
Abstract
Density functional theory (DFT) within the full-potential linearized augmented plane waves (FP-LAPW) method have been employed to investigate the structural, electronic and magnetic properties of a new half-Heusler SiRbCa alloy in its three crystalline phases (α, β and γ) using two alternative approximations; GGA-PBE and mBJ-GGA-PBE that account for spin polarization. For the three phases α, β and γ of half-Heusler SiRbCa alloy, we have observed that the ferromagnetic phase is energetically beneficial relative to the magnetic phase, which is most stable for the three phases. Band structure and density of state calculations using GGA-PBE and mBJ-GGA-PBE are illustrated, the half-Heusler SiRbCa alloy behaves as a semiconductor for the majority of its spins and metal for the minority of its spins, giving a unique half-metallic nature. This approximation maintains the properties of having a direct fundamental gap of X → X.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
New features of materials such as polarization spin, conduction band, symmetry of polarized carriers and magnetism of surfaces are crucial to develop the technology and industry. The use of half metals is a spin-based electronic application. The “half-metallicity” of these substances, in which the conduction electrons are 100% spin polarized at Fermi level is a defined characteristic. As a result, it has very interested in creating materials with this feature and a new class of materials known as ferromagnetic half metals has been predicted [1]. Due to their electronic and magnetic characteristics, half-metallic magnetic materials have garnered attention and suggested for using in spintronic applications. The discovery of materials with two spin polarizations, resulting in 100% spin polarization at Fermi level, has sparked interest in exploring novel ferromagnetic half-metallic compounds [2].
Non-magnetic-based half-metallic (HM) ferromagnetic Heusler alloys have been anticipated according to the research on electronic structure, which is inspired by their peculiar magneto-optical capabilities. Certain Heusler materials exhibit half-metallic ferromagnetism [3, 4], which is a metallic in one spin orientation. De Groot et al. [5,6,7,8,9] have established a categorization scheme by separating three distinct varieties of half-metallic ferromagnetism [10]. In addition, numerous families of Heusler-type alloys have their half-metallicity predicted using first-principles calculations [11,12,13,14,15,16]. Half-Heusler RbSrX (X = C, Si, and Ge) alloys have been investigated for their structural, electronic and magnetic characteristics using FP-LAPW by Rozale et al. [11]. They have found that half-metallic gaps of RbSrC (0.31 eV), RbSrSi (0.14 eV) and RbSrGe (0.12 eV), all support the alloys’ ferromagnetic nature. The structural, elastic, electronic, magnetic and optical characteristics of RbSrX (X = C, Si, and Ge) alloys have been computed using FP-LAPW by Ahmad et al. [12]. They have demonstrated that RbSrC, RbSrSi, and RbSrGe are elastically stable half-metals with indirect gap; 2.01 eV, 1.92 eV and 1.5 eV, respectively. The electronic structure, chemical bonding and magnetism in a variety of half-Heusler XMZ alloys have been studied by Nanda and Dasgupta [13] using FP-LMTO and TB-LMTO ASA methods. Using first-principles calculations to examine the electronic structures of a series of hypothetical alloys with the half-Heusler structure CrTiX (X = Si, Ge, Sn, Pb), Sattar et al. [14] have predicted that these alloys are to be half-metallic. The structural, electronic and magnetic characteristics of XYZ (X = Li, Na, K, and Rb; Y = Mg, Ca, Sr, and Ba; Z = B, Al, and Ga) ferromagnetic half-Heusler alloys have been investigated by Umamaheswari et al. [15] using the linearized muffin-tin orbital approach connected to strong bond method (TB-LMTO-ASA). First-principles calculations by Gao and Wang [16] have projected half-metallicity in full-Heusler Cu2MnAl and RbSrN2 alloys. New Heusler ferromagnetic HM alloys without transition metal ions or rare earth have been studied due to these findings [17,18,19,20,21,22]. In addition, a theoretical investigation of α, β and γ phases of Heusler ternary GeNaZ (where Z = Ca, Sr, and Ba) alloys has been reported by Beldi et al. [22]. They have investigated the half-metallic, elastic and dynamic stability of these alloys. The Fermi level contributions from 3p states of Si atom are largest, as predicted by the density of state and charge density curve for half-Heusler SiRbCa alloy. Theroetcial investigations of half-Heusler alloys including structural, mechanical, thermodynamic, electronic, magnetic and thermoelectric properties are achieved successfully [23,24,25].
The prime novelty is to undertake first-principles calculations for SiRbCa alloy in three distinct phases; α, β and γ due to it is structurally stable to discover novel half-metallic materials that is not available in the literature. Our work is devoted to explore the ferromagnetic and half metallic characters in a new class of half-Heusler alloys that do not contain transition metals. The calculations are predictive and material is predicted. The primary goal is to foretell the structural, electronic and magnetic characteristics of a novel Heusler alloy. This work is divided into three sections. The second is for computational details, followed by the elaborations of results and dicsuiions in the third section. The conclusions are outlined in the fourth section.
2 Computational
Our research focuses on a novel class of half-Heusler alloys that do not include transition metals, therefore, uncharted territory is in terms of their ferromagnetic and half-metallic properties. Half-Heusler SiRbCa alloy’s magnetic moment is an integer, with most of its strength coming from the Si site and the rest is from Rb and Ca sites. The computations in this study are carried out utilizing the full potential linearized augmented plane wave method (FP-LAPW) as implemented in WIEN2K code [26]. The generalized gradient approximation (GGA-PBE) proposed by Perdew, Burke and Ernzerhorf (PBE) [27] is used for handling of potential exchange and correlation. In addition to the aforementioned approximations, mBJ-GGA-PBE approximation is utilized for the electronic characteristics [28, 29]. It provides a more accurate representation of the values for fundamental and half-metallic gaps. This approximation is a hybrid between the exchange potential of TB-mBJ and correlation potential of GGA-PBE. It introduces a novel method for dealing with exchange and correlation potential, similar to Tran and Blaha [29] modification of Beckett-Johnson swap component [25]. It is a viable replacement for GW method or hybrid functional in terms of precision. Several semiconductors and insulators may have their gaps accurately determined by combining TB-mBJ exchange with GGA [29,30,31,32,33,34,35]. You can see the electronic setups and parameters we utilized in Table 1. In this approach, the unit cell is partitioned into a sphere-representing area centered on the nuclei and sphere-free hub region placed in the void between the spheres. The wave function is characterized by plane waves in the interstices but by atomic functions in the spheres of muffin tin. Throughout the un-overlapped spheres surrounding the atomic sites up to l = 10, the basis functions, electronic densities and potentials are formed as a single combination of spherical harmonics, but in the core area with a cut-off radius; RMT × KMAX = 9, they are produced as a Fourier series. (The size of matrix is determined by RMT × KMAX, where Rт is the minimum radius of a muffin tin and KMAX is the standard for greatest wave vector utilized in formulation of one’s own functions in the plane). Samples from the Brillouin zone are taken using a method similar to that proposed by Monkhorst and Pack [36]. For our alloy, we have employed a total 1500 k points in the Brillouin space, which is equivalent to a grid of special points k in the irreducible Brillouin space with dimensions; 11 × 11 × 11 for the three cubic phases (α,β,γ). We have used Ca (4s2), Rb (5s1), and Si (3s2 3p2) valence states in our computations and throughout all phases.
The computational details involving the density functional theory (DFT), specifically the full potential linearized augmented plane wave (FP-LAPW) approach are crucial for accurately simulating the electronic and structural properties of materials. In this method, the interatomic interactions are described by solving Schrödinger’s equation self-consistently.
The FP-LAPW method divides the unit cell into two regions, separating the core and valence electrons, with different treatments. Core electrons are approximated as spherical waves within muffin-tin spheres, while valence electrons are expanded as plane waves outside these spheres. The augmented plane wave basis set allows for precise description of electronic wavefunctions. The integration of this approach with exchange-correlation functionals (GGA-PBE) enables the determination of electronic, structural and magnetic properties with a high accuracy. It is a computationally intensive method, often necessitating high-performance computing clusters for practical application.
For RMT × KMAX, where matrix size and number of k-points in IBZ are determinant fundamental functions, a convergence test of various computation settings is performed. Determining the ground state is the first step in structural optimization. The iterative approach is performed until the calculated total energy converges to less than 0.01 mRyd. Table 2 presents the results of our computation into the structural stability of half-Heusler SiRbCa alloy using the three probable crystal phases (α,β,γ).
While investigating equilibrium structural characteristics, it is important first to make a self-consistent calculation of total energy (self-consistent calculation) and do so for a variety of values of mesh parameter. The second stage is to modify the total energy using Murnaghan’s equation of state [37], provided by the following relation;
where V is volume of cell, V0 is equilibrium volume in ground state, B is bulk modulus and its derivative \( {B^{\prime }}\).
The equilibrium lattice constant is given by the minimum of curve Etot(V), and finding out \( {B^{\prime }}\)s bulk modulus requires knowing the following:
The derivative of bulk modulus \( {B^{\prime }}\) is defined as:
It is necessary to use mBJ-GGA to improve the results of electronic propertied and especially for ferromagnetic materials [38] (Fig 1).

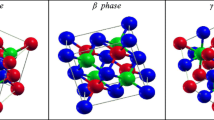
Different crystal structures of half-Heusler SiRbCa alloy used for three phases (a) α, (b) β and (c) γ
3 Results and Discussions
3.1 The Structural Properties of Half-Heusler SiRbCa Alloy
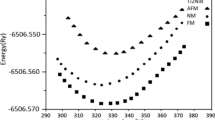
After analyzing the phases of half-Heusler alloy, the crystal structure and magnetic phase with highest stability are established. We have determined the total energy variation as a function of volume for half-Heusler SiRbCa alloy in both non-magnetic (non-spin-polarized) and ferromagnetic (spin-polarized) situations (Table 2). In order to determine the structural stability of half-Heusler SiRbCa alloy, it is investigated in the amorphous, crystalline and agglomerate phases. Murnaghan’s equation of state [37] allows us to determine the equilibrium by examining a plot of total energy versus volume. Figure 2 illustrates the volume-energy-change curve of half-Heusler SiRbCa alloy in its three phases. This figure demonstrates the stability of ferromagnetic phase in half-Heusler SiRbCa alloy. Using both spin polarization (SP) and non-SP computations, we find that the half-Heusler SiRbCa alloy is ferromagnetic throughout all three phases. Table 3 displays the investigated findings derived from with and without SP calculations in the three phases of half-Heusler SiRbCa alloy, which compare these results to other existing theoretical results.
The half-Heusler SiRbCa alloy exhibits intriguing structural properties that make it a subject of significant research interest. It possesses a cubic crystal structure with a space group F-43 m, showcasing excellent crystallographic stability. The alloy is known for its semiconductor characteristics, with a direct bandgap that falls within the visible light range, making it suitable for optoelectronic applications. Its inherent high thermoelectric performance stems from a combination of unique properties such as low thermal conductivity and high electrical conductivity. Furthermore, SiRbCa’s thermal expansion coefficient closely matches that of Si, making it an attractive candidate for silicon-based device integration.
The structural properties of half-Heusler SiRbCa alloy are fascinating. This compound’s crystal lattice combines semiconducting silicon (Si) with metallic rubidium-calcium (RbCa). This amalgamation results in a complex, highly stable structure, enabling enhanced thermoelectric performance. The alloy’s unique electronic configuration, particularly its intricate band structure and high electronic density of states, contributes to its exceptional properties, holding promise for efficient energy conversion and diverse technological applications in the realm of materials science and engineering.

Total energy as a function of volume per formula unit for non-spin-polarized (NSP) and spin-polarized (SP) configurations of SiRbCa half-Heusler alloys calculated for α, β and γ phases using GGA-PBE
3.2 The Magnetic Properties of Half-Heusler SiRbCa Alloy
Table 4 presentes the investigated results of our GGA-PBE and mBJ-PBE calculations for the total and partial magnetic moment of half-Heusler SiRbCa alloy in the three phases α, β and γ. Using GGA-PBE and mBJ-GGA-PBE, we have determine that half-Heusler SiRbCa alloy, which is half-metallic with a total integer magnetic moment equal to 1 µB throughout all three phases. Ferromagnetic half-Heusler alloys; XYZ (X = Li, N’a, Rb and K); (Y = Ca, Mg, Sr and Ba); (Z = B,Al and Ga) have been investigated by Umamaheswari et al. [15] to use the linearized muffin-tin orbital approch connected to the strong bond method. It is found that the magnetic moment value of half-Heusler alloy SiRbCa is an integer that originly comes mainly from the silicon site with a small contribution of Rb and Ca atoms.
The magnetic properties of half-Heusler SiRbCa alloy are a significant focus of research due to their potential for multiapplication. This alloy exhibits interesting magnetism, often characterized by its half-metallic behavior. It is considered a ferromagnetic material, with a high magnetic moment, typically attributed to the partially filled 3d orbitals of transition metal element, such as Fe or Co. The alloy displays robust ferromagnetism, making it valuable for spintronic devices and magnetic storage applications. Moreover, its half-metallic nature means that one spin channel is conducting, while the other is insulating, offering unique opportunities for spin-polarized transport and magnetic tunnel junctions.
The magnetic properties of half-Heusler SiRbCa alloy are captivating. This compound exhibits interesting magnetic behavior, showcasing both ferromagnetic and semiconducting characteristics. Its magnetic structure is influenced by interactions between the spins of constituent elements, offering potential for spintronic applications. The alloy’s tunable magnetic properties, coupled with its promising electronic structure, render it a candidate for novel magnetic devices and spin-based technologies, highlighting its significance in the field of materials science and technology.
3.3 The Electronic Properties of Half-Heusler SiRbCa Alloy
3.3.1 The Band Structure of Half-Heusler SiRbCa Alloy
The band structures of half-Heusler SiRbCa alloy computed using two GGA-PBE and mBJ-GGA-PBE, with spin polarization (for the majority and minority spins) in the most stable phase (in phase α) are shown in Figs. 3 and 4, respectively. The band structures of half-Heusler SiRbCa alloy produced by two GGA-PBE and mBJ-GGA-PBE methods are found to exhibit a half-metallic character (HM) with regard to semiconductor and metal in both majority and minority spin directions. These findings corroborate those published in [22], which examined the same family of half-Heusler alloys composed entirely of non-magnetic elements. Figure 3 illustrates a direct bandgap; Eg=1.17 eV in the direction X → X as predicted using GGA-PBE approximation along the majority spin direction. However, the valence and conduction bands overlap significantly at Fermi level when minority spins are polarized. With a half-metallic gap \( {E}_{g}^{HM}\)=0.25 eV and integer total magnetic moment (µtot = 1 µB), our half-Heusler alloy has been confirmed to be half-metallic (see Table 4). Table 5 displays the basic bandgap; Eg=1.85 eV and half-metallic gap \( {E}_{g}^{HM}\)=0.56 eV are retained using mBJ-GGA-PBE as illustrated in Fig. 4. As far as we know for SiRbCa half-Heusler alloys, there is no theoretical nor experimental research exists. Half-Heusler SiRbCa alloy fundamental bandgap (Eg) and half-metallic gap (HM gap) values are investigated as listed in Table 5. The bandgap is defined as a difference between the Fermi level and valence band maximum, while the HM gap is defined as a difference between the Fermi level and conduction band minimum. Comparing the bandgap values estimated by mBJ-GGA-PBE and GGA, you will see that the former is often larger.
The band structure of half-Heusler SiRbCa alloy plays a pivotal role in understanding its electronic properties. SiRbCa is recognized for its semiconductor behavior, featuring a direct bandgap. The band structure demonstrates a characteristic energy dispersion, which highlights the energy levels of electrons in the crystal lattice. The alloy’s electronic bandgap is typically tailored to fall within the visible light spectrum, making it suitable for optoelectronic applications. The unique combination of its band structure and crystal symmetry results in outstanding electronic properties, including high carrier mobility and excellent thermoelectric performance, adding to its appeal for a wide range of electronic and energy conversion devices.
The band structure of half-Heusler SiRbCa alloy displays a unique electronic configuration. It combines the characteristics of a semiconducting silicon (Si) matrix with the metallic nature of rubidium-calcium (RbCa). This integration creates a complex and diverse band structure, offering a high density of electronic states near the Fermi level. Such a feature not only contributes to its exceptional thermoelectric performance but also holds a potential for various electronic and energy-related applications due to its tailored band properties.

Spin polarized band structures of SiRbCa half-Heusler alloys calculated in stable α phase using GGA-PBE for both (a) majority and (b) minority spin channels. The horizontal dashed line indicates the Fermi level

Spin polarized band structures of half-Heusler SiRbCa alloys calculated in stable α phase using mBJ-GGA-PBE for both (a) majority and (b) minority spin channels. The horizontal dashed line indicates the Fermi level
3.3.2 The Density of States of Half-Heusler SiRbCa Alloy
The hybridization type and the states responsible for the magnetism in this half-Heusler alloy may be better understood by calculating the total density of state (DOS) and partial density of state (PDOS) to get insight into the band structure. Figures 5 and 6 show the computed partial density of state spectra for the two orientations of majority and minority spins of half-Heusler SiRbCa alloy, respectively. Both spectra feature an initial peak distant from the Fermi level that derives from 3s states of Si atoms. The 3p states of Si atoms with a little contribution from 3d states of Ca atoms are primarily responsible for the peaks slightly below and above Fermi level. Ca 3d states contribute less than Si 3p states, so keep that in mind. The density of state reveals a non-zero value at Fermi level for the polarization of minority spins, whereas the density of density suggests a zero value for polarization of majority spins (Figs. 5 and 6), indicating a bandgap in the direction of polarization of majority spins. Moreover, we see that the maxima of Si 3s and Si 3p states at Fermi level change location depending on spin direction. The change in Si 3p states explains a half-metallicity by indicating a full spin polarization. The 3p orbitals of Si atoms are definitive cause of magnetism. The half-metallic (HM) feature of our half-Heusler alloy is shown by a larger peak shift when utilizing mBJ-GGA-PBE (Fig. 6), in contrast with GGA-PBE (Fig. 5) suggested potential avenues for further investigations [38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59]. An attempt has been made to make a detailed theoretical study in order to leave the choice for elaboration of a new spintronic material in future.

Spin polarized total (DOS) and partial (PDOS) densities of states of SiRbCa half-Heusler alloy calculated for stable α phase using GGA-PBE. The vertical dashed lines indicate Fermi level. The positive and negative DOS values correspond to majority and minority spin channels, respectively

Spin polarized total (DOS) and partial (PDOS) densities of states of SiRbCa half-Heusler alloy calculated for stable α phase using mBJ-GGA-PBE. The vertical dashed lines indicate Fermi level. The positive and negative DOS values correspond to majority and minority spin channels, respectively
The density of states (DOS) in half-Heusler SiRbCa alloy is a crucial aspect of its electronic structure. The DOS represents the number of electronic states per unit energy interval and plays a pivotal role in determining the alloy’s electronic and thermal properties. SiRbCa typically exhibits a complex DOS with distinct features, including the presence of band edges and peak-like structures. These features are associated with energy bands and unique electronic configuration of constituent elements. Understanding the DOS is essential for predicting the alloy’s conductivity, thermal behavior and its suitability for applications like thermoelectric devices, where control over electronic states is essential for optimizing efficiency and performance.
The density of states in half-Heusler SiRbCa alloy is a key factor in its unique properties. This compound showcases a diverse electronic structure with a notable density of states that significantly affects its behavior. Its distinctive band structure near the Fermi level contributes to a high density of electronic states, influencing its exceptional thermoelectric properties. The tailored density of states within this alloy holds a promise for applications in energy conversion, electronics and various technological advancements.
4 Conclusions
In this work, the structural and half-metallic stabilities in half Heusler SiRbCa alloy have been examined using FP-LAPW method within GGA-PBE and mBJ-GGA-PBE exchange-correlation. These findings indicate that FM in phase α is preferred FM for this alloy. Its electronic structures are metallic in spin-down channel and semiconducting in spin-up direction, exhibiting a half-metallic behavior. The alloy’s distinctive personality results from the spin polarization of its Si-3p orbitals. The total magnetic moment of SiRbCa is 1 µB as shown by magnetic measurements. Because of its intriguing magnetic properties, we believe this alloy might be used in spintronics.
Data Availability
All used data is embedded in this manuscript and properly referenced where applicable.
References
Fujii S, Okada M, Ishida S, Asano S (2008) High spin polarizaion of ferrimagnetic heusler-type alloys in Mn–Cr–Z system (Z = IIIb, IVb, vb elements). J Phys Soc Jpn 77:074702
Luo H, Zhang H, Zhu Z, Ma L, Xu S, Wu G, Zhu X, Jiang C, Xu H (2008) Half metallic properties for the Mn 2 Fe Z (Z = Al, Ga, Si, Ge, Sb) Heusler alloys: a first principles study. J Appl Phys 103:083908
Kresse G, Hafner J (1993) Ab initio molecular dynamics for open-shell transition metals. Phys Rev B 48:13115
Jolliffe IT (1986) Principal components in regression analysis. Principal component analysis. ed: Springer, pp 129–155
De Groot R, Mueller F, Van Engen P, Buschow K (1983) New class of materials: half metallic ferromagnets. Phys Rev Lett 50:2024
Kübler J, William A, Sommers C (1983) Formation and coupling of magnetic moments in Heusler alloys. Phys Rev B 28:1745
Wurmehl S, Fecher GH, Kandpal HC, Ksenofontov V, Felser C, Lin H-J (2006) Investigation of Co 2 Fe Si: the Heusler compound with highest Curie temperature and magnetic moment. Appl Phys Lett 88:032503
Souza SD, Saxena R, Shreiner W, Zawislak F (1987) Magnetic hyperfine fields in Heusler alloys Co YZ (Y = Ti, Zr; Z = Al, Ga, Sn). Hyperfine Interact 34:431434
De Groot RA, Mueller FM, van Engen PG, Buschow KHJ (1984) Half-metallic ferromagnets and their magneto-optical properties (invited). J Appl Phys 55:2151–2154
Fang CM, De Wijs G, De Groot R (2002) Spin-polarization in half-metals. J Appl Phys 91:8340–8344
Rozale H, Amar A, Lakdja A, Moukadem A, Chahed A (2013) Half-metallicity in the half-Heusler RbSrC, RbSrSi and RbSrGe compounds. J Magn Magn Mater 336:83–87
Ahmad M, Murtaza G, Khenata R, Omran SB, Bouhemadou A (2015) Structural, elastic, electronic, magnetic and optical properties of RbSrX (C, SI, Ge) half-Heusler compounds. J Magn Magn Mater 377:204–210
Nanda B, Dasgupta I (2003) Electronic structure and magnetism in half-Heusler compounds. J Phys: Condens Matter 15:7307
Sattar MA, Rashid M, Rasool MN, Mahmood A, Hashmi MR, Ahmad S, Imran M, Hussain F (2016) Half-metallic ferromagnetism in new half-Heusler compounds: an ab initio study of CrTiX (X = Si, Ge, Sn, Pb). J Supercond Novel Magn 29:931–938
Umamaheswari R, Yogeswari M, Kalpana G (2014) Ab-initio investigation of half metallic ferromagnetism in half-Heusler compounds XYZ (X = Li, na, K and rb; Y = mg, ca, Sr and Ba; Z = B, Al and Ga). J Magn Magn Mater 350:167–173
Gao Y, Wang X (2015) d0 half-metallicity of the bulk and surface (001) of full-heusler alloy RbSrN2: a first-principle study. J Magn Magn Mater 385:394–401
Navdeep Kaur R, Sharma Y, Al-Douri V, Srivastava (2023) Alaa Farag Abd El Rehim Thermodynamic, thermoelectric and optoelectronic performances of Co2MB (M = nb, Ta) full-heusler compounds for solar cell and UV absorber applications. Mater Sci Semiconduct Process 165:107676–107687
Abed Bouadi T, Lantri S, Mesbah M, Houari I, Ameri L, Blaha M, Ameri Y, Al-Douri AF (2022) Abd El-Rehim a new semiconducting full Heusler Li2BeX (X = Si, Ge and Sn): first-principles phonon and boltzmann calculations. Phys Scr 97:105710–105719
Gaid FO, Boufadi FZ, Tayebi N, Ameri M, Mentefa A, Bellagoun L, Odeh AA, Al-Douri Y (2022) Theoretical investigation of structural, electronic, elastic, magnetic, thermodynamic and thermoelectric properties of Ru2MnNb heusler alloy: FP-LMTO method. Emergent Mater 5:1065–1073
Lamia Drici F, Belkharroubi FZ, Boufadi I, Ameri M, Ameri W, Belkilali S, Azzi F, Khelfaoui Y (2022) Al-Douri First-principles calculations of structural, elastic, electronic, and optical properties of CaYP (Y = Cu, Ag) Heusler alloys. Emergent Mater 5:1039–1054
Benaissa Nour-eddine F, Belkharroubi I, Ameri LF, Blaha B, Abdelghani D, Lamia M, Bourdim A, Tebboune MN, Belkaid M, Ameri B, Fassi S, Driz Y, Al-Douri AF (2022) Abd El-Rehim, A. Bouhemadou Effect of 5d state-based full-heusler alloys on the structural, electronic and magnetic properties of new half metallic ferromagnetism. Mater Today Commun 33:104277–104288
Beldi L, Zaoui Y, Obodo KO, Bendaoud H, Bouhafs B (2020) d0 half-metallic ferromagnetism in GeNaZ (Z = ca, Sr, and Ba) Ternary Half-Heusler alloys: an ab initio investigation. J Supercond Novel Magn 33:3121–3132
Kapil J, Pathak A, Shukla P (2022) Computational investigation of half-metallicity in d0 half-Heusler alloys GeKZ (Z = ca, Sr) using first-principles calculations. Comput Mater Sci 213:111665–111672
Kapil J, Shukla P, Pathak A, (2021) A first-principles study of Ru2VGe full-Heusler alloy—pseudopotential approach. Eur Phys J Plus 136:991-999
Mishra AR, Pal S (2021) Ab-initio investigation of structural, mechanical, thermodynamic, electronic, magnetic and thermoelectric properties of half-metallic d0 half-Heusler alloys LiXSi (X = Ca, Sr). J Solid State Chem 304: 122610–122619
Blaha P, Schwarz K, Madsen GKH, Kvasnicka D, Luitz J, Laskowski R, Tran F, Marks LD (2018) WIEN2k, An Augmented Plane Wave + Local Orbitals Program for Calculating Crystal Properties (Karlheinz Schwarz,Techn. Universität Wien, Austria),
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77:3865–3868
Becke AD, Johnson ER (2006) A simple effective potential for exchange. J Chem Phys 124:221101
Tran F, Blaha P (2009) Accurate Band gaps of Semiconductors and insulators with a Semilocal exchange-correlation potential. Phys Rev Lett 102:226401
Koller D, Tran F, Blaha P (2011) Merits and limits of the modified Becke-Johnson exchange potential. Phys Rev B 83:195134
Koller D, Tran F, Blaha P (2012) Improving the modified Becke-Johnson exchange potential. Phys Rev B 85:155109
Chen S, Gong X, Walsh A, Wei S-H (2009) Crystal and electronic band structure of Cu 2 ZnSnX 4 (X = S and Se) photovoltaic absorbers: first-principles insights. Appl Phys Lett 94:041903–041903
Singh DJ (2010) Structure and optical properties of high light output halide scintillators. Phys Rev B 82:155145
Singh DJ, Seo SSA, Lee HN (2010) Optical properties of ferroelectric Bi4Ti3O12. Phys Rev B 82:180103
Pauling L (1960) The nature of the chemical bond and the structure of molecules and crystals: an introduction to modern structural chemistry vol. 18: Cornell University Press,
Monkhorst HJ, Pack JD (1976) Special points for Brillouin-Zone integrations. Phys Rev B 13:5188–5192
Murnaghan F (1944) “The compressibility of media under extreme pressures,“ Proceedings of the National Academy of Sciences, vol. 30, pp. 244–247,
Meinert M (2013) Modified Becke-Johnson potential investigation of half-metallic heusler compounds, Phys. Rev. B, vol. 87, pp. 045103,
Fouad A, Straus S, McDonald D, Heimer F, Xia Q (2023) Optical properties of GaX (X = P, as, sb) under hydrostatic pressure, Experimental and Theoretical NANOTECHNOLOGY, vol. 7, pp. 283–290,
Kojal N, Javec S (2023) Structuraland thermal properties of H6Ta2O17, Experimental and Theoretical NANOTECHNOLOGY, vol. 7, pp. 309–316,
Raman V, Rajan A (2023) Thermodynamic properties of MgFeH3 alloy, Experimental and Theoretical NANOTECHNOLOGY, vol. 7, pp. 381–392,
Radiman S, Rusop M (2023) Investigation of structural and optical properties of In-doped AlSb nanostructures, Experimental and Theoretical NANOTECHNOLOGY, vol. 7, pp. 49–76,
Oula Jabbar AH, Reshak (2023) Structurlectronic, and optoelectronic properties of XYZ2 (X = zn,cd; Y = Si,Sn;Z = pnicogens) Chalcopyrite compounds: first-principles calculations, Experimental and Theoretical NANOTECHNOLOGY, vol. 7, pp. 97–110,
Liu J, Yang R (2023) Structural, dynamical, thermodynamic properties of CdYF3 perovskite, Experimental and Theoretical NANOTECHNOLOGY, vol. 7, pp. 111–126,
Sahnoun R (2023) Gap bowing of in Pb1-xCaxS, Pb1-xCaxSe and Pb1-xCaxTe alloys, Exp Experimental and Theoretical NANOTECHNOLOGY, vol. 7, pp. 149–158,
Samia L, Belkharroubi F, Ibrahim A, Lamia BF, Saim A, Maizia A, Mohammed A, Al-Douri Y (2022) Investigation of structural, elastic, electronic, and magnetic proprieties for X2LuSb (X = mn and ir) full-heusler alloys. Emergent Mater 5:537–551
Benaddi F, Belkharroubi F, Ramdani N, Ameri M, Haouari S, Ameri I, Drici L, Azzi S, Al-Douri Y (2021) Electronic and magnetic investigation of half-metallic ferrimagnetic full-heusler Mn2IrGe. Emergent Mater 4:1745–1760
Ayad M, Belkharroubi F, Boufadi F, Khorsi M, Zoubir M, Ameri M, Ameri I, Al-Douri Y, Bidai K, Bensaid D (2020) First-principles calculations to investigate magnetic and thermodynamic properties of new multifunctional full-Heusler alloy Co 2 TaGa. Indian J Phys 94:767–777
Fadila B, Ameri M, Bensaid D, Noureddine M, Ameri I, Mesbah S, Al-Douri Y (2018) Structural, magnetic, electronic and mechanical properties of full-heusler alloys Co2YAl (Y = fe, Ti): first principles calculations with different exchange-correlation potentials. J Magn Magn Mater 448:208–220
Zoubir MK, Fadila B, Keltoum B, Ibrahim A, Farah BL, Al-Douri Y, Mohammed A (2021) Structural, electronic and thermodynamic investigation of Ag2GdSi, Ag2GdSn and Ag2Gd pb heusler alloys: first-principles calculations. Mater Test 63:537–542
Saim A, Belkharroubi F, Boufadi F, Ameri I, Blaha L, Tebboune A, Belkaid M, Belkilali W, Ameri M, Al-Douri Y (2022) Investigation of the structural, elastic, electronic, and optical properties of half-Heusler CaMgZ (Z = C, Si, Ge, Sn, Pb) compounds. J Electron Mater 51:4014–4028
Semari F, Dahmane F, Baki N, Al-Douri Y, Akbudak S, Uğur G, Uğur Ş, Bouhemadou A, Khenata R, Voon CH (2018) First-principle calculations of structural, electronic and magnetic investigations of Mn2RuGe1-xSnx quaternary Heusler alloys. Chin J Phys 56:567–573
Ameri M, Belkharroubi F, Ameri I, Al-Douri Y, Bouhafs B, Boufadi FZ, Touia A, Boudia K, Mired F et al (2014) “Ab initio calculations of structurlastic, and thermodynamic properties of HoX (X = N, O, S and Se),“ Materials science in semiconductor processing, vol. 26, pp. 205–217,
Fahad J (2022) Energy band estimation of Nitride semiconductors. Experimental and Theoretical NANOTECHNOLOGY 6:443–445
Jandow NN, Khan MM (2022) Combined experimental and theoretical investigation of electronic properties of nitrides. Experimental and Theoretical NANOTECHNOLOGY 6:511–522
Rabia Hani F, Ahmad (2022) Stibniteat modified Becke-Johnson exchange potential, Experimental and Theoretical NANOTECHNOLOGY, vol. 6, pp. 367–386,
Ameri M, Khenata R (2022) Electronic and elastic properties of BN, AlN and GaN, Experimental and Theoretical NANOTECHNOLOGY, vol. 6, pp. 485–490,
Guerrero A, Gulseren O, Gross S (2022) Structural properties of group-III nitrides. Experimental and Theoretical NANOTECHNOLOGY 6:497–504
Baroni S, Ekinci H (2022) Optical and structural properties of III-nitrides compounds. Experimental and Theoretical NANOTECHNOLOGY 6:491–496
Funding
No funding was received.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
We confirm that this work is original and has not been neither published elsewhere nor currently under consideration for publication elsewhere and has ethical issues and no conflict of interest.
Competing interest
We have no financial competing interests. We just want to explore our research productivity through this work.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Nadjia, T., Beldi, L., Mohammed, A. et al. Theoretical Prediction of Structural, Magnetic and Electronic Properties of a New SiRbCa Heusler Alloy. Chemistry Africa 7, 1619–1628 (2024). https://doi.org/10.1007/s42250-023-00836-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42250-023-00836-8