., 2019). Alternately, the adenovirus protein E1B-55 K functions as a SUMO-E3 ligase, promotes SUMOylation of p53 and tethers p53 in PML nuclear bodies, resulting in the inhibition of p53 activity (Pennella et al., 2010). Similar to E1B-55 K, another virus protein K-bZIP also functions as a SUMO-E3 ligase, enhancing the global SUMOylation of cellular proteins in a SUMO-2/3-dependent manner, including p53, leading to an increase of p53-dependent transcriptional activity (Chang et al., 2010). Although SUMO-1 is involved in p53 trafficking, SUMO-2/3 has no effect on the subcellular distribution of p53 (Carter et al., 2007; Stindt et al., 2011). This is probably due to the different dynamics and subcellular distributions of the SUMO isoforms, consequentially resulting in different preference for substrates and distinct regulation and functions (Hecker et al., 2006; Saitoh & Hinchey, 2000; Vertegaal, 2010).
SUMOylation is also a reversible process. SENP1, a SUMO-specific protease, has only been found to directly interact with and de-SUMOylate p53 in cells and in vitro; depletion of SENP1 synergistically induces p53 activation and cell growth inhibition in response to DNA damage (Chauhan et al., 2021). Repression of SENP1 also induces p53-mediated premature senescence in primary human fibroblasts (Yates et al., 2008). Furthermore, SENP2 regulates p53’s function through modulation of MDM2 de-SUMOylation at the PML body (Jiang et al., 2011); SENP6 interacts with and de-SUMOylates TRIM28, thereby suppressing p53 activity (Li, Lu, et al., 2018). Of note, whether there are other direct regulatory proteases de-SUMOylating p53 still need investigation. Another attention is that only K386 in p53 has been reported involved in SUMOylation until now. In one study for global mapping SUMOylation sites of endogenous proteins and SUMOylation signaling networks in cells, the mass spectrometry results suggest that further SUMOylation sites in p53 isoforms have been identified (Hendriks et al., 2014), and their function and regulation are worth exploration.
NEDDylation of p53
In the UBL superfamily, NEDD8 has the highest homology with ubiquitin and is indispensable in various biological processes. Similar to other UBLs, the translation product of NEDD8 is a precursor. Maturation of NEDD8 requires protease, such as ubiquitin C‑terminal hydrolase isozyme 3 (UCHL3) and deneddylase 1 (DEN1), to expose C-terminal di-Gly residues (Wada et al., 1998; Wu et al., 2003). Mature NEDD8 is cascade activated by the specific E1 NEDD8-activating enzyme (NAE), a heterodimer of NAE1 and UBA3, E2-NEDD8 conjugating enzyme UBC12, as well as a dozen of NEDD8-E3 ligases. The majority of NEDD8-E3s also function as ubiquitin E3 ligases and belong to the largest category of cullin-RING ligase family, including c-CBL, FBXO11, IAPs, MDM2, RNF111, TFB3, TRIM40, DCN1, RBX1 and its homologue RBX2 (Enchev et al., 2015; Santonico, 2020). NEDD8 has been reported to form chains through K11, K22, K27, K48, K54 and K60, and participates in NEDD8-related regulatory pathways in transcription, chromatin organization, genomic stability, signal transduction and tumorigenesis (Jeram et al., 2010; Jones et al., 2008; Leidecker et al., 2012; Xirodimas et al., 2008).
It is reported that p53 is a bona fide substrate for NEDDylation, and MDM2 function as a specific NEDD8-E3 ligase can promote p53 NEDDylation in vivo and in vitro (Xirodimas et al., 2004). Although up to six lysine residues in the C-terminus of p53 are known to be ubiquitinated (Nakamura et al., 2000; Rodriguez et al., 2000), the effective NEDDylation requires only three of these lysine residues (K370, K372, and K373). Interestingly, p53 deubiquitinating enzyme HAUSP prevented the formation of high molecular weight p53 species in a NEDDylation pull-down assay, suggesting that these high molecular species are ubiquitin conjugates; therefore, p53 could be simultaneously modified with NEDD8 and ubiquitin (Xirodimas et al., 2004). More recently, K120 of p53 has been identified as a new NEDDylation site in regulating its transactivation activity, and MDM2 is considered as the specific NEDD8-E3 ligase (Bravo-Navas et al., 2021). MDM2 can induce ubiquitination, SUMOylation and NEDDylation of p53, but the order of these modifications in vivo is still unclear. Another report has shown that FBXO11, a novel NEDD8 ligase for p53, promotes NEDD8 conjugation to K320 and K321 of p53 rather than ubiquitination in vitro and in vivo (Abida et al., 2007). In these studies, NEDDylation is found to inhibit p53 transcriptional activity but not to significantly affect its stability (Abida et al., 2007; Xirodimas et al., 2004).
In addition, NEDD8 ultimate buster 1 (NUB1) has been found to decrease p53 NEDDylation but preferentially stimulate its ubiquitination, which results in the cytoplasmic localization and inhibition of transactivity of p53 (Liu & Xirodimas, 2010). Similar to NUB1, TIP60 is capable of selectively inhibiting the MDM2-mediated conjugation of NEDD8 to p53, but it does not affect p53 ubiquitination (Dohmesen et al., 2008). MDM2 itself also can be modified by NEDD8, which markedly increases MDM2’s protein stability, while de-NEDDylation of MDM2 by NEDP1 results in MDM2 destabilization concomitant with p53 activation (Watson et al., 2010; Xirodimas et al., 2004). MDMX is more likely to dimer with MDM2 than MDM2 itself, indicating that MDM2-MDMX complex may play a prominent role in p53 ubiquitination. It is interesting to note that MDMX is also a positive effector for NEDD8 ligase activity of MDM2 in regulating the function of p53 (Marine et al., 2007; Singh et al., 2007). Under growth conditions, MDMX plays a key role in enhancing MDM2-mediated NEDDylation of p53 (Hauck et al., 2017). Moreover, the adenovirus oncoprotein E4orf6 can hijack Cullin 5-based E3 ubiquitin ligase (CRL5) activity via facilitating its NEDDylation, leading to degradation of p53, which efficiently impeding viral replication; while this process can be restricted by the deneddylase DEN1 and NEDD8-activating enzyme inhibitor (Guo et al., 2019). In summary, p53 ubiquitination determines its proteasomal degradation, whereas NEDDylation controls its transcriptional activity. However, the integrated and detailed regulation of NEDDylation and ubiquitination as well as SUMOylation and other PTMs in some cases remains to be elucidated.
ISGylation of p53
Interferon (IFN)-stimulated gene product 15 (ISG15) was the first identified UBL. Its expression and conjugation to targets are induced by viral infection, type I interferons, tumor necrosis factor (TNF), vascular endothelial growth factor (VEGF) and IFN-γ, and other compounds such as poly I:C and lipopolysaccharide (LPS), as well as by DNA damage, ischemia and aging (Chairatvit et al., 2012; Doyle et al., 2002; Haas et al., 1987; Liu et al., 2009; Liu, Gao, et al., 2016; Lou et al., 2009; Nakka et al., 2011; Park et al., 2016; Sadler & Williams, 2008; Sen & Sarkar, 2007; Taylor et al., 1996). ISGylation also requires an enzymatic cascade, involving E1 (ubiquitin-activating enzyme E1-like protein, Ube1L), E2 (ubiquitin-carrier protein H6 or 8, UbcH6 or UbcH8), and E3 enzymes (HECT domain and RCC1-like domain-containing protein 5, HERC5, estrogen-responsive finger protein, EFP, or human homolog of ariadne 1, HHARI) (Kim et al., 2004; Krug et al., 2005; Pitha-Rowe et al., 2004; Zhao et al., 2004). Interestingly, these E1 and E2 enzymes were originally identified in the ubiquitination system. ISGylation has been mainly studied in relation to its antiviral effects, like blocking the entry, replication or release of different intracellular pathogens (Villarroya-Beltri et al., 2017). ISGylation has been found to play an essential role in DNA repair, autophagy, protein translation and exosome secretion (Nakashima et al., 2015; Okumura et al., 2013; Park et al., 2016; Villarroya-Beltri et al., 2016). Unlike ubiquitin, no substrates have been identified to be poly-ISGylated to date.
p53 has been found to be modified by ISG15, which targets misfolded p53 for proteasomal degradation. HERC5 efficiently promotes p53 ISGylation, while USP18 removes this modification. Notably, many lysine residues are modified by ISG15, including 5 N-terminal (K101, K120, K132, K139, and K164) and 6 C-terminal lysines (K291, K292, K320, K321, K351, and K357). Ubiquitination and ISGylation cooperate in the regulation of p53 stability, and inhibition of ISGylation increases p53 ubiquitination. Deletion of ISG15 leads to misfolded p53 accumulation, which results in attenuation of p53’s abilities both in vivo and in vitro (Huang et al., 2014). Furthermore, ISGylation-mediated degradation of p53 is involved in oncogene-mediated cellular transformation. Specifically, the phosphorylation of p53 on Y126 and Y220 by Src can promote p53 ISGylation through enhancing HERC5 binding (Huang & Bulavin, 2014). These observations suggest that ISG15 can act as a signaling molecule to guide ubiquitin-independent proteasome degradation of p53. Subsequently, a 7, 11-disubstituted quinazoline derivative HZ-6d has been screened that can interact with HERC5 and suppress its expression, thereby preventing the ISG15-dependent degradation of p53 (Y. Wang, Ding, et al., 2017). ISG15-conjugating system components ISG15, UBE1L, UBCH8 and EFP all encode p53-responsive elements that can be dramatically induced in response to DNA damage dependent on p53. Contrary to these findings, ISGylation can positively regulate the stability of p53 under DNA damage conditions. The major ISGylation sites of p53 are K291 and K292, and specific ISG15 E3 ligase EFP conducts the ISGylation process, which is reversed by the de-ISGylating enzyme UBP43. EFP, but not HERC5, can interact with p53 and promote DNA damage-induced p53 ISGylation. Additionally, DNA damage-induced ISGylation of p53 dramatically stimulates its acetylation and phosphorylation, thereby activating its transactivity. ISGylation of p53 consequently increases p53’s binding ability to its downstream target promoters to regulate cell cycle and apoptosis, such as p21 and BAX (Park et al., 2016). Despite these reports describing the functional regulation of ISGylation of p53, little is known about how misfolded p53 is recognized and degraded by the proteasome, as well as the interplay between ISGylation and other PTMs, especially the relationship between misfolded/mutant p53 and PTMs. There are a large number of synthesized proteins targeted for ISGylation (Durfee et al., 2010), therefore the degree to which ISGylation controls the synthesis of p53 and the mechanism by which this occurs both invite further investigation.
FATylation of p53
FAT10 is an 18-kD protein discovered by chromosomal sequencing of the human HLA-F locus (Fan et al., 1996), consisting of two ubiquitin-like domains, sharing 29% and 36% identity to ubiquitin, respectively (Wang, Zhu, et al., 2017). Unlike other ubiquitin-like modifiers, FAT10 ends with a free diglycine motif which can immediately form the isopeptide to target proteins; FAT10 also has been found to be the only ubiquitin-like modifier that targets its substrates for proteasomal degradation in a ubiquitin-independent manner (Schmidtke et al., 2014). FATylation is catalyzed by a specific E1 UBA6 and the only E2-like enzyme USE1, however, the FATylation specific E3 ligases as well as the deconjugating enzymes remain unknown. Interestingly, UBA6 can activate both ubiquitin and FAT10, whereas FAT10 binds to UBA6 with a higher affinity than ubiquitin (Chiu et al., 2007; Gavin et al., 2012; Jin et al., 2007). TRIM21 and RanBP2, the ubiquitination and SUMOylation E3 ligases, have been identified as FAT10-modified targets, indicating that they probably participate in the FATylation process (Leng et al., 2014). A growing number of studies have shown that FATylation plays an important role in proteasomal degradation, protein folding, RNA processing, apoptosis, DNA damage response, cell growth, immune response, and tumorigenesis (Aichem et al., 2012; Chen et al., 2018; Leng et al., 2014; Liu et al., 1999; Liu, Chen, et al., 2016; Wang, Zhu, et al., 2017; Xiang et al., 2020).
p53 has also been found to be conjugated to FAT10. Overexpression of FAT10 increases p53 transcriptional activity and alters the conformation of p53, as well as the distribution of PML-NBs in HEK293 cells (Li et al., 2011). Notably, p53 can negatively regulate FAT10 expression, which indicates that activation of p53 by upregulating FAT10 in turn reduces the expression of FAT10 as a feedback regulation to maintain its expression at a low level in normal cells and most tissues (Liu et al., 1999; Zhang et al., 2006). However, overexpression of FAT10 significantly reduces the transcriptional activity of p53 in several cell lines, at either basal level or cytokine stimulated conditions, without alterations of p53 protein level (Choi et al., 2014). In addition, the degradation of FAT10 mediated by p53 requires its transcriptional activity, and this mutually inhibitory regulation makes sense in the context that under inflammatory conditions FAT10 is temporarily induced but quickly returns to its basal level, similar to the double negative regulation of NFκB signaling driven by inflammatory signals (Choi et al., 2014; Zhang et al., 2006). It is well known that ubiquitination is responsible for protein degradation, but how and in what situations FATylation participates in this protein degradation is unclear. Moreover, the sites for p53 FATylation as well as the crosstalk between FATylation and other PTMs remain a mystery, and as such the regulation and function of FATylation in the p53 context are worthy of further investigation.
UFMylation of p53
UFMylation (UFM1 modification) is a recently identified UBL (Komatsu et al., 2004; Tatsumi et al., 2010). The modifier UFM1 is a 9.1-kD protein with low sequence identity but a similar tertiary structure to ubiquitin. It has only one glycine at the C-terminus, which differs from the usual diglycine of other UBLs. Similar to ubiquitin, UFM1 relies on maturation of the UFM1 precursor by specific cysteine proteases (UFSP1 and UFSP2) to expose glycine. UFM1 then conjugates to the substrates via the unique E1- and E2-like enzymes UBA5 and UFC1, and the only known specific E3 ligase UFL1 (Gerakis et al., 2019; Komatsu et al., 2004; Tatsumi et al., 2010). DDRGK1, a regulator for the maintenance of UFL1 ligase activity, plays an essential role in UFMylation process (Yoo et al., 2014). Although only a handful of substrates including DDRGK1, ASC1, H4, MRE11, RPL26, RPN1, p53, and SLC7A11 so far have been described (Lee et al., 2021; Liang et al., 2020; Liu et al., 2020; Qin et al., 2019; Tatsumi et al., 2010; Walczak et al., 2019; Wang, Gong, et al., 2019; Wang, Xu, et al., 2020; Yang et al., 2021; Yoo et al., 2014), the significance of UFMylation is underscored by its critical role in diverse cellular processes ranging from embryonic development, endoplasmic reticulum (ER) and tissue homeostasis maintenance, cell survival and differentiation, vesicle trafficking, autophagy, protein quality control, and DNA damage responses (Cai et al., 2015, 2019; Lee et al., 2021; Lemaire et al., 2011; Li, Yue, et al., 2018; Liang et al., 2020; Liu et al., 2017, 2020; Qin et al., 2019, 2020; Tatsumi et al., 2011; Walczak et al., 2019; Wang, Gong, et al., 2019; Wang, Xu, et al., 2020; Zhang et al., 2015; Zhou et al., 2021; Zhu et al., 2019).
UFMylation contributes to the complexity of p53 regulation as along with the other UBLs. We previously reported that p53 is a novel substrate for UFMylation, which maintains p53 stability by competing with MDM2-mediated ubiquitination either at normal conditions or with DNA damage responses. K351, K357, K370 and K373 are the major lysine residues of p53 that can be UFMylated (Liu et al., 2020), but other sites may be UFMylated, especially under particular physiological and pathological conditions. UFMylated p53 at these lysine residues appears in a mono-UFM1 modification form, whereas ASC1 can be UFMylated with a K69-linked poly-UFM1 chain (Liu et al., 2020; Yoo et al., 2014). Whilst the majority of reported substrates exist in the mono-UFM1 form, no common characteristics of mono-UFM1 modification have been found, and the distinguishing features of the mono-UFM1 and poly-UFM1 modifications are unclear. Besides UFMylation and ubiquitination, K351, K357, K370 and K373 of p53 are subject to acetylation, NEDDylation and ISGylation. Acetylation at K351 and K357 differentially modulate cell cycle arrest and apoptosis but do not affect p53 localization or oligomerization, whereas ISGylation at K351 and K357 contributes to degradation of misfolded p53; NEDDylation at K370 and K373 of p53 inhibits its transcriptional activity; these observations raise an important question regarding the manner in which these different types of PTMs interact with each other as a “molecular switch” to regulate diverse biological processes. One explanation is that under different experimental or physiological conditions, p53 has various PTM preferences thereby selectively binding to the promoters of specific genes, resulting in regulation of diverse biological processes. Notably, several molecules such as IRE1ɑ and p53 have been found to be modulated by UFMylation via proteasomal degradation (Liu et al., 2017, 2020), but it is currently unclear how UFMylation cooperates with ubiquitination in the regulation of proteasomal degradation. The most common explanation is that competitive binding to the same lysine residues regulates the protein levels. Interestingly, our previous results have shown that both UFL1 and DDRGK1 could interact with substantial components of 26S proteasome, especially the non-ATPase subunits family of the 19S regulator lid (Liu et al., 2020), which is responsible for recognizing polyubiquitinated proteins and redirecting them to 20S proteasome for degradation (Glickman & Ciechanover, 2002), providing another possibility that the UFL1-DDRGK1-substrate complex may occupy the binding site of polyubiquitylated proteins and subsequently block ubiquitin-dependent proteasome degradation of the substrates.
Concluding remarks and future perspectives
As an important transcription factor and tumor suppressor, p53 is a highly variable molecule induced under different stresses and signals. The activity of p53 is regulated and finely balanced by a variety of PTMs that not only compete with each other, but also may promote each other. PTMs can alter p53 conformation to enable it to recognize and bind to different sites of downstream target genes, thereby activating the downstream targets and ultimately determining the types of response and cell fate. PTMs can also cause p53 to form a new protein docking site, thereby recruiting different enzymes to induce PTMs at other sites, which greatly increase the complexity of p53 regulation. To date, there are as many as 400 PTMs identified, including more than 10 ubiquitin-like modifications. Interestingly, the majority of these UBLs have been found to be conjugated to p53 except URM1 and FUB1 (Table 2). Questions for further study include whether p53 is a novel target for URM1 or FUB1, whether there are other new PTMs of p53, and what the important biological functions of these potential PTMs are in regulating p53 homeostasis.
At present, the timing and balance of various PTMs of p53 under different stimuli are not completely characterized, and the responses of these PTMs of p53 are variable in different tissues and cell types. What we are certain is that MDM2 is a key node in regulating p53 function at multi-layer across these PTMs (Fig. 3), contributing to the maintenance of p53 homeostasis. A point of clear interest is how these PTMs at different lysine residues of p53 coordinate with each other. The degree to which different stimuli regulate the activity of p53 through these PTMs is still a question vital to attaining a better understanding the regulation of p53. p53 is mutated in 50% of human tumors, and mutant p53 may often play an opposite biological function to wild-type p53. Currently, the full roles and mechanisms of p53 and its mutants in regulating tumor suppression or progression are still unclear. Therefore, the question of how these different PTMs contribute to wild-type and mutant p53’s function may provide clinically valuable insight in p53 mutant cancers. One potential avenue for study is the degree to which these various PTMs are involved in regulating the tumor microenvironment and cell characteristics to influence p53’s function.

The p53–MDM2 axis. p53 is maintained at a low level by MDM2-mediated polyubiquitination in normal condition, whereas monoubiquitination or SUMOylation results in p53 nuclear export. In stressed conditions such as DNA damage, p53 is activated either by MDM2 phosphorylation, or by several PTMs of p53, including phosphorylation, acetylation, methylation, SUMOylation and UFMylation, thereby inducing the expression of different targets with diverse biological functions. MDM2 is one of these targets, which can negatively regulate p53 stability and transcriptional activity. MDM2 also can promote p53 SUMOylation and NEDDylation, leading to inhibition of p53’s function at different context. Ac acetylation, M methylation, Ub ubiquitination, S SUMOylation, N NEDDylation, I ISGylation, U UFMylation are indicated
Owing to an increasing number of studies focus on the regulatory effects of PTMs on p53 behaviors, targeting PTMs of p53 has become a promising strategy for cancer treatment, which includes upregulating/reactivating wild-type p53, restoring tumor suppressive function in mutant p53, and inducing mutant p53 degradation in cancer. Among these strategies, inhibiting the p53-MDM2 interaction is an important approach for activating p53 tumor suppressive function, and a number of p53-MDM2 antagonists have been designed and studied for this purpose. In addition, numerous DUBs, HDACs and sirtuins inhibitors also have become a choice, which can activate p53 and exhibit unique value as antitumor drugs (Liu, Tavana, et al., 2019). Unfortunately, majority of these small molecules have encountered setbacks. The major obstacles include the specificity, therapeutic effects, drug resistance, and side effects of these small molecules. Despite the setbacks, some MDM2 antagonists are still in clinical trials.
In addition to tumor suppression, p53 also impacts the physiology of many important diseases, such as neurodegenerative diseases, metabolic diseases and aging. It is still unclear which exact PTMs of p53 are implicated in these diseases, and the mechanism by which these PTMs of p53 may contribute to these diseases is unknown. These important questions remain to be addressed and the answers to these questions are expected to shed further light on the complexity of the p53 network, leading to new opportunities for therapies for cancer and other diseases.
Change history
06 April 2022
A Correction to this paper has been published: https://doi.org/10.1007/s42764-022-00071-4
References
Abida, W. M., Nikolaev, A., Zhao, W., Zhang, W., & Gu, W. (2007). FBXO11 promotes the neddylation of p53 and inhibits its transcriptional activity. Journal of Biological Chemistry, 282(3), 1797–1804. https://doi.org/10.1074/jbc.M609001200
Aichem, A., Kalveram, B., Spinnenhirn, V., Kluge, K., Catone, N., Johansen, T., & Groettrup, M. (2012). The proteomic analysis of endogenous FAT10 substrates identifies p62/SQSTM1 as a substrate of FAT10ylation. Journal of Cell Science, 125(Pt 19), 4576–4585. https://doi.org/10.1242/jcs.107789
Ashikari, D., Takayama, K., Tanaka, T., Suzuki, Y., Obinata, D., Fujimura, T., Urano, T., Takahashi, S., & Inoue, S. Androgen induces G3BP2 and SUMO-mediated p53 nuclear export in prostate cancer. Oncogene, 36(45), 6272–6281. https://doi.org/10.1038/onc.2017.225
Becker, J., Barysch, S. V., Karaca, S., Dittner, C., Hsiao, H. H., Berriel Diaz, M., Herzig, S., Urlaub, H., & Melchior, F. (2013). Detecting endogenous SUMO targets in mammalian cells and tissues. Nature Structural & Molecular Biology, 20(4), 525–531. https://doi.org/10.1038/nsmb.2526
Brandl, A., Wagner, T., Uhlig, K. M., Knauer, S. K., Stauber, R. H., Melchior, F., Schneider, G., Heinzel, T., & Kramer, O. H. (2012). Dynamically regulated sumoylation of HDAC2 controls p53 deacetylation and restricts apoptosis following genotoxic stress. Journal of Molecular Cell Biology, 4(5), 284–293. https://doi.org/10.1093/jmcb/mjs013
Bravo-Navas, S., Yanez, L., Romon, I., Briz, M., Dominguez-Garcia, J. J., & Pipaon, C. (2021). Map of ubiquitin-like post-translational modifications in chronic lymphocytic leukemia. Role of p53 lysine 120 NEDDylation. Leukemia, 35(12), 3568–3572. https://doi.org/10.1038/s41375-021-01184-7
Brown, J. P., Wei, W., & Sedivy, J. M. (1997). Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science, 277(5327), 831–834. https://doi.org/10.1126/science.277.5327.831
Cai, Y., Pi, W., Sivaprakasam, S., Zhu, X., Zhang, M., Chen, J., Makala, L., Lu, C., Wu, J., Teng, Y., Pace, B., Tuan, D., Singh, N., & Li, H. (2015). UFBP1, a Key Component of the Ufm1 Conjugation System, Is Essential for Ufmylation-Mediated Regulation of Erythroid Development. PLoS Genetics, 11(11), e1005643. https://doi.org/10.1371/journal.pgen.1005643
Cai, Y., Zhu, G., Liu, S., Pan, Z., Quintero, M., Poole, C. J., Lu, C., Zhu, H., Islam, B., Riggelen, J. V., Browning, D., Liu, K., Blumberg, R., Singh, N., & Li, H. (2019). Indispensable role of the Ubiquitin-fold modifier 1-specific E3 ligase in maintaining intestinal homeostasis and controlling gut inflammation. Cell Discov, 5, 7. https://doi.org/10.1038/s41421-018-0070-x
Carr, S. M., Munro, S., & La Thangue, N. B. (2012). Lysine methylation and the regulation of p53. Essays in Biochemistry, 52, 79–92. https://doi.org/10.1042/bse0520079
Carter, S., Bischof, O., Dejean, A., & Vousden, K. H. (2007). C-terminal modifications regulate MDM2 dissociation and nuclear export of p53. Nature Cell Biology, 9(4), 428–435. https://doi.org/10.1038/ncb1562
Chairatvit, K., Wongnoppavich, A., & Choonate, S. (2012). Up-regulation of interferon-stimulated gene15 and its conjugates by tumor necrosis factor-alpha via type I interferon-dependent and -independent pathways. Molecular and Cellular Biochemistry, 368(1–2), 195–201. https://doi.org/10.1007/s11010-012-1360-5
Chang, P. C., Izumiya, Y., Wu, C. Y., Fitzgerald, L. D., Campbell, M., Ellison, T. J., Lam, K. S., Luciw, P. A., & Kung, H. J. (2010). Kaposi’s sarcoma-associated herpesvirus (KSHV) encodes a SUMO E3 ligase that is SIM-dependent and SUMO-2/3-specific. Journal of Biological Chemistry, 285(8), 5266–5273. https://doi.org/10.1074/jbc.M109.088088
Chao, C., Saito, S., Anderson, C. W., Appella, E., & Xu, Y. (2000). Phosphorylation of murine p53 at ser-18 regulates the p53 responses to DNA damage. Proceedings of the National Academy of Sciences of the United States of America, 97(22), 11936–11941. https://doi.org/10.1073/pnas.220252297
Chauhan, K. M., Chen, Y., Liu, A. T., Sun, X. X., & Dai, M. S. (2021). The SUMO-specific protease SENP1 deSUMOylates p53 and regulates its activity. Journal of Cellular Biochemistry, 122(2), 189–197. https://doi.org/10.1002/jcb.29838
Chen, L., & Chen, J. (2003). MDM2-ARF complex regulates p53 sumoylation. Oncogene, 22(34), 5348–5357. https://doi.org/10.1038/sj.onc.1206851
Chen, L., Gilkes, D. M., Pan, Y., Lane, W. S., & Chen, J. (2005). ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. EMBO Journal, 24(19), 3411–3422. https://doi.org/10.1038/sj.emboj.7600812
Chen, L., Li, Z., Zwolinska, A. K., Smith, M. A., Cross, B., Koomen, J., Yuan, Z. M., Jenuwein, T., Marine, J. C., Wright, K. L., & Chen, J. (2010). MDM2 recruitment of lysine methyltransferases regulates p53 transcriptional output. EMBO Journal, 29(15), 2538–2552. https://doi.org/10.1038/emboj.2010.140
Chen, Y. C., Chan, J. Y., Chiu, Y. L., Liu, S. T., Lozano, G., Wang, S. L., Ho, C. L., & Huang, S. M. (2013). Grail as a molecular determinant for the functions of the tumor suppressor p53 in tumorigenesis. Cell Death and Differentiation, 20(5), 732–743. https://doi.org/10.1038/cdd.2013.1
Chen, Y., Wang, Y. G., Li, Y., Sun, X. X., & Dai, M. S. (2017). Otub1 stabilizes MDMX and promotes its proapoptotic function at the mitochondria. Oncotarget, 8(7), 11053–11062. https://doi.org/10.18632/oncotarget.14278
Chen, Z., Zhang, W., Yun, Z., Zhang, X., Gong, F., Wang, Y., Ji, S., & Leng, L. (2018). Ubiquitinlike protein FAT10 regulates DNA damage repair via modification of proliferating cell nuclear antigen. Molecular Medicine Reports, 17(6), 7487–7496. https://doi.org/10.3892/mmr.2018.8843
Chipuk, J. E., Bouchier-Hayes, L., Kuwana, T., Newmeyer, D. D., & Green, D. R. (2005). PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science, 309(5741), 1732–1735. https://doi.org/10.1126/science.1114297
Chiu, Y. H., Sun, Q., & Chen, Z. J. (2007). E1–L2 activates both ubiquitin and FAT10. Molecular Cell, 27(6), 1014–1023. https://doi.org/10.1016/j.molcel.2007.08.020
Cho, J., Park, J., Shin, S. C., Jang, M., Kim, J. H., Kim, E. E., & Song, E. J. (2020). USP47 promotes tumorigenesis by negative regulation of p53 through deubiquitinating ribosomal protein S2. Cancers (basel). https://doi.org/10.3390/cancers12051137
Choi, Y., Kim, J. K., & Yoo, J. Y. (2014). NFkappaB and STAT3 synergistically activate the expression of FAT10, a gene counteracting the tumor suppressor p53. Molecular Oncology, 8(3), 642–655. https://doi.org/10.1016/j.molonc.2014.01.007
Chu, Y., & Yang, X. (2011). SUMO E3 ligase activity of TRIM proteins. Oncogene, 30(9), 1108–1116. https://doi.org/10.1038/onc.2010.462
Chuikov, S., Kurash, J. K., Wilson, J. R., Xiao, B., Justin, N., Ivanov, G. S., McKinney, K., Tempst, P., Prives, C., Gamblin, S. J., Barlev, N. A., & Reinberg, D. (2004). Regulation of p53 activity through lysine methylation. Nature, 432(7015), 353–360. https://doi.org/10.1038/nature03117
Cummins, J. M., Rago, C., Kohli, M., Kinzler, K. W., Lengauer, C., & Vogelstein, B. (2004). Tumour suppression: Disruption of HAUSP gene stabilizes p53. Nature, 428(6982), 1–486. https://doi.org/10.1038/nature02501
Dai, C., & Gu, W. (2010). p53 post-translational modification: Deregulated in tumorigenesis. Trends in Molecular Medicine, 16(11), 528–536. https://doi.org/10.1016/j.molmed.2010.09.002
Montes de Oca Luna, R., Wagner, D. S., & Lozano, G. (1995). Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature, 378(6553), 203–206. https://doi.org/10.1038/378203a0
Dohmesen, C., Koeppel, M., & Dobbelstein, M. (2008). Specific inhibition of Mdm2-mediated neddylation by Tip60. Cell Cycle, 7(2), 222–231. https://doi.org/10.4161/cc.7.2.5185
Dolgin, E. (2017). The most popular genes in the human genome. Nature, 551(7681), 427–431. https://doi.org/10.1038/d41586-017-07291-9
Dornan, D., Wertz, I., Shimizu, H., Arnott, D., Frantz, G. D., Dowd, P., O'Rourke, K., Koeppen, H., & Dixit, V. M. (2004). The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature, 429(6987), 86–92. https://doi.org/10.1038/nature02514
Doyle, S., Vaidya, S., O'Connell, R., Dadgostar, H., Dempsey, P., Wu, T., Rao, G., Sun, R., Haberland, M., Modlin, R., & Cheng, G. (2002). IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity, 17(3), 251–263. https://doi.org/10.1016/s1074-7613(02)00390-4
Durfee, L. A., Lyon, N., Seo, K., & Huibregtse, J. M. (2010). The ISG15 conjugation system broadly targets newly synthesized proteins: Implications for the antiviral function of ISG15. Molecular Cell, 38(5), 722–732. https://doi.org/10.1016/j.molcel.2010.05.002
Enchev, R. I., Schulman, B. A., & Peter, M. (2015). Protein neddylation: Beyond cullin-RING ligases. Nature Reviews Molecular Cell Biology, 16(1), 30–44. https://doi.org/10.1038/nrm3919
Esser, C., Scheffner, M., & Hohfeld, J. (2005). The chaperone-associated ubiquitin ligase CHIP is able to target p53 for proteasomal degradation. Journal of Biological Chemistry, 280(29), 27443–27448. https://doi.org/10.1074/jbc.M501574200
Fan, W., Cai, W., Parimoo, S., Schwarz, D. C., Lennon, G. G., & Weissman, S. M. (1996). Identification of seven new human MHC class I region genes around the HLA-F locus. Immunogenetics, 44(2), 97–103. https://doi.org/10.1007/BF02660056
Fu, S., Shao, S., Wang, L., Liu, H., Hou, H., Wang, Y., Wang, H., Huang, X., & Lv, R. (2017). USP3 stabilizes p53 protein through its deubiquitinase activity. Biochemical and Biophysical Research Communications, 492(2), 178–183. https://doi.org/10.1016/j.bbrc.2017.08.036
Fu, X., Wu, S., Li, B., Xu, Y., & Liu, J. (2020). Functions of p53 in pluripotent stem cells. Protein & Cell, 11(1), 71–78. https://doi.org/10.1007/s13238-019-00665-x
Gannon, H. S., Woda, B. A., & Jones, S. N. (2012). ATM phosphorylation of Mdm2 Ser394 regulates the amplitude and duration of the DNA damage response in mice. Cancer Cell, 21(5), 668–679. https://doi.org/10.1016/j.ccr.2012.04.011
Gavin, J. M., Chen, J. J., Liao, H., Rollins, N., Yang, X., Xu, Q., Ma, J., Loke, H. K., Lingaraj, T., Brownell, J. E., Mallender, W. D., Gould, A. E., Amidon, B. S., & Dick, L. R. (2012). Mechanistic studies on activation of ubiquitin and di-ubiquitin-like protein, FAT10, by ubiquitin-like modifier activating enzyme 6, Uba6. Journal of Biological Chemistry, 287(19), 15512–15522. https://doi.org/10.1074/jbc.M111.336198
Geiss-Friedlander, R., & Melchior, F. (2007). Concepts in sumoylation: A decade on. Nature Reviews Molecular Cell Biology, 8(12), 947–956. https://doi.org/10.1038/nrm2293
Gerakis, Y., Quintero, M., Li, H., & Hetz, C. (2019). The UFMylation system in proteostasis and beyond. Trends in Cell Biology, 29(12), 974–986. https://doi.org/10.1016/j.tcb.2019.09.005
Glickman, M. H., & Ciechanover, A. (2002). The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiological Reviews, 82(2), 373–428. https://doi.org/10.1152/physrev.00027.2001
Gostissa, M., Hengstermann, A., Fogal, V., Sandy, P., Schwarz, S. E., Scheffner, M., & Del Sal, G. (1999). Activation of p53 by conjugation to the ubiquitin-like protein SUMO-1. EMBO Journal, 18(22), 6462–6471. https://doi.org/10.1093/emboj/18.22.6462
Grossman, S. R., Deato, M. E., Brignone, C., Chan, H. M., Kung, A. L., Tagami, H., Nakatani, Y., & Livingston, D. M. (2003). Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science, 300(5617), 342–344. https://doi.org/10.1126/science.1080386
Gu, B., & Zhu, W. G. (2012). Surf the post-translational modification network of p53 regulation. International Journal of Biological Sciences, 8(5), 672–684. https://doi.org/10.7150/ijbs.4283
Gu, W., & Roeder, R. G. (1997). Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell, 90(4), 595–606. https://doi.org/10.1016/s0092-8674(00)80521-8
Guo, H., Shen, S., Li, Y., Bi, R., Zhang, N., Zheng, W., Deng, Y., Yang, Y., Yu, X. F., Wang, C., & Wei, W. (2019). Adenovirus oncoprotein E4orf6 triggers Cullin5 neddylation to activate the CLR5 E3 ligase for p53 degradation. Biochemical and Biophysical Research Communications, 516(4), 1242–1247. https://doi.org/10.1016/j.bbrc.2019.07.028
Haas, A. L., Ahrens, P., Bright, P. M., & Ankel, H. (1987). Interferon induces a 15-kilodalton protein exhibiting marked homology to ubiquitin. Journal of Biological Chemistry, 262(23), 11315–11323.
Hakem, A., Bohgaki, M., Lemmers, B., Tai, E., Salmena, L., Matysiak-Zablocki, E., Jung, Y. S., Karaskova, J., Kaustov, L., Duan, S., Madore, J., Boutros, P., Sheng, Y., Chesi, M., Bergsagel, P. L., Perez-Ordonez, B., Mes-Masson, A. M., Penn, L., Squire, J., Chen, X., Jurisica, I., Arrowsmith, C., Sanchez, O., Benchimol, S., & Hakem, R. (2011). Role of Pirh2 in mediating the regulation of p53 and c-Myc. PLoS Genetics, 7(11), e1002360. https://doi.org/10.1371/journal.pgen.1002360
Hauck, P. M., Wolf, E. R., Olivos, D. J., 3rd, McAtarsney, C. P., & Mayo, L. D. (2017). The fate of murine double minute X (MdmX) is dictated by distinct signaling pathways through murine double minute 2 (Mdm2). Oncotarget, 8(61), 104455–104466. https://doi.org/10.18632/oncotarget.22320
Haupt, Y., Maya, R., Kazaz, A., & Oren, M. (1997). Mdm2 promotes the rapid degradation of p53. Nature, 387(6630), 296–299. https://doi.org/10.1038/387296a0
Hecker, C. M., Rabiller, M., Haglund, K., Bayer, P., & Dikic, I. (2006). Specification of SUMO1- and SUMO2-interacting motifs. Journal of Biological Chemistry, 281(23), 16117–16127. https://doi.org/10.1074/jbc.M512757200
Hendriks, I. A., D’Souza, R. C., Yang, B., Verlaan-de Vries, M., Mann, M., & Vertegaal, A. C. (2014). Uncovering global SUMOylation signaling networks in a site-specific manner. Nature Structural & Molecular Biology, 21(10), 927–936. https://doi.org/10.1038/nsmb.2890
Hock, A. K., Vigneron, A. M., Carter, S., Ludwig, R. L., & Vousden, K. H. (2011). Regulation of p53 stability and function by the deubiquitinating enzyme USP42. EMBO Journal, 30(24), 4921–4930. https://doi.org/10.1038/emboj.2011.419
Honda, R., Tanaka, H., & Yasuda, H. (1997). Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Letters, 420(1), 25–27. https://doi.org/10.1016/s0014-5793(97)01480-4
Hu, W., Feng, Z., & Levine, A. J. (2012). The regulation of multiple p53 stress responses is mediated through MDM2. Genes & Cancer, 3(3–4), 199–208. https://doi.org/10.1177/1947601912454734
Huang, Y. F., & Bulavin, D. V. (2014). Oncogene-mediated regulation of p53 ISGylation and functions. Oncotarget, 5(14), 5808–5818. https://doi.org/10.18632/oncotarget.2199
Huang, Y. F., Wee, S., Gunaratne, J., Lane, D. P., & Bulavin, D. V. (2014). Isg15 controls p53 stability and functions. Cell Cycle, 13(14), 2200–2210. https://doi.org/10.4161/cc.29209
Ito, A., Kawaguchi, Y., Lai, C. H., Kovacs, J. J., Higashimoto, Y., Appella, E., & Yao, T. P. (2002). MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO Journal, 21(22), 6236–6245. https://doi.org/10.1093/emboj/cdf616
Ito, A., Lai, C. H., Zhao, X., Saito, S., Hamilton, M. H., Appella, E., & Yao, T. P. (2001). p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO Journal, 20(6), 1331–1340. https://doi.org/10.1093/emboj/20.6.1331
Jeram, S. M., Srikumar, T., Zhang, X. D., Anne Eisenhauer, H., Rogers, R., Pedrioli, P. G., Matunis, M., & Raught, B. (2010). An improved SUMmOn-based methodology for the identification of ubiquitin and ubiquitin-like protein conjugation sites identifies novel ubiquitin-like protein chain linkages. Proteomics, 10(2), 254–265. https://doi.org/10.1002/pmic.200900648
Jiang, L., Kon, N., Li, T., Wang, S. J., Su, T., Hibshoosh, H., Baer, R., & Gu, W. (2015). Ferroptosis as a p53-mediated activity during tumour suppression. Nature, 520(7545), 57–62. https://doi.org/10.1038/nature14344
Jiang, M., Chiu, S. Y., & Hsu, W. (2011). SUMO-specific protease 2 in Mdm2-mediated regulation of p53. Cell Death and Differentiation, 18(6), 1005–1015. https://doi.org/10.1038/cdd.2010.168
Jin, J., Li, X., Gygi, S. P., & Harper, J. W. (2007). Dual E1 activation systems for ubiquitin differentially regulate E2 enzyme charging. Nature, 447(7148), 1135–1138. https://doi.org/10.1038/nature05902
Jones, J., Wu, K., Yang, Y., Guerrero, C., Nillegoda, N., Pan, Z. Q., & Huang, L. (2008). A targeted proteomic analysis of the ubiquitin-like modifier nedd8 and associated proteins. Journal of Proteome Research, 7(3), 1274–1287. https://doi.org/10.1021/pr700749v
Jones, S. N., Hancock, A. R., Vogel, H., Donehower, L. A., & Bradley, A. (1998). Overexpression of Mdm2 in mice reveals a p53-independent role for Mdm2 in tumorigenesis. Proc Natl Acad Sci U S A, 95(26), 15608–15612. https://doi.org/10.1073/pnas.95.26.15608
Jones, S. N., Roe, A. E., Donehower, L. A., & Bradley, A. (1995). Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature, 378(6553), 206–208. https://doi.org/10.1038/378206a0
Ka, W. H., Cho, S. K., Chun, B. N., Byun, S. Y., & Ahn, J. C. (2018). The ubiquitin ligase COP1 regulates cell cycle and apoptosis by affecting p53 function in human breast cancer cell lines. Breast Cancer, 25(5), 529–538. https://doi.org/10.1007/s12282-018-0849-5
Kahyo, T., Nishida, T., & Yasuda, H. (2001). Involvement of PIAS1 in the sumoylation of tumor suppressor p53. Molecular Cell, 8(3), 713–718. https://doi.org/10.1016/s1097-2765(01)00349-5
Kastenhuber, E. R., & Lowe, S. W. (2017). Putting p53 in context. Cell, 170(6), 1062–1078. https://doi.org/10.1016/j.cell.2017.08.028
Kim, K. I., Giannakopoulos, N. V., Virgin, H. W., & Zhang, D. E. (2004). Interferon-inducible ubiquitin E2, Ubc8, is a conjugating enzyme for protein ISGylation. Molecular and Cellular Biology, 24(21), 9592–9600. https://doi.org/10.1128/MCB.24.21.9592-9600.2004
Ko, L. J., & Prives, C. (1996). p53: Puzzle and paradigm. Genes & Development, 10(9), 1054–1072. https://doi.org/10.1101/gad.10.9.1054
Komander, D. (2009). The emerging complexity of protein ubiquitination. Biochemical Society Transactions, 37(Pt 5), 937–953. https://doi.org/10.1042/BST0370937
Komatsu, M., Chiba, T., Tatsumi, K., Iemura, S., Tanida, I., Okazaki, N., Ueno, T., Kominami, E., Natsume, T., & Tanaka, K. (2004). A novel protein-conjugating system for Ufm1, a ubiquitin-fold modifier. EMBO Journal, 23(9), 1977–1986. https://doi.org/10.1038/sj.emboj.7600205
Krug, R. M., Zhao, C., & Beaudenon, S. (2005). Properties of the ISG15 E1 enzyme UbE1L. Methods in Enzymology, 398, 32–40. https://doi.org/10.1016/S0076-6879(05)98004-X
Kruse, J. P., & Gu, W. (2008). SnapShot: p53 posttranslational modifications. Cell, 133(5), 930-930.e931. https://doi.org/10.1016/j.cell.2008.05.020
Kruse, J. P., & Gu, W. (2009a). Modes of p53 regulation. Cell, 137(4), 609–622. https://doi.org/10.1016/j.cell.2009.04.050
Kruse, J. P., & Gu, W. (2009b). MSL2 promotes Mdm2-independent cytoplasmic localization of p53. Journal of Biological Chemistry, 284(5), 3250–3263. https://doi.org/10.1074/jbc.M805658200
Kubbutat, M. H., Jones, S. N., & Vousden, K. H. (1997). Regulation of p53 stability by Mdm2. Nature, 387(6630), 299–303. https://doi.org/10.1038/387299a0
Kussie, P. H., Gorina, S., Marechal, V., Elenbaas, B., Moreau, J., Levine, A. J., & Pavletich, N. P. (1996). Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science, 274(5289), 948–953. https://doi.org/10.1126/science.274.5289.948
Kwek, S. S., Derry, J., Tyner, A. L., Shen, Z., & Gudkov, A. V. (2001). Functional analysis and intracellular localization of p53 modified by SUMO-1. Oncogene, 20(20), 2587–2599. https://doi.org/10.1038/sj.onc.1204362
Kwon, S. K., Saindane, M., & Baek, K. H. (2017). p53 stability is regulated by diverse deubiquitinating enzymes. Biochimica Et Biophysica Acta - Reviews on Cancer, 1868(2), 404–411. https://doi.org/10.1016/j.bbcan.2017.08.001
Kwon, Y. T., & Ciechanover, A. (2017). The ubiquitin code in the ubiquitin-proteasome system and autophagy. Trends in Biochemical Sciences, 42(11), 873–886. https://doi.org/10.1016/j.tibs.2017.09.002
Lahav-Baratz, S., Kravtsova-Ivantsiv, Y., Golan, S., & Ciechanover, A. (2017). The testis-specific USP26 is a deubiquitinating enzyme of the ubiquitin ligase Mdm2. Biochemical and Biophysical Research Communications, 482(1), 106–111. https://doi.org/10.1016/j.bbrc.2016.10.135
Laine, A., & Ronai, Z. (2007). Regulation of p53 localization and transcription by the HECT domain E3 ligase WWP1. Oncogene, 26(10), 1477–1483. https://doi.org/10.1038/sj.onc.1209924
Lane, D. P. (1992). Cancer.p53, guardian of the genome. Nature, 358(6381), 15–16. https://doi.org/10.1038/358015a0
Le Cam, L., Linares, L. K., Paul, C., Julien, E., Lacroix, M., Hatchi, E., Triboulet, R., Bossis, G., Shmueli, A., Rodriguez, M. S., Coux, O., & Sardet, C. (2006). E4F1 is an atypical ubiquitin ligase that modulates p53 effector functions independently of degradation. Cell, 127(4), 775–788. https://doi.org/10.1016/j.cell.2006.09.031
Lee, E. W., Lee, M. S., Camus, S., Ghim, J., Yang, M. R., Oh, W., Ha, N. C., Lane, D. P., & Song, J. (2009). Differential regulation of p53 and p21 by MKRN1 E3 ligase controls cell cycle arrest and apoptosis. EMBO Journal, 28(14), 2100–2113. https://doi.org/10.1038/emboj.2009.164
Lee, L., Perez Oliva, A. B., Martinez-Balsalobre, E., Churikov, D., Peter, J., Rahmouni, D., Audoly, G., Azzoni, V., Audebert, S., Camoin, L., Mulero, V., Cayuela, M. L., Kulathu, Y., Geli, V., & Lachaud, C. (2021). UFMylation of MRE11 is essential for telomere length maintenance and hematopoietic stem cell survival. Science Advances, 7(39), eabc7371. https://doi.org/10.1126/sciadv.abc7371
Leidecker, O., Matic, I., Mahata, B., Pion, E., & Xirodimas, D. P. (2012). The ubiquitin E1 enzyme Ube1 mediates NEDD8 activation under diverse stress conditions. Cell Cycle, 11(6), 1142–1150. https://doi.org/10.4161/cc.11.6.19559
Lemaire, K., Moura, R. F., Granvik, M., Igoillo-Esteve, M., Hohmeier, H. E., Hendrickx, N., Newgard, C. B., Waelkens, E., Cnop, M., & Schuit, F. (2011). Ubiquitin fold modifier 1 (UFM1) and its target UFBP1 protect pancreatic beta cells from ER stress-induced apoptosis. PLoS ONE, 6(4), e18517. https://doi.org/10.1371/journal.pone.0018517
Leng, L., Xu, C., Wei, C., Zhang, J., Liu, B., Ma, J., Li, N., Qin, W., Zhang, W., Zhang, C., Xing, X., Zhai, L., Yang, F., Li, M., Jin, C., Yuan, Y., Xu, P., Qin, J., Xie, H., He, F., & Wang, J. (2014). A proteomics strategy for the identification of FAT10-modified sites by mass spectrometry. Journal of Proteome Research, 13(1), 268–276. https://doi.org/10.1021/pr400395k
Leng, R. P., Lin, Y., Ma, W., Wu, H., Lemmers, B., Chung, S., Parant, J. M., Lozano, G., Hakem, R., & Benchimol, S. (2003). Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell, 112(6), 779–791. https://doi.org/10.1016/s0092-8674(03)00193-4
Li, F., Han, H., Sun, Q., Liu, K., Lin, N., Xu, C., Zhao, Z., & Zhao, W. (2019). USP28 regulates deubiquitination of histone H2A and cell proliferation. Experimental Cell Research, 379(1), 11–18. https://doi.org/10.1016/j.yexcr.2019.03.026
Li, J., Lu, D., Dou, H., Liu, H., Weaver, K., Wang, W., Yeh, E. T. H., Williams, B. O., Zheng, L., & Yang, T. (2018). Desumoylase SENP6 maintains osteochondroprogenitor homeostasis by suppressing the p53 pathway. Nature Communications, 9(1), 143. https://doi.org/10.1038/s41467-017-02413-3
Li, J., Yue, G., Ma, W., Zhang, A., Zou, J., Cai, Y., Tang, X., Wang, J., Liu, J., Li, H., & Su, H. (2018). Ufm1-specific ligase Ufl1 regulates endoplasmic reticulum homeostasis and protects against heart failure. Circulation. Heart Failure, 11(10), e004917. https://doi.org/10.1161/CIRCHEARTFAILURE.118.004917
Li, M., Brooks, C. L., Kon, N., & Gu, W. (2004). A dynamic role of HAUSP in the p53-Mdm2 pathway. Molecular Cell, 13(6), 879–886. https://doi.org/10.1016/s1097-2765(04)00157-1
Li, M., Brooks, C. L., Wu-Baer, F., Chen, D., Baer, R., & Gu, W. (2003). Mono- versus polyubiquitination: Differential control of p53 fate by Mdm2. Science, 302(5652), 1972–1975. https://doi.org/10.1126/science.1091362
Li, M., Chen, D., Shiloh, A., Luo, J., Nikolaev, A. Y., Qin, J., & Gu, W. (2002). Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature, 416(6881), 648–653. https://doi.org/10.1038/nature737
Li, M., Luo, J., Brooks, C. L., & Gu, W. (2002). Acetylation of p53 inhibits its ubiquitination by Mdm2. Journal of Biological Chemistry, 277(52), 50607–50611. https://doi.org/10.1074/jbc.C200578200
Li, Q., Lin, S., Wang, X., Lian, G., Lu, Z., Guo, H., Ruan, K., Wang, Y., Ye, Z., Han, J., & Lin, S. C. (2009). Axin determines cell fate by controlling the p53 activation threshold after DNA damage. Nature Cell Biology, 11(9), 1128–1134. https://doi.org/10.1038/ncb1927
Li, T., Santockyte, R., Shen, R. F., Tekle, E., Wang, G., Yang, D. C., & Chock, P. B. (2006). Expression of SUMO-2/3 induced senescence through p53- and pRB-mediated pathways. Journal of Biological Chemistry, 281(47), 36221–36227. https://doi.org/10.1074/jbc.M608236200
Li, T., Santockyte, R., Yu, S., Shen, R. F., Tekle, E., Lee, C. G., Yang, D. C., & Chock, P. B. (2011). FAT10 modifies p53 and upregulates its transcriptional activity. Archives of Biochemistry and Biophysics, 509(2), 164–169. https://doi.org/10.1016/j.abb.2011.02.017
Liang, J. R., Lingeman, E., Luong, T., Ahmed, S., Muhar, M., Nguyen, T., Olzmann, J. A., & Corn, J. E. (2020). A genome-wide ER-phagy screen highlights key roles of mitochondrial metabolism and ER-resident UFMylation. Cell, 180(6), 1160-1177.e1120. https://doi.org/10.1016/j.cell.2020.02.017
Liao, P., Bhattarai, N., Cao, B., Zhou, X., Jung, J. H., Damera, K., Fuselier, T. T., Thareja, S., Wimley, W. C., Wang, B., Zeng, S. X., & Lu, H. (2020). Crotonylation at serine 46 impairs p53 activity. Biochemical and Biophysical Research Communications, 524(3), 730–735. https://doi.org/10.1016/j.bbrc.2020.01.152
Linares, L. K., Kiernan, R., Triboulet, R., Chable-Bessia, C., Latreille, D., Cuvier, O., Lacroix, M., Le Cam, L., Coux, O., & Benkirane, M. (2007). Intrinsic ubiquitination activity of PCAF controls the stability of the oncoprotein Hdm2. Nature Cell Biology, 9(3), 331–338. https://doi.org/10.1038/ncb1545
Liu, C., Chang, R., Yao, X., Qiao, W. T., & Geng, Y. Q. (2009). ISG15 expression in response to double-stranded RNA or LPS in cultured fetal bovine lung (FBL) cells. Veterinary Research Communications, 33(7), 723–733. https://doi.org/10.1007/s11259-009-9221-8
Liu, F., Gao, X., Wang, J., Gao, C., Li, X., Gong, X., & Zeng, X. (2016). Transcriptome sequencing to identify transcription factor regulatory network and alternative splicing in endothelial cells under VEGF stimulation. Journal of Molecular Neuroscience, 58(2), 170–177. https://doi.org/10.1007/s12031-015-0653-z
Liu, G., & Xirodimas, D. P. (2010). NUB1 promotes cytoplasmic localization of p53 through cooperation of the NEDD8 and ubiquitin pathways. Oncogene, 29(15), 2252–2261. https://doi.org/10.1038/onc.2009.494
Liu, H., Li, X., Ning, G., Zhu, S., Ma, X., Liu, X., Liu, C., Huang, M., Schmitt, I., Wullner, U., Niu, Y., Guo, C., Wang, Q., & Tang, T. S. (2016). The Machado-Joseph disease deubiquitinase ataxin-3 regulates the stability and apoptotic function of p53. PLoS Biology, 14(11), e2000733. https://doi.org/10.1371/journal.pbio.2000733
Liu, J., Guan, D., Dong, M., Yang, J., Wei, H., Liang, Q., Song, L., Xu, L., Bai, J., Liu, C., Mao, J., Zhang, Q., Zhou, J., Wu, X., Wang, M., & Cong, Y. S. (2020). UFMylation maintains tumour suppressor p53 stability by antagonizing its ubiquitination. Nature Cell Biology, 22(9), 1056–1063. https://doi.org/10.1038/s41556-020-0559-z
Liu, J., Wang, Y., Song, L., Zeng, L., Yi, W., Liu, T., Chen, H., Wang, M., Ju, Z., & Cong, Y. S. (2017). A critical role of DDRGK1 in endoplasmic reticulum homoeostasis via regulation of IRE1alpha stability. Nature Communications, 8, 14186. https://doi.org/10.1038/ncomms14186
Liu, J., Zhang, C., Hu, W., & Feng, Z. (2019). Tumor suppressor p53 and metabolism. Journal of Molecular Cell Biology, 11(4), 284–292. https://doi.org/10.1093/jmcb/mjy070
Liu, X., Chen, L., Ge, J., Yan, C., Huang, Z., Hu, J., Wen, C., Li, M., Huang, D., Qiu, Y., Hao, H., Yuan, R., Lei, J., Yu, X., & Shao, J. (2016). The ubiquitin-like protein FAT10 stabilizes eEF1A1 expression to promote tumor proliferation in a complex manner. Cancer Research, 76(16), 4897–4907. https://doi.org/10.1158/0008-5472.CAN-15-3118
Liu, X., Tan, Y., Zhang, C., Zhang, Y., Zhang, L., Ren, P., Deng, H., Luo, J., Ke, Y., & Du, X. (2016). NAT10 regulates p53 activation through acetylating p53 at K120 and ubiquitinating Mdm2. EMBO Reports, 17(3), 349–366
Liu, Y. C., Pan, J., Zhang, C., Fan, W., Collinge, M., Bender, J. R., & Weissman, S. M. (1999). A MHC-encoded ubiquitin-like protein (FAT10) binds noncovalently to the spindle assembly checkpoint protein MAD2. Proceedings of the National Academy of Sciences of the United States of America, 96(8), 4313–4318. https://doi.org/10.1073/pnas.96.8.4313
Liu, Y., Tavana, O., & Gu, W. (2019). p53 modifications: Exquisite decorations of the powerful guardian. Journal of Molecular Cell Biology, 11(7), 564–577. https://doi.org/10.1093/jmcb/mjz060
Lou, Z., Wei, J., Riethman, H., Baur, J. A., Voglauer, R., Shay, J. W., & Wright, W. E. (2009). Telomere length regulates ISG15 expression in human cells. Aging (albany NY), 1(7), 608–621. https://doi.org/10.18632/aging.100066
Luo, J., Lu, Z., Lu, X., Chen, L., Cao, J., Zhang, S., Ling, Y., & Zhou, X. (2013). OTUD5 regulates p53 stability by deubiquitinating p53. PLoS ONE, 8(10), e77682. https://doi.org/10.1371/journal.pone.0077682
Luo, J., Su, F., Chen, D., Shiloh, A., & Gu, W. (2000). Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature, 408(6810), 377–381. https://doi.org/10.1038/35042612
Marine, J. C., Dyer, M. A., & Jochemsen, A. G. (2007). MDMX: From bench to bedside. Journal of Cell Science, 120(Pt 3), 371–378. https://doi.org/10.1242/jcs.03362
Marx, J. (1993). How p53 suppresses cell growth. Science, 262(5140), 1644–1645. https://doi.org/10.1126/science.8259506
Maya, R., Balass, M., Kim, S. T., Shkedy, D., Leal, J. F., Shifman, O., Moas, M., Buschmann, T., Ronai, Z., Shiloh, Y., Kastan, M. B., Katzir, E., & Oren, M. (2001). ATM-dependent phosphorylation of Mdm2 on serine 395: Role in p53 activation by DNA damage. Genes & Development, 15(9), 1067–1077. https://doi.org/10.1101/gad.886901
Meek, D. W., & Anderson, C. W. (2009). Posttranslational modification of p53: Cooperative integrators of function. Cold Spring Harbor Perspectives in Biology, 1(6), a000950. https://doi.org/10.1101/cshperspect.a000950
Nakamura, S., Roth, J. A., & Mukhopadhyay, T. (2000). Multiple lysine mutations in the C-terminal domain of p53 interfere with MDM2-dependent protein degradation and ubiquitination. Molecular and Cellular Biology, 20(24), 9391–9398. https://doi.org/10.1128/MCB.20.24.9391-9398.2000
Nakashima, H., Nguyen, T., Goins, W. F., & Chiocca, E. A. (2015). Interferon-stimulated gene 15 (ISG15) and ISG15-linked proteins can associate with members of the selective autophagic process, histone deacetylase 6 (HDAC6) and SQSTM1/p62. Journal of Biological Chemistry, 290(3), 1485–1495. https://doi.org/10.1074/jbc.M114.593871
Nakka, V. P., Lang, B. T., Lenschow, D. J., Zhang, D. E., Dempsey, R. J., & Vemuganti, R. (2011). Increased cerebral protein ISGylation after focal ischemia is neuroprotective. Journal of Cerebral Blood Flow and Metabolism, 31(12), 2375–2384. https://doi.org/10.1038/jcbfm.2011.103
Nelson, V., Davis, G. E., & Maxwell, S. A. (2001). A putative protein inhibitor of activated STAT (PIASy) interacts with p53 and inhibits p53-mediated transactivation but not apoptosis. Apoptosis, 6(3), 221–234. https://doi.org/10.1023/a:1011392811628
Nie, Q., Chen, H., Zou, M., Wang, L., Hou, M., Xiang, J. W., Luo, Z., Gong, X. D., Fu, J. L., Wang, Y., Zheng, S. Y., Xiao, Y., Gan, Y. W., Gao, Q., Bai, Y. Y., Wang, J. M., Zhang, L., Tang, X. C., Hu, X., Gong, L., Liu, Y., & Li, D. W. (2021). The E3 ligase PIAS1 regulates p53 sumoylation to control stress-induced apoptosis of lens epithelial cells through the proapoptotic regulator bax. Front Cell Dev Biol, 9, 660494. https://doi.org/10.3389/fcell.2021.660494
Oda, E., Ohki, R., Murasawa, H., Nemoto, J., Shibue, T., Yamashita, T., Tokino, T., Taniguchi, T., & Tanaka, N. (2000). Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science, 288(5468), 1053–1058. https://doi.org/10.1126/science.288.5468.1053
Ofir-Rosenfeld, Y., Boggs, K., Michael, D., Kastan, M. B., & Oren, M. (2008). Mdm2 regulates p53 mRNA translation through inhibitory interactions with ribosomal protein L26. Molecular Cell, 32(2), 180–189. https://doi.org/10.1016/j.molcel.2008.08.031
Okumura, F., Okumura, A. J., Uematsu, K., Hatakeyama, S., Zhang, D. E., & Kamura, T. (2013). Activation of double-stranded RNA-activated protein kinase (PKR) by interferon-stimulated gene 15 (ISG15) modification down-regulates protein translation. Journal of Biological Chemistry, 288(4), 2839–2847. https://doi.org/10.1074/jbc.M112.401851
Owerbach, D., McKay, E. M., Yeh, E. T., Gabbay, K. H., & Bohren, K. M. (2005). A proline-90 residue unique to SUMO-4 prevents maturation and sumoylation. Biochemical and Biophysical Research Communications, 337(2), 517–520. https://doi.org/10.1016/j.bbrc.2005.09.090
Pan, M., & Blattner, C. (2021). Regulation of p53 by E3s. Cancers (basel). https://doi.org/10.3390/cancers13040745
Park, J. H., Yang, S. W., Park, J. M., Ka, S. H., Kim, J. H., Kong, Y. Y., Jeon, Y. J., Seol, J. H., & Chung, C. H. (2016). Positive feedback regulation of p53 transactivity by DNA damage-induced ISG15 modification. Nature Communications, 7, 12513. https://doi.org/10.1038/ncomms12513
Park, S. Y., Phorl, S., Jung, S., Sovannarith, K., Lee, S. I., Noh, S., Han, M., Naskar, R., Kim, J. Y., Choi, Y. J., & Lee, J. Y. (2017). HDAC6 deficiency induces apoptosis in mesenchymal stem cells through p53 K120 acetylation. Biochemical and Biophysical Research Communications, 494(1–2), 51–56. https://doi.org/10.1016/j.bbrc.2017.10.087
Pearce, M. J., Mintseris, J., Ferreyra, J., Gygi, S. P., & Darwin, K. H. (2008). Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science, 322(5904), 1104–1107. https://doi.org/10.1126/science.1163885
Pennella, M. A., Liu, Y., Woo, J. L., Kim, C. A., & Berk, A. J. (2010). Adenovirus E1B 55-kilodalton protein is a p53-SUMO1 E3 ligase that represses p53 and stimulates its nuclear export through interactions with promyelocytic leukemia nuclear bodies. Journal of Virology, 84(23), 12210–12225. https://doi.org/10.1128/JVI.01442-10
Pereg, Y., Shkedy, D., de Graaf, P., Meulmeester, E., Edelson-Averbukh, M., Salek, M., Biton, S., Teunisse, A. F., Lehmann, W. D., Jochemsen, A. G., & Shiloh, Y. (2005). Phosphorylation of Hdmx mediates its Hdm2- and ATM-dependent degradation in response to DNA damage. Proc Natl Acad Sci U S A, 102(14), 5056–5061. https://doi.org/10.1073/pnas.0408595102
Pitha-Rowe, I., Hassel, B. A., & Dmitrovsky, E. (2004). Involvement of UBE1L in ISG15 conjugation during retinoid-induced differentiation of acute promyelocytic leukemia. Journal of Biological Chemistry, 279(18), 18178–18187. https://doi.org/10.1074/jbc.M309259200
Qin, B., Yu, J., Nowsheen, S., Wang, M., Tu, X., Liu, T., Li, H., Wang, L., & Lou, Z. (2019). UFL1 promotes histone H4 ufmylation and ATM activation. Nature Communications, 10(1), 1242. https://doi.org/10.1038/s41467-019-09175-0
Qin, B., Yu, J., Nowsheen, S., Zhao, F., Wang, L., & Lou, Z. (2020). STK38 promotes ATM activation by acting as a reader of histone H4 ufmylation. Science Advances, 6(23), eaxx8214. https://doi.org/10.1126/sciadv.aax8214
Rodriguez, M. S., Desterro, J. M., Lain, S., Lane, D. P., & Hay, R. T. (2000). Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Molecular and Cellular Biology, 20(22), 8458–8467. https://doi.org/10.1128/MCB.20.22.8458-8467.2000
Rodriguez, M. S., Desterro, J. M., Lain, S., Midgley, C. A., Lane, D. P., & Hay, R. T. (1999). SUMO-1 modification activates the transcriptional response of p53. EMBO Journal, 18(22), 6455–6461. https://doi.org/10.1093/emboj/18.22.6455
Rodriguez-Bravo, V., Maciejowski, J., Corona, J., Buch, H. K., Collin, P., Kanemaki, M. T., Shah, J. V., & Jallepalli, P. V. (2014). Nuclear pores protect genome integrity by assembling a premitotic and Mad1-dependent anaphase inhibitor. Cell, 156(5), 1017–1031. https://doi.org/10.1016/j.cell.2014.01.010
Russo, A., & Russo, G. (2017). Ribosomal Proteins Control or Bypass p53 during Nucleolar Stress. International Journal of Molecular Sciences. https://doi.org/10.3390/ijms18010140
Ryan, S. D., Britigan, E. M., Zasadil, L. M., Witte, K., Audhya, A., Roopra, A., & Weaver, B. A. (2012). Up-regulation of the mitotic checkpoint component Mad1 causes chromosomal instability and resistance to microtubule poisons. Proceedings of the National Academy of Sciences of the United States of America, 109(33), E2205-2214. https://doi.org/10.1073/pnas.1201911109
Ryu, H. W., Shin, D. H., Lee, D. H., Choi, J., Han, G., Lee, K. Y., & Kwon, S. H. (2017). HDAC6 deacetylates p53 at lysines 381/382 and differentially coordinates p53-induced apoptosis. Cancer Letters, 391, 162–171. https://doi.org/10.1016/j.canlet.2017.01.033
Saadatzadeh, M. R., Elmi, A. N., Pandya, P. H., Bijangi-Vishehsaraei, K., Ding, J., Stamatkin, C. W., Cohen-Gadol, A. A., & Pollok, K. E. (2017). The role of MDM2 in promoting genome stability versus instability. International Journal of Molecular Sciences. https://doi.org/10.3390/ijms18102216
Sadler, A. J., & Williams, B. R. (2008). Interferon-inducible antiviral effectors. Nature Reviews Immunology, 8(7), 559–568. https://doi.org/10.1038/nri2314
Saitoh, H., & Hinchey, J. (2000). Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. Journal of Biological Chemistry, 275(9), 6252–6258. https://doi.org/10.1074/jbc.275.9.6252
Sanford, J. D., Yang, J., Han, J., Tollini, L. A., Jin, A., & Zhang, Y. (2021). MDMX is essential for the regulation of p53 protein levels in the absence of a functional MDM2 C-terminal tail. BMC Mol Cell Biol, 22(1), 46. https://doi.org/10.1186/s12860-021-00385-3
Santonico, E. (2020). Old and new concepts in ubiquitin and NEDD8 recognition. Biomolecules. https://doi.org/10.3390/biom10040566
Scheffner, M., Huibregtse, J. M., Vierstra, R. D., & Howley, P. M. (1993). The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell, 75(3), 495–505. https://doi.org/10.1016/0092-8674(93)90384-3
Schmidt, D., & Muller, S. (2002). Members of the PIAS family act as SUMO ligases for c-Jun and p53 and repress p53 activity. Proceedings of the National Academy of Sciences of the United States of America, 99(5), 2872–2877. https://doi.org/10.1073/pnas.052559499
Schmidtke, G., Aichem, A., & Groettrup, M. (2014). FAT10ylation as a signal for proteasomal degradation. Biochimica Et Biophysica Acta, 1843(1), 97–102. https://doi.org/10.1016/j.bbamcr.2013.01.009
Sen, G. C., & Sarkar, S. N. (2007). The interferon-stimulated genes: Targets of direct signaling by interferons, double-stranded RNA, and viruses. Current Topics in Microbiology and Immunology, 316, 233–250. https://doi.org/10.1007/978-3-540-71329-6_12
Shen, J., Li, P., Shao, X., Yang, Y., Liu, X., Feng, M., Yu, Q., Hu, R., & Wang, Z. (2018). The E3 ligase RING1 targets p53 for degradation and promotes cancer cell proliferation and survival. Cancer Research, 78(2), 359–371. https://doi.org/10.1158/0008-5472.CAN-17-1805
Sheng, Y., Laister, R. C., Lemak, A., Wu, B., Tai, E., Duan, S., Lukin, J., Sunnerhagen, M., Srisailam, S., Karra, M., Benchimol, S., & Arrowsmith, C. H. (2008). Molecular basis of Pirh2-mediated p53 ubiquitylation. Nature Structural & Molecular Biology, 15(12), 1334–1342. https://doi.org/10.1038/nsmb.1521
Sheren, J. E., & Kassenbrock, C. K. (2013). RNF38 encodes a nuclear ubiquitin protein ligase that modifies p53. Biochemical and Biophysical Research Communications, 440(4), 473–478. https://doi.org/10.1016/j.bbrc.2013.08.031
Shi, D., Pop, M. S., Kulikov, R., Love, I. M., Kung, A. L., & Grossman, S. R. (2009). CBP and p300 are cytoplasmic E4 polyubiquitin ligases for p53. Proceedings of the National Academy of Sciences of the United States of America, 106(38), 16275–16280. https://doi.org/10.1073/pnas.0904305106
Shieh, S. Y., Ikeda, M., Taya, Y., & Prives, C. (1997). DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell, 91(3), 325–334. https://doi.org/10.1016/s0092-8674(00)80416-x
Shloush, J., Vlassov, J. E., Engson, I., Duan, S., Saridakis, V., Dhe-Paganon, S., Raught, B., Sheng, Y., & Arrowsmith, C. H. (2011). Structural and functional comparison of the RING domains of two p53 E3 ligases, Mdm2 and Pirh2. Journal of Biological Chemistry, 286(6), 4796–4808. https://doi.org/10.1074/jbc.M110.157669
Singh, R. K., Iyappan, S., & Scheffner, M. (2007). Hetero-oligomerization with MdmX rescues the ubiquitin/Nedd8 ligase activity of RING finger mutants of Mdm2. Journal of Biological Chemistry, 282(15), 10901–10907. https://doi.org/10.1074/jbc.M610879200
Stevenson, L. F., Sparks, A., Allende-Vega, N., Xirodimas, D. P., Lane, D. P., & Saville, M. K. (2007). The deubiquitinating enzyme USP2a regulates the p53 pathway by targeting Mdm2. EMBO Journal, 26(4), 976–986. https://doi.org/10.1038/sj.emboj.7601567
Stindt, M. H., Carter, S., Vigneron, A. M., Ryan, K. M., & Vousden, K. H. (2011). MDM2 promotes SUMO-2/3 modification of p53 to modulate transcriptional activity. Cell Cycle, 10(18), 3176–3188. https://doi.org/10.4161/cc.10.18.17436
Sun, L., Shi, L., Li, W., Yu, W., Liang, J., Zhang, H., Yang, X., Wang, Y., Li, R., Yao, X., Yi, X., & Shang, Y. (2009). JFK, a Kelch domain-containing F-box protein, links the SCF complex to p53 regulation. Proceedings of the National Academy of Sciences of the United States of America, 106(25), 10195–10200. https://doi.org/10.1073/pnas.0901864106
Sun, L., Shi, L., Wang, F., Huangyang, P., Si, W., Yang, J., Yao, Z., & Shang, Y. (2011). Substrate phosphorylation and feedback regulation in JFK-promoted p53 destabilization. Journal of Biological Chemistry, 286(6), 4226–4235. https://doi.org/10.1074/jbc.M110.195115
Sun, X. X., Challagundla, K. B., & Dai, M. S. (2012). Positive regulation of p53 stability and activity by the deubiquitinating enzyme Otubain 1. EMBO Journal, 31(3), 576–592. https://doi.org/10.1038/emboj.2011.434
Swatek, K. N., & Komander, D. (2016). Ubiquitin modifications. Cell Research, 26(4), 399–422. https://doi.org/10.1038/cr.2016.39
Takayama, K. I., Suzuki, T., Tanaka, T., Fujimura, T., Takahashi, S., Urano, T., Ikeda, K., & Inoue, S. (2018). TRIM25 enhances cell growth and cell survival by modulating p53 signals via interaction with G3BP2 in prostate cancer. Oncogene, 37(16), 2165–2180. https://doi.org/10.1038/s41388-017-0095-x
Tang, Y., Zhao, W., Chen, Y., Zhao, Y., & Gu, W. (2008). Acetylation is indispensable for p53 activation. Cell, 133(4), 612–626. https://doi.org/10.1016/j.cell.2008.03.025
Tatsumi, K., Sou, Y. S., Tada, N., Nakamura, E., Iemura, S., Natsume, T., Kang, S. H., Chung, C. H., Kasahara, M., Kominami, E., Yamamoto, M., Tanaka, K., & Komatsu, M. (2010). A novel type of E3 ligase for the Ufm1 conjugation system. Journal of Biological Chemistry, 285(8), 5417–5427. https://doi.org/10.1074/jbc.M109.036814
Tatsumi, K., Yamamoto-Mukai, H., Shimizu, R., Waguri, S., Sou, Y. S., Sakamoto, A., Taya, C., Shitara, H., Hara, T., Chung, C. H., Tanaka, K., Yamamoto, M., & Komatsu, M. (2011). The Ufm1-activating enzyme Uba5 is indispensable for erythroid differentiation in mice. Nature Communications, 2, 181. https://doi.org/10.1038/ncomms1182
Taylor, J. L., D’Cunha, J., Tom, P., O’Brien, W. J., & Borden, E. C. (1996). Production of ISG-15, an interferon-inducible protein, in human corneal cells. Journal of Interferon and Cytokine Research, 16(11), 937–940. https://doi.org/10.1089/jir.1996.16.937
Ulrich, H. D. (2008). The fast-growing business of SUMO chains. Molecular Cell, 32(3), 301–305. https://doi.org/10.1016/j.molcel.2008.10.010
Unger, T., Juven-Gershon, T., Moallem, E., Berger, M., Vogt Sionov, R., Lozano, G., Oren, M., & Haupt, Y. (1999). Critical role for Ser20 of human p53 in the negative regulation of p53 by Mdm2. EMBO Journal, 18(7), 1805–1814. https://doi.org/10.1093/emboj/18.7.1805
Vertegaal, A. C. (2010). SUMO chains: Polymeric signals. Biochemical Society Transactions, 38(Pt 1), 46–49. https://doi.org/10.1042/BST0380046
Villarroya-Beltri, C., Baixauli, F., Mittelbrunn, M., Fernandez-Delgado, I., Torralba, D., Moreno-Gonzalo, O., Baldanta, S., Enrich, C., Guerra, S., & Sanchez-Madrid, F. (2016). (2016). ISGylation controls exosome secretion by promoting lysosomal degradation of MVB proteins. Nature Communications, 7, 13588. https://doi.org/10.1038/ncomms13588
Villarroya-Beltri, C., Guerra, S., & Sanchez-Madrid, F. (2017). ISGylation—A key to lock the cell gates for preventing the spread of threats. Journal of Cell Science, 130(18), 2961–2969. https://doi.org/10.1242/jcs.205468
Vousden, K. H., & Prives, C. (2009). Blinded by the light: the growing complexity of p53. Cell, 137(3), 413–431. https://doi.org/10.1016/j.cell.2009.04.037
Wada, H., Kito, K., Caskey, L. S., Yeh, E. T., & Kamitani, T. (1998). Cleavage of the C-terminus of NEDD8 by UCH-L3. Biochemical and Biophysical Research Communications, 251(3), 688–692. https://doi.org/10.1006/bbrc.1998.9532
Walczak, C. P., Leto, D. E., Zhang, L., Riepe, C., Muller, R. Y., DaRosa, P. A., Ingolia, N. T., Elias, J. E., & Kopito, R. R. (2019). Ribosomal protein RPL26 is the principal target of UFMylation. Proc Natl Acad Sci U S A, 116(4), 1299–1308. https://doi.org/10.1073/pnas.1816202116
Wan, J., Block, S., Scribano, C. M., Thiry, R., Esbona, K., Audhya, A., & Weaver, B. A. (2019). Mad1 destabilizes p53 by preventing PML from sequestering MDM2. Nature Communications, 10(1), 1540. https://doi.org/10.1038/s41467-019-09471-9
Wang, L., Xu, Y., Rogers, H., Saidi, L., Noguchi, C. T., Li, H., Yewdell, J. W., Guydosh, N. R., & Ye, Y. (2020). UFMylation of RPL26 links translocation-associated quality control to endoplasmic reticulum protein homeostasis. Cell Research, 30(1), 5–20. https://doi.org/10.1038/s41422-019-0236-6
Wang, Y., Ding, Q., Xu, T., Li, C. Y., Zhou, D. D., & Zhang, L. (2017). HZ-6d targeted HERC5 to regulate p53 ISGylation in human hepatocellular carcinoma. Toxicology and Applied Pharmacology, 334, 180–191. https://doi.org/10.1016/j.taap.2017.09.011
Wang, Z., Gong, Y., Peng, B., Shi, R., Fan, D., Zhao, H., Zhu, M., Zhang, H., Lou, Z., Zhou, J., Zhu, W. G., Cong, Y. S., & Xu, X. (2019). MRE11 UFMylation promotes ATM activation. Nucleic Acids Research, 47(8), 4124–4135. https://doi.org/10.1093/nar/gkz110
Wang, Z., Zhu, W. G., & Xu, X. (2017). Ubiquitin-like modifications in the DNA damage response. Mutation Research, 803–805, 56–75. https://doi.org/10.1016/j.mrfmmm.2017.07.001
Watson, I. R., Li, B. K., Roche, O., Blanch, A., Ohh, M., & Irwin, M. S. (2010). Chemotherapy induces NEDP1-mediated destabilization of MDM2. Oncogene, 29(2), 297–304. https://doi.org/10.1038/onc.2009.314
Weger, S., Hammer, E., & Heilbronn, R. (2005). Topors acts as a SUMO-1 E3 ligase for p53 in vitro and in vivo. FEBS Letters, 579(22), 5007–5012. https://doi.org/10.1016/j.febslet.2005.07.088
Wei, T., Biskup, E., Gjerdrum, L. M., Niazi, O., Odum, N., & Gniadecki, R. (2016). Ubiquitin-specific protease 2 decreases p53-dependent apoptosis in cutaneous T-cell lymphoma. Oncotarget, 7(30), 48391–48400. https://doi.org/10.18632/oncotarget.10268
Wei, W., Yang, P., Pang, J., Zhang, S., Wang, Y., Wang, M. H., Dong, Z., She, J. X., & Wang, C. Y. (2008). A stress-dependent SUMO4 sumoylation of its substrate proteins. Biochemical and Biophysical Research Communications, 375(3), 454–459. https://doi.org/10.1016/j.bbrc.2008.08.028
Wen, W., Peng, C., Kim, M. O., Ho Jeong, C., Zhu, F., Yao, K., Zykova, T., Ma, W., Carper, A., Langfald, A., Bode, A. M., & Dong, Z. (2014). Knockdown of RNF2 induces apoptosis by regulating MDM2 and p53 stability. Oncogene, 33(4), 421–428. https://doi.org/10.1038/onc.2012.605
Wu, K., Yamoah, K., Dolios, G., Gan-Erdene, T., Tan, P., Chen, A., Lee, C. G., Wei, N., Wilkinson, K. D., Wang, R., & Pan, Z. Q. (2003). DEN1 is a dual function protease capable of processing the C terminus of Nedd8 and deconjugating hyper-neddylated CUL1. Journal of Biological Chemistry, 278(31), 28882–28891. https://doi.org/10.1074/jbc.M302888200
Wu, X., Bayle, J. H., Olson, D., & Levine, A. J. (1993). The p53-mdm-2 autoregulatory feedback loop. Genes & Development, 7(7A), 1126–1132. https://doi.org/10.1101/gad.7.7a.1126
Xenaki, G., Ontikatze, T., Rajendran, R., Stratford, I. J., Dive, C., Krstic-Demonacos, M., & Demonacos, C. (2008). PCAF is an HIF-1alpha cofactor that regulates p53 transcriptional activity in hypoxia. Oncogene, 27(44), 5785–5796. https://doi.org/10.1038/onc.2008.192
Xiang, S., Shao, X., Cao, J., Yang, B., He, Q., & Ying, M. (2020). FAT10: Function and relationship with cancer. Current Molecular Pharmacology, 13(3), 182–191. https://doi.org/10.2174/1874467212666191113130312
Xirodimas, D. P., Saville, M. K., Bourdon, J. C., Hay, R. T., & Lane, D. P. (2004). Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell, 118(1), 83–97. https://doi.org/10.1016/j.cell.2004.06.016
Xirodimas, D. P., Sundqvist, A., Nakamura, A., Shen, L., Botting, C., & Hay, R. T. (2008). Ribosomal proteins are targets for the NEDD8 pathway. EMBO Reports, 9(3), 280–286. https://doi.org/10.1038/embor.2008.10
Yang, J., Zhou, Y., Xie, S., Wang, J., Li, Z., Chen, L., Mao, M., Chen, C., Huang, A., Chen, Y., Zhang, X., Khan, N. U. H., Wang, L., & Zhou, J. (2021). Metformin induces ferroptosis by inhibiting UFMylation of SLC7A11 in breast cancer. Journal of Experimental & Clinical Cancer Research, 40(1), 206. https://doi.org/10.1186/s13046-021-02012-7
Yates, K. E., Korbel, G. A., Shtutman, M., Roninson, I. B., & DiMaio, D. (2008). Repression of the SUMO-specific protease Senp1 induces p53-dependent premature senescence in normal human fibroblasts. Aging Cell, 7(5), 609–621. https://doi.org/10.1111/j.1474-9726.2008.00411.x
Yoo, H. M., Kang, S. H., Kim, J. Y., Lee, J. E., Seong, M. W., Lee, S. W., Ka, S. H., Sou, Y. S., Komatsu, M., Tanaka, K., Lee, S. T., Noh, D. Y., Baek, S. H., Jeon, Y. J., & Chung, C. H. (2014). Modification of ASC1 by UFM1 is crucial for ERalpha transactivation and breast cancer development. Molecular Cell, 56(2), 261–274. https://doi.org/10.1016/j.molcel.2014.08.007
Yu, M., Shi, X., Ren, M., Liu, L., Qi, H., Zhang, C., Zou, J., Qiu, X., Zhu, W. G., Zhang, Y. E., Wang, W., & Luo, J. (2020). SIRT7 deacetylates STRAP to regulate p53 activity and stability. International Journal of Molecular Sciences. https://doi.org/10.3390/ijms21114122
Yuan, J., Luo, K., Zhang, L., Cheville, J. C., & Lou, Z. (2010). USP10 regulates p53 localization and stability by deubiquitinating p53. Cell, 140(3), 384–396. https://doi.org/10.1016/j.cell.2009.12.032
Zhang, D. W., Jeang, K. T., & Lee, C. G. (2006). p53 negatively regulates the expression of FAT10, a gene upregulated in various cancers. Oncogene, 25(16), 2318–2327. https://doi.org/10.1038/sj.onc.1209220
Zhang, M., Zhu, X., Zhang, Y., Cai, Y., Chen, J., Sivaprakasam, S., Gurav, A., Pi, W., Makala, L., Wu, J., Pace, B., Tuan-Lo, D., Ganapathy, V., Singh, N., & Li, H. (2015). RCAD/Ufl1, a Ufm1 E3 ligase, is essential for hematopoietic stem cell function and murine hematopoiesis. Cell Death and Differentiation, 22(12), 1922–1934. https://doi.org/10.1038/cdd.2015.51
Zhang, S., & Sun, Y. (2020). Cullin RING Ligase 5 (CRL-5): Neddylation Activation and Biological Functions. Advances in Experimental Medicine and Biology, 1217, 261–283. https://doi.org/10.1007/978-981-15-1025-0_16
Zhang, X., Berger, F. G., Yang, J., & Lu, X. (2011). USP4 inhibits p53 through deubiquitinating and stabilizing ARF-BP1. EMBO Journal, 30(11), 2177–2189. https://doi.org/10.1038/emboj.2011.125
Zhang, Y., Cui, N., & Zheng, G. (2020). Ubiquitination of P53 by E3 ligase MKRN2 promotes melanoma cell proliferation. Oncology Letters, 19(3), 1975–1984. https://doi.org/10.3892/ol.2020.11261
Zhao, C., Beaudenon, S. L., Kelley, M. L., Waddell, M. B., Yuan, W., Schulman, B. A., Huibregtse, J. M., & Krug, R. M. (2004). The UbcH8 ubiquitin E2 enzyme is also the E2 enzyme for ISG15, an IFN-alpha/beta-induced ubiquitin-like protein. Proc Natl Acad Sci U S A, 101(20), 7578–7582. https://doi.org/10.1073/pnas.0402528101
Zhou, Y., Ye, X., Zhang, C., Wang, J., Guan, Z., Yan, J., Xu, L., Wang, K., Guan, D., Liang, Q., Mao, J., Zhou, J., Zhang, Q., Wu, X., Wang, M., Cong, Y. S., & Liu, J. (2021). Ufl1 deficiency causes kidney atrophy associated with disruption of endoplasmic reticulum homeostasis. Journal of Genetics and Genomics, 48(5), 403–410. https://doi.org/10.1016/j.jgg.2021.04.006
Zhu, H., Bhatt, B., Sivaprakasam, S., Cai, Y., Liu, S., Kodeboyina, S. K., Patel, N., Savage, N. M., Sharma, A., Kaufman, R. J., Li, H., & Singh, N. (2019). Ufbp1 promotes plasma cell development and ER expansion by modulating distinct branches of UPR. Nature Communications, 10(1), 1084. https://doi.org/10.1038/s41467-019-08908-5
Zou, Q., Jin, J., Hu, H., Li, H. S., Romano, S., Xiao, Y., Nakaya, M., Zhou, X., Cheng, X., Yang, P., Lozano, G., Zhu, C., Watowich, S. S., Ullrich, S. E., & Sun, S. C. (2014). USP15 stabilizes MDM2 to mediate cancer-cell survival and inhibit antitumor T cell responses. Nature Immunology, 15(6), 562–570. https://doi.org/10.1038/ni.2885
Acknowledgements
We thank Dr. Yu-sheng Cong for critical reading of the manuscript. This work was supported by grants from the National Natural Science Foundation of China (31871416, 81871116).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
Additional information
The original online version of this article was revised: Table 2 was missing from the original article and has now been added. Furthermore, the title of Table 1 has been corrected to “The effects of p53 ubiquitination”.
Rights and permissions
About this article

Cite this article
Wang, Y., Zhang, C., Wang, J. et al. p53 regulation by ubiquitin and ubiquitin-like modifications. GENOME INSTAB. DIS. 3, 179–198 (2022). https://doi.org/10.1007/s42764-022-00067-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42764-022-00067-0