
Abstract
Telomeres are specialized structures located at the ends of chromosomes that are critical for maintaining genomic integrity. Telomeres are shortened during each cycle of cell division because chromosomes are not able to completely replicate, a phenomenon known as the end-replication problem. Telomere shortening or dysfunction causes genome instability and is implicated in a variety of diseases, including cancer, cardiovascular disease and neurodegenerative disorders. Here, we discuss recent advances in basic and clinical research into telomere regulation and maintenance, and highlight how dysfunctional telomeres influence aging and age-related diseases.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Telomeres are specialized structures located at the ends of chromosomes. Structurally, telomeres are composed of tandem-repeat sequences (TTAGGG) in vertebrates and are terminated by a single-stranded overhang of the G-rich strand (Fig. 1). Telomeres protect the end of chromosome from degradation and DNA repair activities (de Lange, 2009). Because of a phenomenon known as the ‘end-replication problem’, which was introduced by Olovnikov and Watson about 50 years ago (Olovnikov, 1973; Watson, 1972), DNA polymerase cannot replicate sequences at 3’ ends and telomeres lose TTAGGG repeats with each cell division. Once telomere shortening reaches a critical length, referred to as the “Hayflick limit” (Harley et al., 1990), cells undergo growth arrest and become senescent. Therefore, the length of the telomere is highly correlated to the number of cell division, and telomeres are regarded as the ‘molecular clock’ of cellular aging. Telomeres have many other important functions, including the regulation of gene expression by the telomere position effect (Gottschling et al., 1990). Telomere chromatin contains telomeric repeat-containing RNA (TERRA), which has been implicated in telomerase regulation, organization of heterochromatin at telomeres and in the regulation of gene expression (Porro et al., 2014). Moreover, telomeres are critical to ensure proper chromosome separation during mitosis (Canudas & Smith, 2009).

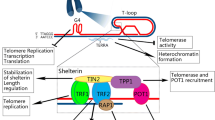
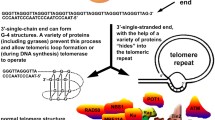
Telomere structure. A Telomeres are composed of tandem-repeat sequences (TTAGGG). Telomeric DNA is largely double-stranded, but ends in a short single-stranded G-rich 3’ overhang. B The ends of telomeres are protected by invasion of the terminal single-stranded DNA overhang into duplex TTAGGG repeats to form the T-loop and D-loop. The shelterin complex (TRF1, green; TRF2, blue; TPP1, red; POT1, orange; TIN2, yellow and RAP1, gray) prevents the ends of telomeres from being recognized as damaged DNA and activation of DNA damage response. C Structure of the telomere G quadruplex
Telomere dysfunction causes chromosomal instability, which is associated with many age-related diseases. In highly proliferating tissues such as the skin, gastrointestinal tract and hematopoietic system, progressive telomere attrition, due to the low level of telomerase and the continuous renewal of the tissues, trigger DNA damage responses, whereas ROS-induced damage of telomere sequences causes attrition and uncapping of telomere in lowly proliferative tissue such as heart, brain, and liver (Chakravarti et al., 2021). Telomere dysfunction not only causes genome instability, but also contributes to inflammatory responses in aging-related diseases including atherosclerosis (Hagg, 2018), type 2 diabetes (Cheng et al., 2020), osteoarthritis (Bekaert et al., 2005), and Parkinson’s (Chen & Zhan, 2021) and Alzheimer’s diseases (Lukens et al., 2009).
In this review, we provide an overview of current telomere research. We will describe the structure of telomeres, then introduce the mechanisms of telomere maintenance and lengthening and finally highlight the role of telomere dysfunction in cardiovascular disease, neurological disorder and cancer.
Telomere structure and maintenance
The average length of telomere DNA varies by species. In humans, telomeres are typically 10–15 kb in length and in mice are about 25–40 kb in length (Blackburn, 2001; Blasco, 2005; Moyzis et al., 1988). The telomere is covered by the shelterin complex (de Lange, 2005, 2018), a multimer composed of six protein subunits: TRF1, TRF2, TPP1, POT1, TIN2, and RAP1 (Fig. 1A, B). TRF1 and TRF2 directly bind to duplex TTAGGG repeats, and TRF2 forms a heterodimer with RAP1 (de Lange, 2018; Janouskova et al., 2015). POT1 recognizes single-strand TTAGGG overhangs and binds to TPP1 (Wu et al., 2012). TIN2 connects POT1–TPP1 to TRF1 and TRF2 to form a large complex (Hu et al., 2017). Shelterin interacts with both double-stranded and single-stranded telomeric DNA and is sufficiently abundant to bind all telomeric DNA (Takai et al., 2010). This higher-order structure can fold back and form a T-loop (Griffith et al., 1999). When the guanine-rich single chain folds back to invade the double-stranded region, the two chains of the double-stranded region are separated, and the three chains form a D-loop structure (Fig. 1B). Telomeres can also form another specialized structure, in which the four adjacent TTAGGG repeats can form a four-stranded structure, known as a guanine quadruplex (G quadruplex) (Burge et al., 2006). A square plane is formed by the four guanine residues of the telomere quadruplex; each guanine residue is a hydrogen bond donor and acceptor, meaning that Hoogsteen hydrogen bond pairing occurs between guanine residues (Fig. 1C). It has been proposed that G-quadruplexes can sequester the 3’ end of the telomere and prevent it from being extended by telomerase (Rhodes & Lipps, 2015).
Telomeres are susceptible to the actions of DNA damage response pathways (de Lange, 2018). The shelterin complex has fundamental roles in the regulation of telomere length and telomere maintenance. This complex is composed of six core proteins including TRF1, TRF2, TPP1, POT1, TIN2, and RAP1 (de Lange, 2018). TRF1 negatively regulates telomere length and participates in telomere DNA replication (Sfeir et al., 2009; Steensel & Lange, 1997). TRF2 and POT1 control the formation of the 3’ overhang at telomeres, which is critical for T-loop formation and telomere protection (Doksani et al., 2013; Griffith et al., 1999; Wu et al., 2012). The T-loop protects chromosome ends from degradation and deleterious effects of DNA damage responses to contribute to normal telomere function (Griffith et al., 1999). POT1 binds to single-stranded DNA with high specificity and affinity, thereby inhibiting the ATR damage signal and inhibiting homologous recombination (Denchi & Lange, 2007). The POT1–TPP1 complex increases the affinity of POT1 binding to single-stranded DNA, which stabilizes chromosome ends (Wang et al., 2007; Xin et al., 2007). When telomeres are in an open state, such as during replication or extension, POT1–TPP1 recruits telomerase to accelerate the process of telomere extension (Xin et al., 2007). TIN2 acts as a bridge connecting the POT1–TPP1 complex with TRF1 and TRF2, and stabilizes binding between TRF1 and TRF2 and double-stranded DNA. The RAP1–TRF2 complex inhibits non-homologous end joining of telomeres (Sfeir et al., 2010). The dysfunction of shelterin complex due to critically shortened telomeres and/or mutations in shelterin components triggers DNA damage response (DDR) at chromosome ends.
Mechanisms of telomere lengthening
In normal human somatic cells, telomeres are shortened 50–200 bases every cell cycle. However, in tumor cells, the telomere length can be maintained usually by telomerase. Telomerase is a reverse transcriptase that synthesizes telomeric DNA and is composed of the telomerase RNA component (TERC) and the telomerase reverse transcriptase (TERT) (Schmidt & Cech, 2015). Telomerase activity is usually absent in normal human somatic cells due to transcriptional repression of hTERT gene during embryonic differentiation, except in some high proliferative tissues, such as male germ cells, stem cell populations and activated lymphocytes, the enzyme remains active. However, during the cellular immortalization and transformation process, telomerase is reactivated by diverse and complex mechanisms (Cong et al., 2002). Recent studies demonstrated that TERT promoter mutations are the most common non-coding mutations in human cancer (Fredriksson et al., 2014; Rheinbay et al., 2020). Re-activation telomerase plays a critical role in the initiation and development cancer by maintaining telomere length and replicative capacity of cancer cells (Bodnar et al., 1998; Yuan et al., 2019).
The alternative lengthening of telomeres (ALT) is another mechanism of telomere maintenance and is observed in ~ 10–15% of cancer cells (Cesare & Reddel, 2010). ALT telomeres are characterized by chronic replication stress and are susceptible to double-strand breaks (DSBs) (Cesare & Reddel, 2010), which can result in a break-induced telomere synthesis through break-induced replication (BIR) mechanism, a type of homology-directed repair (Kramara et al., 2018; McEachern and Haber, 2006). ALT is naturally present in tumors and many immortal cell lines without reactivating telomerase, but it can also be induced by inhibiting telomere replication or by depleting several factors needed for telomere integrity like the shelterin complex (Bryan et al., 1995; Epum & Haber, 2021). ALT-associated replication defects trigger RAD52-dependent mitotic DNA synthesis (MiDAS) (Min et al., 2017). Cancer cells that rely on the ALT mechanism have a very variable and rapidly changing telomere length. For example, 61% of human pancreatic neuroendocrine tumors have variable telomeres and display characteristics of ALT (Heaphy et al., 2011). In addition to maintaining telomere length in telomerase-deficient mammalian cells, ALT has several additional characteristics. The C-circle, which is an extrachromosomal circle with an intact CCCTAA (C-rich) strand, is ALT specific (Epum & Haber, 2021; Nabetani & Ishikawa, 2009). ALT is often associated with promyelocytic leukemia (PML) body, referred to as ALT-associated PML body (APB) (Yeager et al., 1999). APB contains PML protein, telomeric DNA, and the telomere-specific binding proteins hTRF1 and hTRF2 (Yeager et al., 1999). Immunostaining showed that some proteins involved in DNA synthesis and recombination have been found in APBs, including replication factor A, RAD51, and RAD52 (Yeager et al., 1999). Furthermore, both ATRX and DAXX gene mutations are significantly correlated with ALT positivity (Heaphy et al., 2011). Studies have shown that ATRX affects telomeric double-strand break (DSB) repair through telomere cohesion and a DAXX-dependent pathway (Lovejoy et al., 2020).
TERRA (telomeric repeat-containing RNA) is a long non-coding RNA that is transcribed from subtelomere and telomere-derived sequences and contains (UUAGGG)n repeat sequences (Azzalin et al., 2007). Recent studies have shown that all eukaryotic cells examined express TERRA (Azzalin et al., 2007; Luke et al., 2008; Schoeftner & Blasco, 2008). The length of TERRA ranges from 100 bp to more than 9 kb in mammals (Azzalin et al., 2007). Since TERRA has a unique G-rich (UUAGGG)n sequence, it has the potential to generate TERRA–telomere DNA hybrids that create R-loop structures (Aguilera & Garcia-Muse, 2012; Graf et al., 2017). The shortening of telomeres induces TERRA expression and TERRA R-loops formation, which in turn activates the DNA damage response (DDR) and homology directed repair (HDR) at critically short telomeres (Graf et al., 2017). In addition, TERRA might also fold into G quadruplexes (Xu et al., 2008, 2010), which has been shown to inhibit telomerase activity (Mei et al., 2021; Rocca et al., 2017).
Telomere crisis and genome instability
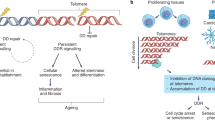
Critically shortened telomeres or dysfunctional telomeres can be recognized as a double-stranded DNA break and trigger a permanent growth arrest known as cellular senescence (Maciejowski et al., 2017). In the absence of tumor suppressor pathways such as the p53 and/or Rb pathways that prevent cell cycle arrest induced by telomere shortening, cells continue to divide with further telomere loss, which ultimately leads to the genome instability. This causes the cells to enter into a period called telomere crisis (Maciejowski & Lange, 2017; Shay & Wright, 2005). Telomere crisis occurs during the early stages of tumorigenesis (Dewhurst, 2020). During telomere crisis, unprotected chromosome ends generate end-to-end fusions and dicentric chromosomes, resulting in breakage–fusion–bridge cycles, aneuploidy and tetraploidization, translocations, and amplifications (Hayashi et al., 2015). These processes eventually lead to various forms of genome instability through kataegis (localized hypermutations) and chromothripsis (clustered chromosomal rearrangements) during mitosis (Artandi & DePinho, 2010; Maciejowski & Lange, 2017; Maciejowski et al., 2015) (Fig. 2).

Telomere crisis and genome instability. In the presence of tumor suppressor pathways (such as p53/p21 and/or Rb/p16 pathways), critically shortened or dysfunctional telomeres induce leads to cellular senescence, whereas in the absence of tumor suppressor pathways, unprotected chromosome ends can generate end-to-end fusions and dicentric chromosomes. These dicentric chromosomes generate anaphase bridges with subsequent chromosome breakage and the breakage–fusion–bridge (BFB) cycle, which promote genome instability and eventually tumorigenesis. ROS reactive oxygen species, ALT alternative lengthening of telomeres
The breakage–fusion–bridge cycle occurs when a bicentric chromosome (including a chromosome formed by telomere fusion) breaks, followed by a secondary fusion of the broken ends in the daughter cells (Eisfeldt et al., 2019; Tang et al., 2018). The fusion between sister chromatids can also lead to end deletions or inverted end repeats, which can be amplified in subsequent breakage–fusion–bridge cycles (Maciejowski & Lange, 2017). Many studies indicate that the breakage–fusion–bridge cycle is closely related to cancer outcomes such as loss of heterozygosity, translocation, and gene amplification (Maciejowski et al., 2015).
Recently, it has been shown that chromothripsis occurs as a result of telomere crisis. Chromothripsis describes a pattern of genome rearrangement in which fragmented or broken chromosomes are randomly reconnected to the derived chromosome arrangement (Cleal & Baird, 2020; Stephens et al., 2011). Thus, chromothripsis could promote tumorgenesis in a variety of ways. Chromothriptic breakpoints are frequently associated with kataegis mutation clusters (Maciejowski & Lange, 2017)—hypermutated patterns of clustered C > T and C > G changes at TpC dinucleotides associated with APOBEC-mediated mutagenesis (Maciejowski et al., 2015). Whole-genome sequence analysis revealed that kataegis occurs at the break sites of chromothriptic rearrangements (Maciejowski et al., 2015).
Dysfunctional telomeres in age-related diseases
The special properties of telomeres not only protect the ends of linear chromosomes from degradation and repair activities to ensure genome stability, but are also critically involved in a number of cellular processes contributing to the pathology of aging and cancer. Telomeres are protected and maintained by the shelterin complex; therefore, deficiency of any shelterin component may result in telomere dysfunction, which triggers DNA damage response. Recent studies using mouse models suggest that the deficiency of shelterin complex has been implicated in cancer susceptibility and aging-related pathologies (Martinez & Blasco, 2017).
Numerous studies have implicated telomere dysfunction as a major driver of age-related diseases, such as dyskeratosis congenita (DC), aplastic anemia (AA), and idiopathic pulmonary fibrosis (IPF) (Armanios, 2013). DC is associated with germline mutations in genes involved in telomere biology (Mitchell et al., 1999). DKC1 encodes the nuclear protein dyskerin, which is as an essential component of the telomerase holoenzyme, and is the first gene found mutated in DC (Holme et al., 2012; Mitchell et al., 1999). The classic phenotype of DC consists of the mucocutaneous triad of dysplastic finger and toenails, oral leukoplakia, and lacy, reticular skin pigmentation (Niewisch & Savage, 2019). AA is a common complication in DC patients, and was later characterized as an independent telomere biology disorder (Savage & Bertuch, 2010). About 10–20% of patients with acquired AA have short telomeres (Dolberg & Levy, 2014). Moreover, IPF is considered the most prevalent manifestation of telomere disorders (Armanios, 2012). Studies have shown that mutations in TERT or TERC confer a dramatic increase in susceptibility to adult-onset IPF (Tsakiri et al., 2007). These findings provide strong evidences that telomere length is a determinant or a predictor for some of specific diseases.
A recent study highlighted that there is a strong correlation between whole-blood telomere length and the telomere length of various tissues, and showed that the telomere length of blood cells is a biomarker of human aging and disease (Demanelis et al., 2020). The researchers collected 6391 tissue samples across more than 20 tissue types from 952 individuals, and characterized the variability of telomere length of each tissue sample. Generally, telomere length differs across human tissue types, but is correlated among tissue types. Whole blood telomere length was positively correlated with tissue-specific telomere length measurements, and leukocyte blood telomere length is used as a proxy for telomere length in many tissue types (Demanelis et al., 2020). The positive correlations of telomere length among tissue types are likely due to the fact that the initial telomere length in the zygote affects telomere length in all adult tissues through mitotic inheritance. This finding provides support for the validity of epidemiological studies based on blood telomere length. Shortened telomeres are associated with an increased risk of cardiovascular disease, type 2 diabetes, neurodegenerative disorders, and cancer (Table 1).
Cardiovascular disease is the leading cause of death worldwide. Several studies indicate that telomere dysfunction is associated with coronary heart disease, atherosclerosis, myocardial infarction, heart failure and stroke (Willeit et al., 2010). Compared with age-matched healthy controls, individuals with early myocardial infarction have shorter leukocyte telomere length (Brouilette et al., 2003). Telomeres are shorter in leukocytes from patients with severe heart failure (van der Harst et al., 2007), and there is a significant independent association between shorter telomeres and cardiovascular risk (Brouilette et al., 2007; D'Mello et al., 2016).
Similarly, telomere shortening is associated with the development of type 2 diabetes and related clinical conditions such as insulin resistance, impaired glucose tolerance, obesity and inflammation. A study conducted over 5 years indicated that diabetic patients have significantly shorter leukocyte telomere length (Zhao et al., 2014). Patients with type 2 diabetes also had shorter telomeres in peripheral monocytes and endothelial cells (Sampson et al., 2006). Meanwhile, two GWAS studies have indicated that type 2 diabetes is strongly linked to telomere shortening (Liu et al., 2014; Saxena et al., 2014).
Alzheimer's disease is one of the most common neurodegenerative diseases. Defects in telomere maintenance accelerate aging in mice and humans (Blasco et al., 1997; Herrera et al., 1999; Lopez-Otin et al., 2013). Many studies have showed that leukocyte telomere length is shorter in patients with Alzheimer's disease than in control individuals (Forero et al., 2016), and that telomere length in peripheral blood mononuclear cells may help predict disease progression (Tedone et al., 2015). Telomere shortening is also involved in the pathogenesis of the neurological disorder Parkinson’s disease (Eitan et al., 2014).
A number of studies have explored the relationship between telomere length and cancer risk or prognosis (Aviv et al., 2017). Telomeres in breast, colon, and prostate tumor tissues are shorter than those in adjacent tissue from the same patient (Fasching, 2018). Furthermore, in telomerase-positive tumors, short telomeres are usually associated with cancer progression and reduced survival rate (Fordyce et al., 2006; Valls et al., 2011). However, in lung cancer, patients with adenocarcinoma have longer telomeres than controls, while patients with squamous cell carcinoma have shorter telomeres than controls (Sanchez-Espiridion et al., 2014). Whether telomere length can be used to predict cancer risk prediction requires further investigation.
Telomeres in osteoblasts and mesenchymal stem cells are shortened with age. The first large-scale epidemiology study by Valdes et al. showed that there is a significant correlation between leukocyte telomere length and the bone mineral density of the spine and forearm (Boccardi et al., 2013). In a prospective study of elderly population (age range 71–86 years), shorter leukocyte telomere length was associated with bone loss in several distal forearm locations (Bekaert et al., 2005). Experiments in mice showed that the replicative senescence of osteoblast precursors promotes bone loss and senile osteoporosis (Saeed et al., 2011). A recent study suggests that short and dysfunctional telomeres contribute to age-associated renal fibrosis by influencing the epithelial-to-mesenchymal transition program (Saraswati et al., 2021).
Telomeres and the external environment
Telomere length serves as a ‘molecular clock’ for human aging. Strong evidence indicates that telomere length in parental germ cells affects telomere length in offspring cells (Delgado et al., 2019). Besides genetic factors, lifestyle factors (diet, physical activity, cigarette smoking) and environmental exposures (including radiation) also affect telomere length (Chakravarti et al., 2021; Fasching, 2018). A meta-analysis showed a positive correlation between adherence to the Mediterranean diet and maintenance of telomere length (Canudas et al., 2020). A study conducted on 5309 adults from the USA with no history of diabetes or cardiovascular disease showed that people who consumed sugar-sweetened beverages had a shorter leukocyte telomere length (Leung et al., 2014). Telomere length correlates with body mass index; obese individuals have shorter telomeres and weight loss positively correlates with telomere length (Wulaningsih et al., 2018). Increased physical activity positively correlates with telomere length, and people who engage in higher levels of physical activity have longer telomeres than sedentary individuals (Ludlow et al., 2008). The NASA Twins Study, in which one twin went to the International Space Station for 340 days while the other identical twin remained on Earth, observed the effects of the Earth and space environments on physiological functions (Garrett-Bakelman et al., 2019). Both twins had similar telomere lengths at the start of the study. The telomere length of the twin on Earth remained relatively stable, whereas telomere length increased significantly in the other twin during the space flight and then shortened rapidly upon return to Earth. Interestingly, within 6 months after return to Earth, telomere length stabilized to near preflight averages, but increased numbers of short telomeres were observed. Together, these studies strongly support the notion that telomere length is carefully regulated and is subject to changes in lifestyle and environment factors.
Conclusions and future perspectives
In conclusion, telomeres have a crucial role in maintaining genome stability. Detailed understanding of the mechanisms of telomere maintenance and regulations will provide insights into the role of telomere attrition in disease development and progression, and opportunities for prognosis and therapeutic intervention. Basic and clinical research showed that telomere length can be used as an indicator of aging and age-related diseases. The telomerase repair therapy has been explored as a potential anti-aging or anti-tumor strategies. Several small molecules such as TA-65 and the histone deacetylase inhibitors targeting TERT have been identified (Bernardes de Jesus et al., 2011; Yu et al., 2018). Jesus et al. show that TA-65 is capable of increasing average telomere length and decreasing the percentage of critically short telomeres in haploinsufficient mouse embryonic fibroblasts (Bernardes de Jesus et al., 2011). Won et al. suggest that the HDAC complex plays an important role in the regulation of hTERT in various proliferation conditions such as normal cycling, senescent, and tumor cells (Won et al., 2004). Future research will reveal the complex biological mechanisms between telomere function and diseases. This research may be translated into more accurate predictors of age-related disease risks and more effective treatment strategies that may ultimately be useful in monitoring and intervening in the aging process.
References
Aguilera, A., & Garcia-Muse, T. (2012). R loops: From transcription byproducts to threats to genome stability. Molecular Cell, 46(2), 115–124. https://doi.org/10.1016/j.molcel.2012.04.009
Armanios, M. (2012). Telomerase and idiopathic pulmonary fibrosis. Mutation Research, 730(1–2), 52–58. https://doi.org/10.1016/j.mrfmmm.2011.10.013
Armanios, M. (2013). Telomeres and age-related disease: how telomere biology informs clinical paradigms. The Journal of Clinical Investigation, 123(3), 996–1002. https://doi.org/10.1172/JCI66370
Artandi, S. E., & DePinho, R. A. (2010). Telomeres and telomerase in cancer. Carcinogenesis, 31(1), 9–18. https://doi.org/10.1093/carcin/bgp268
Aviv, A., Anderson, J. J., & Shay, J. W. (2017). Mutations, cancer and the telomere length paradox. Trends Cancer, 3(4), 253–258. https://doi.org/10.1016/j.trecan.2017.02.005
Azzalin, C. M., Reichenbach, P., Khoriauli, L., Giulotto, E., & Lingner, J. (2007). Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science, 318(5851), 798–801. https://doi.org/10.1126/science.1147182
Bekaert, S., Van Pottelbergh, I., De Meyer, T., Zmierczak, H., Kaufman, J. M., Van Oostveldt, P., & Goemaere, S. (2005). Telomere length versus hormonal and bone mineral status in healthy elderly men. Mechanisms of Ageing and Development, 126(10), 1115–1122. https://doi.org/10.1016/j.mad.2005.04.007
Bernardes de Jesus, B., Schneeberger, K., Vera, E., Tejera, A., Harley, C. B., & Blasco, M. A. (2011). The telomerase activator TA-65 elongates short telomeres and increases health span of adult/old mice without increasing cancer incidence. Aging Cell, 10(4), 604–621. https://doi.org/10.1111/j.1474-9726.2011.00700.x
Blackburn, E. H. (2001). Switching and signaling at the telomere. Cell, 106(6), 661–673. https://doi.org/10.1016/s0092-8674(01)00492-5
Blasco, M. A. (2005). Telomeres and human disease: ageing, cancer and beyond. Nature Reviews Genetics, 6(8), 611–622. https://doi.org/10.1038/nrg1656
Blasco, M. A., Lee, H. W., Hande, M. P., Samper, E., Lansdorp, P. M., DePinho, R. A., & Greider, C. W. (1997). Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell, 91(1), 25–34. https://doi.org/10.1016/s0092-8674(01)80006-4
Boccardi, V., Barbieri, M., Rizzo, M. R., Marfella, R., Esposito, A., Marano, L., & Paolisso, G. (2013). A new pleiotropic effect of statins in elderly: modulation of telomerase activity. The FASEB Journal, 27(9), 3879–3885. https://doi.org/10.1096/fj.13-232066
Bodnar, A. G., Ouellette, M., Frolkis, M., Holt, S. E., Chiu, C. P., Morin, G. B., & Wright, W. E. (1998). Extension of life-span by introduction of telomerase into normal human cells. Science, 279(5349), 349–352. https://doi.org/10.1126/science.279.5349.349
Brouilette, S., Singh, R. K., Thompson, J. R., Goodall, A. H., & Samani, N. J. (2003). White cell telomere length and risk of premature myocardial infarction. Arteriosclerosis, Thrombosis, and Vascular Biology, 23(5), 842–846. https://doi.org/10.1161/01.ATV.0000067426.96344.32
Brouilette, S. W., Moore, J. S., McMahon, A. D., Thompson, J. R., Ford, I., Shepherd, J., & West of Scotland Coronary Prevention Study. (2007). Telomere length, risk of coronary heart disease, and statin treatment in the West of Scotland Primary Prevention Study: a nested case-control study. Lancet, 369(9556), 107–114. https://doi.org/10.1016/S0140-6736(07)60071-3
Bryan, T. M., Englezou, A., Gupta, J., Bacchetti, S., & Reddel, R. R. (1995). Telomere elongation in immortal human cells without detectable telomerase activity. EMBO Journal, 14(17), 4240–4248.
Burge, S., Parkinson, G. N., Hazel, P., Todd, A. K., & Neidle, S. (2006). Quadruplex DNA: sequence, topology and structure. Nucleic Acids Research, 34(19), 5402–5415. https://doi.org/10.1093/nar/gkl655
Canudas, S., & Smith, S. (2009). Differential regulation of telomere and centromere cohesion by the Scc3 homologues SA1 and SA2, respectively, in human cells. Journal of Cell Biology, 187(2), 165–173. https://doi.org/10.1083/jcb.200903096
Canudas, S., Becerra-Tomas, N., Hernandez-Alonso, P., Galie, S., Leung, C., Crous-Bou, M., & Salas-Salvado, J. (2020). Mediterranean diet and telomere length: a systematic review and meta-analysis. Advances in Nutrition, 11(6), 1544–1554. https://doi.org/10.1093/advances/nmaa079
Cesare, A. J., & Reddel, R. R. (2010). Alternative lengthening of telomeres: models, mechanisms and implications. Nature Reviews Genetics, 11(5), 319–330. https://doi.org/10.1038/nrg2763
Chakravarti, D., LaBella, K. A., & DePinho, R. A. (2021). Telomeres: history, health, and hallmarks of aging. Cell, 184(2), 306–322. https://doi.org/10.1016/j.cell.2020.12.028
Chen, R., & Zhan, Y. (2021). Association between telomere length and Parkinson’s disease: a Mendelian randomization study. Neurobiology of Aging, 97, 144e149–144e111. https://doi.org/10.1016/j.neurobiolaging.2020.07.019
Cheng, F., Carroll, L., Joglekar, M. V., Januszewski, A. S., Wong, K. K., Hardikar, A. A., & Ma, R. C. W. (2020). Diabetes, metabolic disease, and telomere length. The Lancet Diabetes and Endocrinology. https://doi.org/10.1016/S2213-8587(20)30365-X
Cleal, K., & Baird, D. M. (2020). Catastrophic endgames: emerging mechanisms of telomere-driven genomic instability. Trends in Genetics, 36(5), 347–359. https://doi.org/10.1016/j.tig.2020.02.001
Cong, Y. S., Wright, W. E., & Shay, J. W. (2002). Human telomerase and its regulation. Microbiology and Molecular Biology Reviews, 66(3), 407–425. https://doi.org/10.1128/MMBR.66.3.407-425.2002
de Lange, T. (2005). Shelterin: the protein complex that shapes and safeguards human telomeres. Genes & Development, 19(18), 2100–2110. https://doi.org/10.1101/gad.1346005
de Lange, T. (2009). How telomeres solve the end-protection problem. Science, 326(5955), 948–952. https://doi.org/10.1126/science.1170633
de Lange, T. (2018). Shelterin-mediated telomere protection. Annual Review of Genetics, 52, 223–247. https://doi.org/10.1146/annurev-genet-032918-021921
Delgado, D. A., Zhang, C., Gleason, K., Demanelis, K., Chen, L. S., Gao, J., & Pierce, B. L. (2019). The contribution of parent-to-offspring transmission of telomeres to the heritability of telomere length in humans. Human Genetics, 138(1), 49–60. https://doi.org/10.1007/s00439-018-1964-2
Demanelis, K., Jasmine, F., Chen, L. S., Chernoff, M., Tong, L., Delgado, D., & Pierce, B. L. (2020). Determinants of telomere length across human tissues. Science. https://doi.org/10.1126/science.aaz6876
Denchi, E. L., & de Lange, T. (2007). Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature, 448(7157), 1068–1071. https://doi.org/10.1038/nature06065
Dewhurst, S. M. (2020). Chromothripsis and telomere crisis: engines of genome instability. Current Opinion in Genetics & Development, 60, 41–47. https://doi.org/10.1016/j.gde.2020.02.009
D’Mello, M. J. J., Ross, S. A., Anand, S. S., Gerstein, H., McQueen, M., Yusuf, S., & Pare, G. (2016). Telomere length and risk of myocardial infarction in a multiethnic population: the INTERHEART study. Journal of the American College of Cardiology, 67(15), 1863–1865. https://doi.org/10.1016/j.jacc.2016.01.061
Doksani, Y., Wu, J. Y., de Lange, T., & Zhuang, X. (2013). Super-resolution fluorescence imaging of telomeres reveals TRF2-dependent T-loop formation. Cell, 155(2), 345–356. https://doi.org/10.1016/j.cell.2013.09.048
Dolberg, O. J., & Levy, Y. (2014). Idiopathic aplastic anemia: diagnosis and classification. Autoimmunity Reviews, 13(4–5), 569–573. https://doi.org/10.1016/j.autrev.2014.01.014
Eisfeldt, J., Pettersson, M., Vezzi, F., Wincent, J., Kaller, M., Gruselius, J., & Lindstrand, A. (2019). Comprehensive structural variation genome map of individuals carrying complex chromosomal rearrangements. PLoS Genetics, 15(2), e1007858. https://doi.org/10.1371/journal.pgen.1007858
Eitan, E., Hutchison, E. R., & Mattson, M. P. (2014). Telomere shortening in neurological disorders: an abundance of unanswered questions. Trends in Neurosciences, 37(5), 256–263. https://doi.org/10.1016/j.tins.2014.02.010
Epum, E. A., & Haber, J. E. (2021). DNA replication: the recombination connection. Trends in Cell Biology. https://doi.org/10.1016/j.tcb.2021.07.005
Fasching, C. L. (2018). Telomere length measurement as a clinical biomarker of aging and disease. Critical Reviews in Clinical Laboratory Sciences, 55(7), 443–465. https://doi.org/10.1080/10408363.2018.1504274
Fitzpatrick, A. L., Kronmal, R. A., Gardner, J. P., Psaty, B. M., Jenny, N. S., Tracy, R. P., Walston, J., Kimura, M., & Aviv, A. (2006). Leukocyte telomere length and cardiovascular disease in the cardiovascular health study. American Journal of Epidemiology 165(1), 14–21. https://doi.org/10.1093/aje/kwj346
Fordyce, C. A., Heaphy, C. M., Bisoffi, M., Wyaco, J. L., Joste, N. E., Mangalik, A., & Griffith, J. K. (2006). Telomere content correlates with stage and prognosis in breast cancer. Breast Cancer Research and Treatment, 99(2), 193–202. https://doi.org/10.1007/s10549-006-9204-1
Forero, D. A., Gonzalez-Giraldo, Y., Lopez-Quintero, C., Castro-Vega, L. J., Barreto, G. E., & Perry, G. (2016). Meta-analysis of telomere length in Alzheimer’s disease. Journals of Gerontology. Series a, Biological Sciences and Medical Sciences, 71(8), 1069–1073. https://doi.org/10.1093/gerona/glw053
Fredriksson, N. J., Ny, L., Nilsson, J. A., & Larsson, E. (2014). Systematic analysis of noncoding somatic mutations and gene expression alterations across 14 tumor types. Nature Genetics, 46(12), 1258–1263. https://doi.org/10.1038/ng.3141
Garrett-Bakelman, F. E., Darshi, M., Green, S. J., Gur, R. C., Lin, L., Macias, B. R., & Turek, F. W. (2019). The NASA twins study: a multidimensional analysis of a year-long human spaceflight. Science. https://doi.org/10.1126/science.aau8650
Gottschling, D. E., Aparicio, O. M., Billington, B. L., & Zakian, V. A. (1990). Position effect at S. cerevisiae telomeres: reversible repression of Pol II transcription. Cell, 63(4), 751–762. https://doi.org/10.1016/0092-8674(90)90141-z
Graf, M., Bonetti, D., Lockhart, A., Serhal, K., Kellner, V., Maicher, A., & Luke, B. (2017). Telomere length determines TERRA and R-loop regulation through the cell cycle. Cell, 170(1), 72-85e14. https://doi.org/10.1016/j.cell.2017.06.006
Griffith, J. D., Comeau, L., Rosenfield, S., Stansel, R. M., Bianchi, A., Moss, H., & de Lange, T. (1999). Mammalian telomeres end in a large duplex loop. Cell, 97(4), 503–514. https://doi.org/10.1016/s0092-8674(00)80760-6
Hagg, S. (2018). Telomere length dynamics and atherosclerotic disease. Circulation Research, 122(4), 546–547. https://doi.org/10.1161/CIRCRESAHA.118.312567
Hammadah, M., Al Mheid, I., Wilmot, K., Ramadan, R., Abdelhadi, N., Alkhoder, A., Obideen, M., Pimple, P. M., Levantsevych, O., Kelli, H. M., Shah, A., Sun, Y. V., Pearce, B., Kutner, M., Long, Q., Ward, L., Ko, Y. A., Hosny Mohammed, K., Lin, J., Zhao, J., Bremner, J. D., Kim, J., Waller, E. K., Raggi, P., Sheps, D., Quyyumi, A. A., & Vaccarino, V. (2017). Telomere shortening, regenerative capacity, and cardiovascular outcomes. Circulation Research, 120(7), 1130–1138. https://doi.org/10.1161/CIRCRESAHA.116.309421
Harley, C. B., Futcher, A. B., & Greider, C. W. (1990). Telomeres shorten during ageing of human fibroblasts. Nature, 345(6274), 458–460. https://doi.org/10.1038/345458a0
Hayashi, M. T., Cesare, A. J., Rivera, T., & Karlseder, J. (2015). Cell death during crisis is mediated by mitotic telomere deprotection. Nature, 522(7557), 492–496. https://doi.org/10.1038/nature14513
Heaphy, C. M., Fleet, T. M., Treat, E. G., Lee, S. J., Smith, A. Y., Davis, M. S., Griffith, J. K., Fischer, E. G., & Bisoffi, M. (2010). Organ-wide telomeric status in diseased and disease-free prostatic tissues. Prostate, 70(13), 1471–1479. https://doi.org/10.1002/pros.21182
Heaphy, C. M., de Wilde, R. F., Jiao, Y., Klein, A. P., Edil, B. H., Shi, C., & Meeker, A. K. (2011). Altered telomeres in tumors with ATRX and DAXX mutations. Science, 333(6041), 425. https://doi.org/10.1126/science.1207313
Herrera, E., Samper, E., Martin-Caballero, J., Flores, J. M., Lee, H. W., & Blasco, M. A. (1999). Disease states associated with telomerase deficiency appear earlier in mice with short telomeres. EMBO Journal, 18(11), 2950–2960. https://doi.org/10.1093/emboj/18.11.2950
Holme, H., Hossain, U., Kirwan, M., Walne, A., Vulliamy, T., & Dokal, I. (2012). Marked genetic heterogeneity in familial myelodysplasia/acute myeloid leukaemia. British Journal of Haematology, 158(2), 242–248. https://doi.org/10.1111/j.1365-2141.2012.09136.x
Hu, C., Rai, R., Huang, C., Broton, C., Long, J., Xu, Y., & Chen, Y. (2017). Structural and functional analyses of the mammalian TIN2-TPP1-TRF2 telomeric complex. Cell Research, 27(12), 1485–1502. https://doi.org/10.1038/cr.2017.144
Janouskova, E., Necasova, I., Pavlouskova, J., Zimmermann, M., Hluchy, M., Marini, V., & Hofr, C. (2015). Human Rap1 modulates TRF2 attraction to telomeric DNA. Nucleic Acids Research, 43(5), 2691–2700. https://doi.org/10.1093/nar/gkv097
Kramara, J., Osia, B., & Malkova, A. (2018). Break-Induced Replication: The Where, The Why, and The How. Trends in Genetics, 34(7), 518–531. https://doi.org/10.1016/j.tig.2018.04.002
Leung, C. W., Laraia, B. A., Needham, B. L., Rehkopf, D. H., Adler, N. E., Lin, J., & Epel, E. S. (2014). Soda and cell aging: associations between sugar-sweetened beverage consumption and leukocyte telomere length in healthy adults from the National Health and Nutrition Examination Surveys. American Journal of Public Health, 104(12), 2425–2431. https://doi.org/10.2105/AJPH.2014.302151
Liu, Y., Cao, L., Li, Z., Zhou, D., Liu, W., Shen, Q., & Shi, Y. (2014). A genome-wide association study identifies a locus on TERT for mean telomere length in Han Chinese. PLoS ONE, 9(1), e85043. https://doi.org/10.1371/journal.pone.0085043
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M., & Kroemer, G. (2013). The hallmarks of aging. Cell, 153(6), 1194–1217. https://doi.org/10.1016/j.cell.2013.05.039
Lovejoy, C. A., Takai, K., Huh, M. S., Picketts, D. J., & de Lange, T. (2020). ATRX affects the repair of telomeric DSBs by promoting cohesion and a DAXX-dependent activity. PLoS Biology, 18(1), e3000594. https://doi.org/10.1371/journal.pbio.3000594
Ludlow, A. T., Zimmerman, J. B., Witkowski, S., Hearn, J. W., Hatfield, B. D., & Roth, S. M. (2008). Relationship between physical activity level, telomere length, and telomerase activity. Medicine and Science in Sports and Exercise, 40(10), 1764–1771. https://doi.org/10.1249/MSS.0b013e31817c92aa
Luke, B., Panza, A., Redon, S., Iglesias, N., Li, Z., & Lingner, J. (2008). The Rat1p 5’ to 3’ exonuclease degrades telomeric repeat-containing RNA and promotes telomere elongation in Saccharomyces cerevisiae. Molecular Cell, 32(4), 465–477. https://doi.org/10.1016/j.molcel.2008.10.019
Lukens, J. N., Van Deerlin, V., Clark, C. M., Xie, S. X., & Johnson, F. B. (2009). Comparisons of telomere lengths in peripheral blood and cerebellum in Alzheimer’s disease. Alzheimer’s & Dementia, 5(6), 463–469. https://doi.org/10.1016/j.jalz.2009.05.666
Maciejowski, J., & de Lange, T. (2017). Telomeres in cancer: tumour suppression and genome instability. Nature Reviews Molecular Cell Biology, 18(3), 175–186. https://doi.org/10.1038/nrm.2016.171
Maciejowski, J., Li, Y., Bosco, N., Campbell, P. J., & de Lange, T. (2015). Chromothripsis and kataegis induced by telomere crisis. Cell, 163(7), 1641–1654. https://doi.org/10.1016/j.cell.2015.11.054
Martinez, P., & Blasco, M. A. (2017). Telomere-driven diseases and telomere-targeting therapies. Journal of Cell Biology, 216(4), 875–887. https://doi.org/10.1083/jcb.201610111
McEachern, M. J., & Haber, J. E. (2006). Break-induced replication and recombinational telomere elongation in yeast. Annual Review of Biochemistry, 75(1), 111–135. https://doi.org/10.1146/annurev.biochem.74.082803.133234
Mei, Y., Deng, Z., Vladimirova, O., Gulve, N., Johnson, F. B., Drosopoulos, W. C., & Lieberman, P. M. (2021). TERRA G-quadruplex RNA interaction with TRF2 GAR domain is required for telomere integrity. Science and Reports, 11(1), 3509. https://doi.org/10.1038/s41598-021-82406-x
Min, J., Wright, W. E., & Shay, J. W. (2017). Alternative lengthening of telomeres mediated by mitotic dna synthesis engages break-induced replication processes. Molecular and Cellular Biology. https://doi.org/10.1128/MCB.00226-17
Mitchell, J. R., Wood, E., & Collins, K. (1999). A telomerase component is defective in the human disease dyskeratosis congenita. Nature, 402(6761), 551–555. https://doi.org/10.1038/990141
Moyzis, R. K., Buckingham, J. M., Cram, L. S., Dani, M., Deaven, L. L., Jones, M. D., & Wu, J. R. (1988). A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proceedings of the National Academy of Sciences USA, 85(18), 6622–6626. https://doi.org/10.1073/pnas.85.18.6622
Nabetani, A., & Ishikawa, F. (2009). Unusual telomeric DNAs in human telomerase-negative immortalized cells. Molecular and Cellular Biology, 29(3), 703–713. https://doi.org/10.1128/MCB.00603-08
Nakamura, K., Furugori, E., Esaki, Y., Arai, T., Sawabe, M., Okayasu, I., Fujiwara, M., Kammori, M., Mafune, K., Kato, M., Oshimura, M., Sasajima, K., & Takubo, K. (2000). Correlation of telomere lengths in normal and cancers tissue in the large bowel. Cancer Letters, 158(2), 179–184. https://doi.org/10.1016/s0304-3835(00)00521-8
Niewisch, M. R., & Savage, S. A. (2019). An update on the biology and management of dyskeratosis congenita and related telomere biology disorders. Expert Review of Hematology, 12(12), 1037–1052. https://doi.org/10.1080/17474086.2019.1662720
Olovnikov, A. M. (1973). A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. Journal of Theoretical Biology, 41(1), 181–190. https://doi.org/10.1016/0022-5193(73)90198-7
Porro, A., Feuerhahn, S., Delafontaine, J., Riethman, H., Rougemont, J., & Lingner, J. (2014). Functional characterization of the TERRA transcriptome at damaged telomeres. Nature Communications, 5, 5379. https://doi.org/10.1038/ncomms6379
Rheinbay, E., Nielsen, M. M., Abascal, F., Wala, J. A., Shapira, O., Tiao, G., & Consortium P. (2020). Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature, 578(7793), 102–111. https://doi.org/10.1038/s41586-020-1965-x
Rhodes, D., & Lipps, H. J. (2015). G-quadruplexes and their regulatory roles in biology. Nucleic Acids Research, 43(18), 8627–8637. https://doi.org/10.1093/nar/gkv862
Rocca, R., Moraca, F., Costa, G., Nadai, M., Scalabrin, M., Talarico, C., & Richter, S. N. (2017). Identification of G-quadruplex DNA/RNA binders: structure-based virtual screening and biophysical characterization. Biochimica Et Biophysica Acta - General Subjects, 1861(5 Pt B), 1329–1340. https://doi.org/10.1016/j.bbagen.2016.12.023
Saeed, H., Abdallah, B. M., Ditzel, N., Catala-Lehnen, P., Qiu, W., Amling, M., & Kassem, M. (2011). Telomerase-deficient mice exhibit bone loss owing to defects in osteoblasts and increased osteoclastogenesis by inflammatory microenvironment. Journal of Bone and Mineral Research, 26(7), 1494–1505. https://doi.org/10.1002/jbmr.349
Sampson, M. J., Winterbone, M. S., Hughes, J. C., Dozio, N., & Hughes, D. A. (2006). Monocyte telomere shortening and oxidative DNA damage in type 2 diabetes. Diabetes Care, 29(2), 283–289. https://doi.org/10.2337/diacare.29.02.06.dc05-1715
Sanchez-Espiridion, B., Chen, M., Chang, J. Y., Lu, C., Chang, D. W., Roth, J. A., & Gu, J. (2014). Telomere length in peripheral blood leukocytes and lung cancer risk: a large case-control study in Caucasians. Cancer Research, 74(9), 2476–2486. https://doi.org/10.1158/0008-5472.CAN-13-2968
Saraswati, S., Martínez, P., Graña-Castro, O., & Blasco, M. A. (2021). Short and dysfunctional telomeres sensitize the kidneys to develop fibrosis. Nature Aging, 1(3), 269–283. https://doi.org/10.1038/s43587-021-00040-8
Savage, S. A., & Bertuch, A. A. (2010). The genetics and clinical manifestations of telomere biology disorders. Genetics in Medicine, 12(12), 753–764. https://doi.org/10.1097/GIM.0b013e3181f415b5
Saxena, R., Bjonnes, A., Prescott, J., Dib, P., Natt, P., Lane, J., & Sanghera, D. K. (2014). Genome-wide association study identifies variants in casein kinase II (CSNK2A2) to be associated with leukocyte telomere length in a Punjabi Sikh diabetic cohort. Circulation Cardiovascular Genetics, 7(3), 287–295. https://doi.org/10.1161/circgenetics.113.000412
Schmidt, J. C., & Cech, T. R. (2015). Human telomerase: biogenesis, trafficking, recruitment, and activation. Genes & Development, 29(11), 1095–1105. https://doi.org/10.1101/gad.263863.115
Schoeftner, S., & Blasco, M. A. (2008). Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II. Nature Cell Biology, 10(2), 228–236. https://doi.org/10.1038/ncb1685
Sfeir, A., Kosiyatrakul, S. T., Hockemeyer, D., MacRae, S. L., Karlseder, J., Schildkraut, C. L., & de Lange, T. (2009). Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell, 138(1), 90–103. https://doi.org/10.1016/j.cell.2009.06.021
Sfeir, A., Kabir, S., van Overbeek, M., Celli, G. B., & de Lange, T. (2010). Loss of Rap1 induces telomere recombination in the absence of NHEJ or a DNA damage signal. Science, 327(5973), 1657–1661. https://doi.org/10.1126/science.1185100
Shay, J. W., & Wright, W. E. (2005). Senescence and immortalization: role of telomeres and telomerase. Carcinogenesis, 26(5), 867–874. https://doi.org/10.1093/carcin/bgh296
Stephens, P. J., Greenman, C. D., Fu, B., Yang, F., Bignell, G. R., Mudie, L. J., & Campbell, P. J. (2011). Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell, 144(1), 27–40. https://doi.org/10.1016/j.cell.2010.11.055
Takai, K. K., Hooper, S., Blackwood, S., Gandhi, R., & de Lange, T. (2010). In vivo stoichiometry of shelterin components. Journal of Biological Chemistry, 285(2), 1457–1467. https://doi.org/10.1074/jbc.M109.038026
Tang, Z., Yang, J., Wang, X., Zeng, M., Wang, J., Wang, A., & Chen, J. (2018). Active DNA end processing in micronuclei of ovarian cancer cells. BMC Cancer, 18(1), 426. https://doi.org/10.1186/s12885-018-4347-0
Tedone, E., Arosio, B., Colombo, F., Ferri, E., Asselineau, D., Piette, F., & Mari, D. (2015). Leukocyte telomere length in Alzheimer’s disease patients with a different rate of progression. Journal of Alzheimer’s Disease, 46(3), 761–769. https://doi.org/10.3233/JAD-142808
Tsakiri, K. D., Cronkhite, J. T., Kuan, P. J., Xing, C., Raghu, G., Weissler, J. C., & Garcia, C. K. (2007). Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proceedings of the National Academy of Sciences USA, 104(18), 7552–7557. https://doi.org/10.1073/pnas.0701009104
Valls, C., Pinol, C., Rene, J. M., Buenestado, J., & Vinas, J. (2011). Telomere length is a prognostic factor for overall survival in colorectal cancer. Colorectal Disease, 13(11), 1265–1272. https://doi.org/10.1111/j.1463-1318.2010.02433.x
van der Harst, P., van der Steege, G., de Boer, R. A., Voors, A. A., Hall, A. S., Mulder, M. J., & Group, M. H. S. (2007). Telomere length of circulating leukocytes is decreased in patients with chronic heart failure. Journal of the American College of Cardiology, 49(13), 1459–1464. https://doi.org/10.1016/j.jacc.2007.01.027
van Steensel, B., & de Lange, T. (1997). Control of telomere length by the human telomeric protein TRF1. Nature, 385(6618), 740–743. https://doi.org/10.1038/385740a0
Wang, F., Podell, E. R., Zaug, A. J., Yang, Y., Baciu, P., Cech, T. R., & Lei, M. (2007). The POT1–TPP1 telomere complex is a telomerase processivity factor. Nature, 445(7127), 506–510. https://doi.org/10.1038/nature05454
Watson, J. D. (1972). Origin of concatemeric T7 DNA. Nature New Biology, 239(94), 197–201. https://doi.org/10.1038/newbio239197a0
Willeit, P., Willeit, J., Brandstatter, A., Ehrlenbach, S., Mayr, A., Gasperi, A., & Kiechl, S. (2010). Cellular aging reflected by leukocyte telomere length predicts advanced atherosclerosis and cardiovascular disease risk. Arteriosclerosis, Thrombosis, and Vascular Biology, 30(8), 1649–1656. https://doi.org/10.1161/ATVBAHA.110.205492
Won, J., Chang, S., Oh, S., & Kim, T. K. (2004). Small-molecule-based identification of dynamic assembly of E2F-pocket protein-histone deacetylase complex for telomerase regulation in human cells. Proceedings of the National Academy of Sciences USA, 101(31), 11328–11333. https://doi.org/10.1073/pnas.0401801101
Wu, P., Takai, H., & de Lange, T. (2012). Telomeric 3’ overhangs derive from resection by Exo1 and Apollo and fill-in by POT1b-associated CST. Cell, 150(1), 39–52. https://doi.org/10.1016/j.cell.2012.05.026
Wulaningsih, W., Kuh, D., Wong, A., & Hardy, R. (2018). Adiposity, Telomere Length, and Telomere Attrition in Midlife: The 1946 British Birth Cohort. Journals of Gerontology. Series A Biological Sciences and Medical Sciences, 73(7), 966–972. https://doi.org/10.1093/gerona/glx151
Xin, H., Liu, D., Wan, M., Safari, A., Kim, H., Sun, W., & Songyang, Z. (2007). TPP1 is a homologue of ciliate TEBP-beta and interacts with POT1 to recruit telomerase. Nature, 445(7127), 559–562. https://doi.org/10.1038/nature05469
Xu, Y., Kaminaga, K., & Komiyama, M. (2008). G-quadruplex formation by human telomeric repeats-containing RNA in Na+ solution. Journal of the American Chemical Society, 130(33), 11179–11184. https://doi.org/10.1021/ja8031532
Xu, Y., Suzuki, Y., Ito, K., & Komiyama, M. (2010). Telomeric repeat-containing RNA structure in living cells. Proceedings of the National Academy of Sciences USA, 107(33), 14579–14584. https://doi.org/10.1073/pnas.1001177107
Yeager, T. R., Neumann, A. A., Englezou, A., Huschtscha, L. I., Noble, J. R., & Reddel, R. R. (1999). Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Research, 59(17), 4175–4179.
Yu, Y., Zhou, L., Yang, Y., & Liu, Y. (2018). Cycloastragenol: An exciting novel candidate for age-associated diseases. Experimental and Therapeutic Medicine, 16(3), 2175–2182. https://doi.org/10.3892/etm.2018.6501
Yuan, X., Larsson, C., & Xu, D. (2019). Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: old actors and new players. Oncogene, 38(34), 6172–6183. https://doi.org/10.1038/s41388-019-0872-9
Zhao, J., Zhu, Y., Lin, J., Matsuguchi, T., Blackburn, E., Zhang, Y., & Howard, B. V. (2014). Short leukocyte telomere length predicts risk of diabetes in american indians: the strong heart family study. Diabetes, 63(1), 354–362. https://doi.org/10.2337/db13-0744
Acknowledgements
This work was supported by grants from the Hangzhou Science and Technology Bureau (20182014B01) and the National Natural Science Foundation of China (31730020).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Xu, L., Zhang, K. & Cong, YS. The role of telomere dysfunction in genomic instability and age-related diseases. GENOME INSTAB. DIS. 2, 292–301 (2021). https://doi.org/10.1007/s42764-021-00052-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42764-021-00052-z