
Abstract
The genus Pseudomonas is well known biocontrol agent established for its diverse role and antagonistic properties against phytopathogens due its ability of producing antibiotics. The studies were conducted to characterize Pseudomonas spp. for antibiotic production on the basis of gene specific primer. 150 isolates of Pseudomonas spp. were isolated from different agricultural fields of Jammu on King’s B medium using plate dilution method. Out of the total, thirty isolates were selected. The antibiotic i.e. 2,4-Diacetylphloroglucinol (2,4-DAPG) extracted from the various isolates showed spots having Rf values in the range of 0.7–0.89 which is very close to the Rf value of synthetic phloroglucinol (0.89). 2,4-DAPG positive isolates were tested against Fusarium oxysporum using dual culture assay and they were found to inhibit the mycelial growth of F. oxysporum by 52.76–65.45%. PCR analysis with 2,4-DAPG gene (phlD) specific primer indicated that a DNA fragment of approximately 750 bp in size was obtained in all the strains except PS 3 and 5 which is closely corroborated with the already reported size of 2,4-DAPG gene in literature. This confirms that 28 isolates out of 30 isolates have the genetic ability for the production of 2,4-DAPG antibiotic and thus these isolates can be promoted as potential biocontrol agents. The genetic diversity of anti-fungal producing rhizobacteria Pseudomonas spp. was investigated using amplified 16S rDNA restriction analysis. 16S rDNA was amplified using PCR technique and digestion with restriction endonuclease HaeIII and dendrogram analysis was carried out by the SHAN clustering UPGMA method (NTSYS 2.1). ARDRA analysis revealed the variability of the Pseudomonas spp. based on the restriction sites and the amplified 16S rDNA distinguished into 18 clusters/types including twelve minor groups and six major groups.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The genus Pseudomonas is a very large and important group of non-fermenting, gram negative bacteria, living as saprophytes in soils, sediments and fresh water (Bossis et al. 2000). Pseudomonas spp. forms an important group of beneficial organisms with several important traits like biological control of plant pathogens, plant growth promotional activity, nutrient mobilization etc. Some of the species in this genus are already known to improve plant growth and health and are implicated in the natural suppression of various soil borne diseases like bacterial wilt of potato, Fusarium and Verticilium wilt of vegetable crops whereas others are involved in the biodegradation of natural or man-made toxic chemical compounds (Bossis et al. 2000). Strains of Pseudomonas spp. that have the ability to produce the antibiotic 2,4-Diacetylphloroglucinol (2,4-DAPG) are responsible for the natural suppression of take-all diseases of wheat (Gaeumannomyces graminis var. tritici), known as take-all decline (TAD) (Weller et al. 2002, 2007). 2,4-DAPG producers that occur in soils worldwide have a significant role in plant defense, the genotypic and phenotypic diversity of these strains has been studied extensively by De La Fuente et al. (2006). Phylogenetic comparisons on the basis of amplified rRNA gene restriction analysis of 16S rRNA genes revealed three distinct lineages among 2,4-DAPG-producing Pseudomonas spp. strains (McSpadden Gardener et al. 2000). 2,4-DAPG producer have been used synonymously because the detection of phlD correlates with the production of 2,4-DAPG by Pseudomonas spp. (Weller et al. 2007). The broad-spectrum antibiotic DAPG is one of the most important antibiotics that is produced by biocontrol Pseudomonas spp. and is involved in the natural suppression of several fungal root diseases and in suppressive soils (Ramette et al. 2003; Weller et al. 2003). The success of BCAs may be attributed to their high reproductive capacity, ability to survive under very unfavourable conditions, efficiency in the utilization of nutrients, capacity to modify the rhizosphere, strong aggressiveness against phytopathogenic fungi and efficiency in promoting plant growth and defense mechanisms (Gupta et al. 2016a). Biological control using potential microorganisms having strong antifungal activity is coming up as an alternative strategy for plant disease management (Gupta et al. 2016b). Two main phenotypic groups that can be differentiated on the basis of their antibiotic production. One group that has the capacity to produce only DAPG and HCN and the other group produces pyoluteorin and, in some cases pyrrolnitrin in addition (Keel et al. 1996). While members of this last group are known to be genetically very similar to the first group (De La Fuente et al. 2004; Keel et al. 1996; Sharifi-Tehrani et al. 1998; McSpadden Gardener et al. 2000), extensive genomic diversity was highlighted in DAPG + PLT-Pseudomonas by different fingerprinting methods such as ARDRA, RAPD and BOX-PCR (Mavrodi et al. 2001; McSpadden Gardener et al. 2000; Keel et al. 1996). This led to the subdivision of first group of DAPG-producing Pseudomonas in as many as 17 different genomic groups (Landa et al. 2002; Mazzola et al. 2005; McSpadden Gardener et al. 2000). This diversity among DAPG producing Pseudomonas is relevant to biocontrol, because strains belonging to certain genotypes can have better biocontrol capacities, such as superior root colonizing abilities (Landa et al. 2002; Mavrodi et al. 2002) or higher antibiotic production (Ramette et al. 2001). In this study, we described diversity of antifungal producing Pseudomonas spp. isolated from agricultural fields of Jammu employing ARDRA and gene specific characterization through amplification of known antibiotic coding gene phlD.
Materials and methods
Collection of soil samples
Soil was collected from four different agricultural fields of Chatha (Jammu) situated at 33°-55′ North latitude and 74°-58′ East longitude with an altitude of 332 m above mean sea level where different crop types viz. pulses, flowers, vegetables and cereals were grown. From each experimental plot, ten soil samples based on random sampling were taken from approximately 15 cm depth. A custom made soil auger of 1 m length with 5.0 cm dia (AIC make) was pushed into the soil up to approximately 15 cm deep and pulled out. A steel handle was used to push out the soil core and was separated out from the tubular auger with a sterilized blade or knife. From this soil core, 100 g of soil samples were taken out and placed into labelled containers, covered with lids and brought to the laboratory and kept at 4 °C for further analysis.
Isolation of Pseudomonas spp
Soil of field samples was processed for soil dilution plating. The soil components from 10 cardinal points per reference field were homogenously mixed and 10 g soil per field was taken and placed into a 100 ml Duran Schott bottle filled with 90 ml of sterile 0.01% Distilled Water Agar (DWA). The Duran Schott bottle was placed on an orbital shaker at 210 rpm for 20 min. The bottle was then placed in a laminar flow and left to stand for 10 min for heavy particles to settle down. 1 ml of soil dilution was pipetted out using a sterile pipette tip into a 25 ml glass vial filled with 9 ml of 0.01% DWA and shaken. This dilution was labelled as 10−2. The process was repeated up to a dilution of 10−8. Isolation of Pseudomonas spp. was made following the serial dilutions and pour plate method using the specific King’s B medium (Kings et al. 1954). The individual colonies were picked up with sterile loop and transferred to fresh King’s B slants and the pure cultures so obtained were stored in refrigerator at 4 °C for further use.
Extraction of 2,4-diacetylphloroglucinol (2,4-DAPG)
2,4-DAPG antibiotic was extracted from isolates of Pseudomonas spp. by using the methodology of Rosales et al. (1995). Bacterial cultures were grown in 5 ml of pigment production medium (peptone, 20 g; glycerol, 20 ml; NaCl, 5 g; KNO3, 1 g; distilled water, 1 L; pH 7.2) for 4 days on a rotary shaker at room temperature (28 ± 2 °C). The culture was centrifuged at 3500 rpm for 5 min, the supernatant collected, acidified to pH 2 with 1 N HCl and then extracted with an equal volume of ethyl acetate. The ethyl acetate extract was reduced to dryness in vacuum, and the residue was dissolved in methanol.
Preparation of TLC plates and thin layer chromatography of 2,4-DAPG
An applicator was used to uniform spread slurry of silica gel G (25 g of silica gel G powder with 50 ml of water) to a thickness of 250 µm on glass plate (20 × 20 cm and 0.5 cm thickness). The plates were airdried to allow the binder to set. The plates were then activated at 120 °C for 1 h just before use. 20 µl samples were applied to thin layer chromatography plates coated with a 250 µm layer of silica gel and developed in acetonitrile/methanol/water (1:1:1) solvent system. Spots were visualized by spraying with diazotized sulphanilic acid. Rf values of the spots were compared with synthetic 2,4-DAPG (50 mg ml−1 methanol).
Preparative thin layer chromatography of 2,4-DAPG
Precoated Aluminium silica gel plates (silica gel G 60 F254, Merck India) was performed using the 2,4-DAPG extract from different isolates of Pseudomonas spp. Spots were visualized under ultra violet light at 254 nm and 360 nm. Rf values of the spots were compared with synthetic 2,4-diacetylphloroglucinol (50 mg ml−1 methanol).
Screening of antagonistic activity of 2,4-DAPG positive isolates of Pseudomonas spp. against Fusarium oxysporum using dual culture assay
The antagonistic activity of 2,4-DAPG positive isolates of Pseudomonas spp. against Fusarium oxysporum was tested by dual culture technique (Azadeh et al. 2010). Bacterial isolates were streaked at one side of petri dish (one cm away from the edge) containing PDA. Nine mm mycelial disc from 7 days old PDA culture of F. oxysporum was placed at the opposite side of each petri dish perpendicular to the bacterial streak respectively and incubated at 27 ± 2 °C for 5–7 days. Petri dishes inoculated with fungal discs alone served as control. Three replications were maintained for each isolate. Observations of width of inhibition of mycelial growth of test pathogen were recorded and per cent inhibition of pathogen growth was calculated by using the formula given by Vincent (1927).
Isolation of genomic DNA
The genomic DNA of the Pseudomonas spp. was isolated using the modified methodology of Stahl and Flesher (1987). The test strains grown overnight in 25 ml of Luria broth at 30 °C were centrifuged at 10,000 rpm at 4 °C for 10 min and the pellet was resuspended in 2.5 ml of T10 NaCl100 (10 mM Tris and 100 mM sodium chloride, pH 8.0). The suspension was centrifuged at 10,000 rpm for 10 min at 4 °C. The supernatant was discarded and the pellet was resuspended in 2.5 ml of T10E1 (10 mM Tris and 1 mM EDTA) containing lysozyme at 1 mg/ml. It was incubated at 37 °C for 20 min. To this, RNase was added at 0.2 mg/ml and incubated at 37 °C for 10 min. It was treated with 2.5 ml of 2% sarkosyl prepared in T50E20 (50 mm Tris and 20 mM EDTA) and incubated at 50 °C for 45 min. To this suspension, proteinase K was added at 0.5 mg/ml and incubated at 55 °C for 15 min. The DNA lysate was then extracted using phenol. The lysate was treated with equal volume of phenol and mixed well to form milky emulsion. It was spun at 15,000 rpm for 20 min at 4 °C to separate organic and aqueous phases. The upper aqueous phase was collected in a fresh tube, equal volume of phenol: chloroform: isoamyl alcohol (25:24:1) was added, centrifuged and the aqueous phase was collected in a fresh tube. To this, equal volume of chloroform: isoamyl alcohol (24:1) was added.
The components were mixed well and centrifuged at 15,000 rpm for 20 min at 4 °C. The aqueous phase was finally subjected to ethanol precipitation. To the aqueous phase, 1/10th volume of 3 M sodium acetate was added and incubated on ice for 20 min. 2 volumes of ice-cold ethanol was added, mixed well and centrifuged at 10,000 rpm for 10 min at 4 °C. The thick DNA pellet obtained was washed with 70% alcohol, dried and dissolved in 100 µl of T10 E1 (10 mM Tris and 1 mM EDTA) and stored at 4 °C for further analysis.
PCR amplification of 2,4-DAPG gene
PCR amplification of phlD gene which codes for 2,4-DAPG was performed using the forward primer phlD F (5´-GAG TAC GTC GAA GAC CAC CA-3´) and reverse primer phld R (5´-ACC TCA GCA TCG TGT ATG AG-3´), which were designed from the phlD sequence of Pseudomonas available in the NCBI data base using Primer 3 software. DNA amplification was carried out in PCR tubes containing 25 µl reaction mixture. Reaction mixture contained 2.5 µl of template DNA (25 ng/µl), 2.5 µl of 10× PCR Buffer, MgCl2 (4 mM), 0.2 mM of each dTTPs, dGTPs, dCTPs and dATPs, primer (2 pmol), 2.5 units Taq DNA polymerase. PCR tubes containing master mix and DNA template were thoroughly mixed and subjected to the thermal profile. The amplification reaction was carried out in a Mastercycler (Eppendorf). An initial denaturation step of 2 min was programmed in the thermocycler, followed by a loop of 30 cycles each consisting of denaturation (at 94 °C for 30 s), annealing (at 65 °C for 30 s) and extention (at 72 °C for 1 min). The final extension was performed at 72 °C for 10 min. The PCR products were then stored at 4 °C. The amplification products were then subjected to electrophoretic separation using horizontal agarose gel electrophoresis. 1.5% agarose, TBE (1×) gel was prepared with ethidium bromide (3 µl) in it. In each PCR tube, 7 μl of loading dye was added and then loaded to separate wells. 100 bp DNA ladder was used as a molecular weight marker for determining the molecular weights of PCR product. Electrophoresis was carried out at 60 V for 3 h. The gel was visually examined under UV and photographed and documented using gel documentation system (Biometra).
16S rDNA amplification
DNA coding for 16S rDNA of each isolates of Pseudomonas spp. was amplified with primer 63F (5′-CAG GCC TAA CAC ATG CAA GTC-3′) and 1387R (5′-GGG CGG WGT GTA CAA GGC-3′). These primers amplifies approximately 1300 bp of the 16S rDNA gene, specific to the consensus regions that are considered as universal bacterial domains. PCR reactions were done in a total reaction mixture of 50 µl contains 2.0 µl of template DNA (25 ng/µl), 5 µl of 10× PCR Buffer, MgCl2 (4 mM), 0.2 mM of each dTTPs, dGTPs, dCTPs and dATPs, primer (2 pmol) of each forward and reverse primer, 2.5 units Taq DNA polymerase. PCR tubes containing master mix and DNA template were thoroughly mixed and subjected to the thermal profile. The amplification reaction was carried out in a Mastercycler (Eppendorf). An initial denaturation step of 2 min was programmed in the thermocycler, initial denaturation (at 94 °C for 4 min) followed by a loop of 30 cycles each consisting of denaturation (at 94 °C for 2 min), annealing (at 55 °C for 1 min) and extention (at 72 °C for 1 min 30 s). The final extension was performed at 72 °C for 10 min. The PCR products were then stored at 4 °C. The amplification products were then subjected to electrophoretic separation using horizontal agarose gel electrophoresis.
Amplified ribosomal DNA restriction analysis (ARDRA)
The purified amplification PCR products were singly digested with the restriction endonuclease Hae III (5U/μl). Enzyme digestion was carried out at 37 °C for overnight. The restriction fragments were analyzed by electrophoresis at 2% (w/v) agarose gel in 1% TBE buffer at 100 V for 50 min. DNA 1 kb ladder was used as a DNA molecular size marker. Restriction profiles were classified according to the presence or absence of fragments. Similarity among strains was estimated from the endonuclease restriction data, using the Jaccards Coefficient, Sim Qual in similarity to compute the distance matrix using Numerical Taxonomy and Multivariate analysis system (NTSYS) software 2.1. The SHAN clustering Unweighted Pair Group Method with Arithmetic Mean (UPGMA) method was used to build the similarity dendrogram from the distance matrix using the NTSYS software.
Results and discussion
The biological control of soil-borne plant pathogens by antagonistic bacteria, particularly Pseudomonas spp. belonging to Plant Growth Promoting Rhizobacteria (PGPR), has received prominent attention because of the dual role of these bacteria in plant-growth promotion and disease control (Zehnder et al. 2001). When identifying potential biocontrol agents, antifungal metabolites produced by them or the ability of these agents to induce antifungal compounds in plants are important factors to be taken into account. In the present study, 150 isolations of Pseudomonas spp. were made from the soil samples collected from agricultural fields where different crop types viz. pulses, flowers, vegetables and cereals were grown. Representative samples from each crop type were taken for isolations. Thirty isolates (PS 1–PS 30) which showed fluorescence under UV light were selected for further studies. The isolates formed irregular and non-spreading type of colonies with slimy cream colour and yellow-green diffusible pigment of variable intensities on Pseudomonas Specific Media i.e. King’s B media. The selected isolates showed negative results on Gram staining. Microscopic observations revealed them to be rod shaped and these isolates fluoresced under UV light.
Extraction of 2,4-DAPG and its conformation through TLC and PTLC
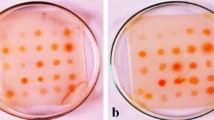
Antibiotic-producing PGPR have been studied intensively during the last decade and special attention has been given to 2,4-DAPG-producing Pseudomonas spp. because of their ability to control a wide variety of soil-borne plant pathogens (Sharifi-Tehrani et al. 1998). In our studies, all the isolates produced 2,4-DAPG. Distinct spots of 2,4-DAPG extracted from the Pseudomonas isolates having Rf in the range of 0.7–0.89 which was close to the Rf value of synthetic 2,4-DAPG (0.89). Similar results were obtained by Rosales et al. (1995) who reported distinct spots at Rf of 0.88 that appeared on the TLC plate after spraying diazotized sulphanilic acid (Figs. 1, 2). The antibiotic 2,4-DAPG produced by the isolates of Pseudomonas spp. were quantified and found to be in the range of 56–2152.70 µg ml−1. Thus, on the basis of their 2,4-DAPG production potential, they were classified as high 2,4-DAPG producers and low 2,4-DAPG producers.

Visualization of spots of 2,4-DAPG extracted from isolates of Pseudomonas spp. after spraying TLC plate with diazotized Sulphanilic acid. Lane 1: synthetic 2,4-DAPG. Lane 2–7: 2,4-DAPG extracted from isolates of Pseudomonas spp. (PS 1–PS 6)

Visualization of spots of 2,4-DAPG extracted from isolates of Pseudomonas spp. on PTLC plate under UV light (254 nm). Lane 1: Synthetic 2,4-DAPG. Lane 2–11: 2,4-DAPG extracted from isolates of Pseudomonas spp. (PS 7–PS 15)
Antagonistic activity of 2,4-DAPG positive isolates of Pseudomonas spp. against F. oxysporum using dual culture assay
The antifungal activity of 2,4-DAPG positive isolates of Pseudomonas spp. was tested against F. oxysporum by dual culture assay. The results of the dual culture technique indicated that the isolates of P. fluorescens inhibited the mycelial growth of F. oxysporum by 52.76–65.45% (Table 1). PS 1 isolate exhibited maximum per cent inhibition of mycelial growth of F. oxysporum i.e. 65.45% where as low 2,4-DAPG producers (PS 3 and PS 5) showed 52.45 and 51.46% inhibition of mycelial growth. This suggests that the low DAPG producing bacteria may be good producer of other known antibiotics secreted by rhizosphere bacteria viz. Phenazine, Pyrrolnitrin, Pyocyanin. Similar results were obtained by Kumar et al. (2014) who reported Pseudomonas fluorescens strain P.f 07 to be most effective with the highest antagonistic activity against three fungal pathogens and showed maximum inhibition of mycelial growth of Fusarium moniliforme (65.45%), Rhizoctonia solani (68.23%), and Alternaria alternata (48.13%).
PCR amplification of 2,4-DAPG gene
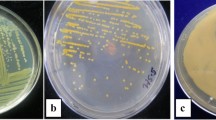
Phloroglucinol, one of the antibiotics produced by Pseudomonas spp., has been shown to have known antiviral, antibacterial, antifungal and anthelminthic properties (Keel et al. 1992). In the present study, we used PCR analysis to detect 2,4-DAPG-producing Pseudomonas populations based on the amplification of phlD gene sequence. The results of the PCR analysis with 2,4-DAPG gene-specific primer phlD indicated that a DNA fragments of approximately 750 bp in size were obtained in all the strains except PS 3 and PS 5 (Fig. 3). It was quantified and concentration of DNA in amplified product was found to be in the range of 0.05–0.5 µg/µl. Some non-specific amplications were also found in all the isolates and this may be due to the formation of primer-dimer. Occurrence of the phlD gene was demonstrated through the detection of approximately 750 bps DNA fragment in 28 isolates out of thirty isolates used in the present study. These results corresponded to Ahmadzadeh et al. (2006) who reported a DNA fragment approximately 745 bp in size in three strains P-5, P-32 and P-47.

Amplified products of phlD gene of DNA of isolates Pseudomonas spp. Lane M: 1 kb ladder, Lane 1–30: Amplified bands of phlD gene from isolates of Pseudomonas spp. (PS 1–PS 30)
Amplified 16S rDNA and ARDRA pattern
The 16S rDNA of 30 isolates of Pseudomonas spp. were amplified. Different banding patterns were obtained when digested with restriction endonuclease enzyme Hae III which distinguished the isolate one from another. In this study, digestion of 16S rDNA of 30 Pseudomonas isolates with restriction enzyme produced 3–5 bands of different sizes (Fig. 4). 16S rDNA sequencing is particularly important for accurate bacterial identification in case of bacterial isolates and it reveals bacterial community development in the rhizosphere.

rDNA restriction fragments of isolates Pseudomonas spp. (PS1–PS 30) digested with HaeIII. Lane M: 1 kb ladder
ARDRA based cluster analysis
The results of ARDRA using Hae III were subjected to cluster analysis. UPGMA method was used to construct the dendrogram of bands resulting from the digestion with restriction enzyme Hae III. The ARDRA clustering revealed that there is considerable genetic diversity among these 30 isolates (Fig. 5). Data analysis showed that the isolates could be assigned to twelve minor groups, ARDRA type PS 5, ARDRA type PS 25, ARDRA type PS 6, ARDRA type PS 2, ARDRA type PS 3, ARDRA type PS 4, ARDRA type PS 9, ARDRA type PS 13, ARDRA type PS 26, ARDRA type PS 27, ARDRA type PS 24, ARDRA type PS 30 and six major groups: (i) ARDRA types PS 1 PS 7 PS 11 PS 15 PS 16, (ii) ARDRA types PS 8 PS 10 (iii) ARDRA types PS 17 PS 21, (iv) ARDRA types PS 18 PS 19 PS 20 PS 22, (v) ARDRA types PS 23 PS 29 PS 28 and ARDRA types PS 12 PS 14. The isolates that occupy the same cluster/type are sharing the same attributes.

Dendrogram based on the rDNA restriction fragments of isolates Pseudomonas spp. (PS1–PS 30) digested with HaeIII restriction enzyme
ARDRA of Pseudomonas spp. indicated six groups with supposedly high genetic diversity. Genetic diversity play a very important role in the adaptability and survival of a species because when there is change in the organism’s environment, there is slight change in the gene variations that is necessary to produce changes in the organism that enable it to survive and adapt. A species that has a high degree of genetic diversity among its populations will have more variations from which to choose the most fit. Knowledge on the diversity within a group of strains that share a common antifungal production which provide a new and important approach for the identification of the isolates that are superior with respect to its ability to suppress the soilborne fugal pathogens and rhizosphere competence (Raaijmakers and Wellers 2001). Keel et al. (1996) studied the cluster analysis of restriction patterns of amplified DNA coding for 16S rRNA of 13 fluorescent Pseudomonads isolates that phenotypically produced antibiotics, DAPG, pyuluteorin and hydrogen cyanide from almost all samples from United States, Europe and Africa were genetically highly similar. Moreover, this ARDRA showed more diversity at the genotypic level. However, for discrimination of species that are closely related, sequencing of the entire 16S rRNA gene is essential.
It is evident that antibiotic-producing PGPR have been studied intensively during the last decade and special attention has been given to 2,4-DAPG-producers. The present study revealed that 28 out of 30 isolates have the genetic ability for the production of 2,4-DAPG antibiotic. The present study on 2,4-DAPG producing ability of PGPRs indicates their potential role as bio control agents. Further field studies needs to be conducted for further confirming their possible role as bio control agents.
References
Ahmadzadeh M, Afsharmanesh H, Javan-Nikkhah M, Sharifi-Tehrani A (2006) Identification of some molecular traits in fluorescent Pseudomonads with antifungal activity. Iran J Biotechnol 4:245–253
Azadeh BF, Sariah M, Wong MY (2010) Characterization of Burkholderia cepacia genom over as a potential bio-control agent of Ganoderma boninense in oil palm. Afr J Biotechnol 9(24):3542–3548
Bossis E, Lemanceau P, Latour X, Gardan L (2000) The taxonomy of Pseudomonas and Pseudomonas putida: current status and need for revision. Agronomie 20:51–63
De La Fuente L, Thomashow LS, Weller DM, Bajsa N, Quagliotto L, Chernin L, Arias A (2004) Pseudomonas fluorescens UP61 isolated from birdsfoot trefoil rhizosphere produces multiple antibiotics and exerts a broad spectrum of biocontrol activity. Eur J Plant Pathology 110:671–681
De La Fuente L, Mavrodi DV, Landa BB, Thomashow LS, Weller DM (2006) phlD-based genetic diversity and detection of genotypes of 2,4-diacetylphloroglucinol-producing Pseudomonas. FEMS Microbiol Ecol 56:64–78
Gupta M, Mahajan S, Sharma D, Gupta S, Mallick SA (2016a) Agrowastes as substrates for hydrolytic enzymes and biomass production of native isolates of Trichoderma spp. Indian Phytopath 69(4s):558–564
Gupta S, Mahajan S, Gupta M, Sharma D (2016b) Screening of native isolates of Trichoderma spp. of Jammu for their biocontrol potential through hydrolytic enzyme activities. Indian Phytopath 69:173–180
Keel C, Schnider U, Maurhofer M, Voisard D, Laville J, Burger U, Wirthner P, Haas P, Defago G (1992) Suppression of root diseases by Pseudomonas CHAO, importance of the bacterial secondary metabolite, 2,4-diacetylphloroglucinol. Mol Plant Microbe Interact 5:4–13
Keel C, Weller DM, Natsch A, Defago G, Cook RJ, Thomashow LS (1996) Conservation of the 2,4-diacetylphloroglucinol biosynthesis locus among fluorescent Pseudomonas strains from diverse geographic locations. Appl Environ Microbiol 62:552–563
King EO, Wood MK, Raney DE (1954) Two simple media for the demonstration of pyocyanin and luorescein. J Lab Clin Med 44(2):301–307
Kumar MM, Singh R, Tomer A (2014) In vitro evaluation of antagonistic activity of Pseudomonas fluorescens against fungal pathogen. J Biopest 7(1):43–46
Landa BB, Mavrodi OV, Raaijmakers JM, McSpadden Gardener BB, Thomashow LS, Weller DM (2002) Differential ability of genotypes of 2,4-diacetylphloroglucinol-producing Pseudomonas strains to colonize the roots of pea plants. Appl Environ Microbiol 68:3226–3237
Mavrodi DV, Bonsall RF, Delaney SM, Soule MJ, Phillips G, Thomashow LS (2001) Functional analysis of genes for biosynthesis of pyocyanin and phenazine-1-carboxamide from Pseudomonas aeruginosa PAO1. J Bacteriol 183:6454–6465
Mavrodi DV, Mavrodi OV, McSpadden-Gardener BB, Landa BB, Weller DM, Thomashow LS (2002) Identification of differences in genome content among phlD-positive Pseudomonas strains by using PCR-based subtractive hybridization. Appl Environ Microbiol 68:5170–5176
Mazzola M, Funnell DL, Raaijmakers JM (2005) Weath cultivar-specific selection of 2,4-diacethylphloroglucinol-producing fluorescent Pseudomonas species from resident soil populations. Microb Ecol 48:338–348
McSpadden Gardener BB, Schroeder KL, Kalloger SE, Raaijmakers JM, Thomashow LS, Weller DM (2000) Genotypic and phenotypic diversity of phlD-containing Pseudomonas strains isolated from the rhizosphere of wheat. Appl Environ Microbiol 66:1939–1946
Raaijmakers JM, Weller DM (2001) Exploiting genotypic diversity of 2,4-diacetylphloroglucinol-producing Pseudomonas spp.: characterization of superior root-colonizing P. fluorescens strain Q8r1-96. Appl Environ Microbiol 67:2545–2554
Ramette A, Moenne-Loccoz Y, Defago G (2001) Polymorphism of the polyketide synthase gene phlD in biocontrol fluorescent Pseudomonads producing 2,4-diacetylphloroglucinol and comparison of PhlD with plant polyketide synthases. Mol Plant-Microbe Interact 14:639–652
Ramette A, Moenne-Loccoz Y, Defago G (2003) Prevalence of fluorescents pseudomonads producing antifungal phloroglucinols and/or hydrogen cyanide in soils naturally suppressive or conductive to tobacco black root rot. FEMS Microbial Ecol 44:35–43
Rosales AM, Thomashow L, Cook RJ, Mew TW (1995) Isolation and identification of antifungal metabolites produced by rice-associated antagonistic Pseudomonas spp. Phytopathology 85:1028–1032
Sharifi-Tehrani A, Zala M, Natsch A, Moenne-Loccoz Y, Defago G (1998) Biocontrol of soil-borne fungal plant diseases by 2,4-diacetylphloroglucinol-producing fluorescent Pseudomonads with different restriction profiles of amplified 16S rDNA. Eur J Plant Pathol 104:631–643
Stahl DA, Flesher B (1987) Application of ribosomal RNA sequencing to studies of microbial ecology: Gray Freshwater Biological Institute Summer Course. Urbana, III
Vincent JM (1927) Distortion of fungal hyphae in the presence of some inhibitors. Nature 159:850
Weller DM, Raaijmakers JM, Gardener BB, Thomashow LS (2002) Microbial populations responsible for specific soil suppressiveness to plant pathogens. Annu Rev Phytopathol 40:309–348
Weller DM, Landa BB, Mavrodi OV, Schroeder KL, De La Fuente L, Bankhead SB, Molar RA, Bonsall RF, Mavrodi DV, Thomashow LS (2007) Role of 2,4-diacetylphloroglucinol-producing fluorescent Pseudomonas spp. in the defense of plant roots. Plant Biol 9:4–20
Zehnder GW, Murphy JF, Sikora EJ, Kloepper JW (2001) Application of rhizobacteria for induced resistance. Eur J Plant Pathol 107:39–50
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sharma, D., Gupta, M., Gupta, S. et al. Antibiotic gene specific characterization and ARDRA analysis of native isolates of Pseudomonas spp. from Jammu, India. Indian Phytopathology 71, 225–233 (2018). https://doi.org/10.1007/s42360-018-0028-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42360-018-0028-9