
Abstract
The synthesis, structural chemistry, spectroscopic studies, optical absorption and photoluminescence, density functional theory (DFT), thermal study (TGA–DSC) and dielectric measurement of the new hybrid bis(4-amino-2-chloropyridinium) tetrachloromercurate (II) monohydrate, (C5H6N2Cl)2[HgCl4]•H2O, (abbreviated (CAP)2[HgCl4]•H2O), were described and discussed in this research paper. The result of the crystallographic study at room temperature revealed that it crystallises in the centrosymmetric triclinic system. The 13C NMR study demonstrated the presence of ten resonance signals, proving consistency with the X-ray structure, which corroborates the purity of the produced mixture. With a zero-dimensional lattice, this structure is stabilized through the presence of classical and non-classical C–H…Cl hydrogen bonds, and offset as well as π–π stacking interactions between identical antiparallel (aromatic-aromatic) CAPs. These interactions were further investigated through the use of Hirshfeld surface analysis. The theoretical results displayed a good convergence with the experimental results, using the time-dependent DFT (TD-DFT) approach. Moreover, the simulated spectrum satisfactorily coincided with the experimental UV–visible spectrum. Photoluminescence (PL) was characterized by two bands at 346 and 381 nm. The thermal properties were confirmed by TGA–DSC analysis. The studies conducted on electronic and dielectric properties unveiled that the conduction mechanism in this new salt was due to an H+ proton hopping process.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
In recent years, hybrid materials have whetted an extensive interest and drew the widest attention among scientists and researchers. In this respect, they have been the subject of several previous studies due to the outstanding combination of their inorganic and organic properties [1, 2]. This type of material exhibits promising properties such as magnetic, ferroelectric, electrical, conductivity, non-linear optics (NLO), catalysis and luminescence [3, 3, 4, 6], which implies that it can be used in a wide range of scientific fields.
Multiple hybrid materials were prepared based on the specific properties of the organic molecule, often with a prorogated amine [5,6,7]. Organic and inorganic solids were kept attached through interactions via various bonds such as hydrogen contacts, π–π functionalization and Van der Waals interactions [8, 9].
One of the most prominent properties of hybrid materials including organics based on pyridine and its derivatives, which has spilled much ink, is the photochemical property [10,11,12,13]. The inorganic part of the hybrids rely on transition metals and poor metals such as mercury, copper, tin, cadmium, lead and others. These materials display interesting physical properties [14,15,16,17]. As for the hybrid materials based on mercury halide and organic cations, they present a semiconductor behavior [18, 21], in addition to a wide structural diversity [19].
Complexes of halomercurates (II) with alkali metal ions and aliphatic or aromatic ammonium ions yielded significant results in several fields [23]. In this regard, our team is interested in examing such physical properties as optics, conductivity and luminescence of the new hybrid materials, which have been the subject of several articles [14,15,16,17,18,19,20,21, 23].
In order to explore new properties of organic–inorganic materials, this study is devoted to exploring the properties of this new compound bis(4-amino-2-chloropyridinium) tetrachloromercurate (II) monohydrate, (C5H6N2Cl)2[HgCl4]•H2O, (abbreviated (CAP)2[HgCl4]•H2O), using various significant techniques such as X-ray diffraction, FT-IR, Raman, solid-state NMR studies and UV–Visible spectroscopy via DFT calculations (B3LYP/LanL2DZ basis set). To quantify the intermolecular interactions involved in stabilising the crystal structure, analyses of the three-dimensional Hirshfeld surface and the molecular electrostatic potential (MEP) surface were carried out. Eventually, photoluminescence (PL), thermal analysis (DSC-TGA) and dielectric studies were carried out to grain a deeper insight into the conduction phenomenon in this new material.
2 Experimental
2.1 Preparation of (CAP)2[HgCl4]•H2O Compound
To obtain our new hybrid compound, (C5H6N2Cl)2[HgCl4]•H2O, we used the following starting materials, from an equimolar specified proportions (2:1): 4-amino-2-chloropyridine (CAP) (0.398 g; 3.09 mmol); mercury chloride (HgCl2) (0.199 g; 0.733 mmol) and hydrochloric acid (HCl) (38%; 5 mL). Using the slow evaporation method, this compound was prepared at room temperature. The above products were dissolved separately in hydrochloric acid, then mixed together in an approximate volume of 10 ml of water with magnetic stirring until complete dissolution of the solutes. Colourless crystals appeared with a prismatic-shaped salt having parallelogram bases (Fig. 1a, b, c). These crystals were washed with ether, carefully filtered and left for a day to dry. Subsequently, they were collected and used in various physical and chemical analyzes.

a Typical EDX spectrum, b photograph (magnification *10) c and SEM image of the crystals of the compound(CAP)2[HgCl4]•H2O having the same shape as prismatic
2.2 Scanning Electron Microscopic Study of the Sample
Quantitative analysis under a scanning electron microscope performed on this material reveals the presence of all atoms other than hydrogen atoms: Hg, C, N, O and Cl without setting their percentage. The result of this analysis is indicated on the following spectrum Fig. 1a. The morphology of bis(4-amino-2-chloropyridinium) tetrachloromercurate (II) monohydrate crystals obtained with the scanning electron microscopy (SEM) is shown in Fig. 1b, c show an assemblage of crystal fragments having uniform distribution, a flat surface, regular edges that indicate good crystalline quality are clearly visible in the images.
2.3 Characterization
2.3.1 Crystal Data and Structure Determination
A Bruker Kappa Apex II CCD diffractometer equipped with a monochromatic MoKα graphite beam (0.71073 Å) was used for the crystalline study of a well-chosen single crystal of dimensions (0.39 × 0.32 × 0.26) mm3. Intensity data were used at room temperature. The final unit cell parameters and crystal orientation matrix were obtained by a least-square fit of 4021 reflections and the structure was solved and refined using the SHELXS program which is included in the WINGX software [21]. Firstly, direct methods located the heavy atoms Hg and Cl. N, C and O atoms were next located using successive Fourier difference maps. Hydrogen atoms were fixed geometrically. The final refinement led to the following reliability factors: R = 0.048 and Rw = 0.216. Table 1 illustrates the experimental details of the structure determination.
The graphical illustrations were generated using the following programs: ORTEP [22], DIAMAND 2.1 [23] and MERCURY3.1 [24].
2.3.2 Spectroscopy Equipment
FT-IR spectrum of the sample was recorded within the range 400–4000 cm−1 using a Perkin Elmer L120-00 spectrometer. The Raman spectrum of sample was recorded in the range 50–4000 cm−1 HORIBA JOBIN–YVON (T64000) spectrometer triple monochromator instrument using the 514.5 nm line spectra-physics argon ion laser.
13C NMR experiment was conducted on a Bruker MSL 300 spectrometer operating at 75.48 MHz for 13C. The powdered sample was packed in a 4 mm diameter rotor and allowed to rotate at speeds up to 10 kHz in a Doty MAS probehead. The spectra were acquired by the use of cross-polarization for proton with 5 ms contact time.
Optical absorption spectrum by spin-coating of the films (compound in powder form (result of grinding the crystals)) was deduced from direct transmission measurements carried out using a conventional UV–Vis spectrophotometer (Hitachi, U3300) that has a dual-beam monochromator, covering a spectrum from 200 to 2400 nm and using two sources: (i) Xenon lamp for the UV–Visible domain, (ii) Halogen lamp for the infrared range.
2.3.3 Hirshfeld Surface Analysis
The 3–D Hirshfeld surfaces [29] in the crystal structure were constructed resting on the electron distribution calculated as the sum of spherical atom electron densities. For each point on the 3–D Hirshfeld surface, we determined both de (which is the distance from the 3–D Hirshfeld surface to the nearest nucleus outside the surface) and di (which is the corresponding distance to the nearest nucleus inside the surface). For further information about the intermolecular interactions in the crystal structure, they were estimated utilizing the 3–D Hirshfeld surface analysis [25, 26] whereas the associated 2–D fingerprint plots were specified via the Crystal Explorer 3.1 [27] which is based on the structure of the CIF file. The mapped Hirshfeld surfaces were made transparent to allow the visualization of the molecular moiety, in a similar orientation for the crystal. In particular, the Hirshfeld surface mapped on a dnorm clearly highlighting, with the spots, in addition to the circular depressions (dark red) the donor (de > di) and the acceptor (de < di), which are indicative of intermolecular interactions of hydrogen bonding contacts. We can therefore assert that, the π–π stacking interactions are verified by the existing evidence of the adjacent red and blue triangles on the shape index surface.
The normalized contact distance (dnorm) resting on both de and di, and the van der Waals (VdW) radii of the atom, indicated by Eq. (1) allowed the identification of the regions of particular importance to intermolecular interactions [28].
With rivdw and revdw are the van der Waals radii of the atoms.
The value of dnorm was highlighted using a red–white–blue color scheme corresponding to the negative value (red regions), zero (white regions) and positive (blue regions) for shorter contacts and for contacts around the VDW separation and for longer contacts, respectively.
The 2–D fingerprint plots appeared as expanded two-dimensional with the (de) and (di) distance scales installed on the graph axes. They can be decomposed to highlight particular atom pairs in close contacts [34].
The void surface was located where the sum of the promolecule density in the unit cell is less than a user definable threshold, where the void surface met the boundary of the unit cell, and capping faces were generated to create an enclosed volume. In addition to visualizing voids, Crystal Explorer equally reported the volume of the void and its surface area.
2.3.4 Computational Methods
The fully optimized ground state geometry and harmonic vibrational wavenumbers were computed at the DFT level using the Lee–Yang–Parr correlation fuctional (B3LYP) in conjunction with the LanL2DZ basis set [29, 36] by means of Gaussien 09 software package [30]. On this basis, all the parameters were allowed to relax and all the calculations converged to an optimized geometry, which corresponds to a minimum of energy as revealed by the lack of imaginary values in the calculated frequency. An empirical scaling factor of 0.961 was used to offset the systematic error [38] by basis set incompleteness, neglect of electron correlation and vibrational anharmonicity. The calculated vibrational wave numbers were assigned deploying the GaussView 05 program [31] in order to generate visual presentations and verify the normal mode assignments. The molecular electrostatic potentials (MEPs) were calculated to investigate the reactive sites of synthesized crystal. To account for the chemical selectivity or reactivity sites, the electron density-based local reactivity descriptors such as Fukui functions were computed [32]. The electronic properties such as HOMO and LUMO energies and absorption wavelengths were determined by time-dependent DFT (TD-DFT) approach [33] at the same level of theory. It is necessary to clarify that the sample was also optimized in ethanol solvent.
2.3.5 Thermal Analysis
The thermal behavior was examined under an air flow in Mettler Toledo DSC 822e calorimeter and Perkin Elmer Pyris 6 TGA equipment, at a heating rate of 5 K.min−1.
2.3.6 Electrical Measurements
Electrical measurements of the (CAP)2[HgCl4]·H2O sample, which was pressed into pellets 8 mm in diameter and 1.3 mm thick, were carried out with the TEGAM 3550 ALF automatic bridge controlled by a microcomputer between 318 and 408 K in a frequency range from 209 Hz to 46 MHz.
3 Results and Discussion
3.1 X-ray Diffraction Analysis
The sample (CAP)2[HgCl4]·H2O was characterized by single-crystal X-ray diffraction at room temperature. It revealed that this new hybrid belongs to the triclinic system with the space group Pī, whose elementary point displays two unitary forms.
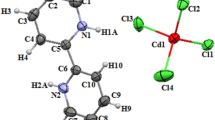
The asymmetric unit contains two monoprotonated cationic entities (4-amino-2-chloropyridinium), an anionic entity [HgCl4]2− in the form of an isolated tetrad and a water molecule H2O, drawn with a 50% probability of thermal ellipsoids, as depicted in Fig. 2a. This unit contains both an anionic and a cationic charge, ensuring the electrical neutrality of the crystal structure. At this point, the best possible geometry was undertaken using DFT/B3LYP/LanL2DZ basis set, which accepts a structure input file in its crystallographic data. It is indicated in Fig. 2b in accordance with the experimental structure as well as the relaxation of all parameters; that the calculations converge to optimize geometries, which correspond to true energy minima.

Atom numbering scheme for the structure a experimental results and b theoretical optimized geometry
The projection of this structure along the a-axis (Fig. 3) revealed that the organic cations are positioned between the infinite chains of the inorganic part and the water molecules, which have a different orientation. Projection along the b axis demonstrated that the organic molecules are parallel along the a-axis and equidistant. This assembly was then built by a succession of cationic dimers and isolated tetrahedra in addition to water molecules.

Projections of the (CAP)2[HgCl4]•H2O compound: a along the a-axis and b along the b axis
However, in this structure, the organic part consists of two cations presenting a regular spatial configuration with experimental distances (Table 2) the C–C and C–N bond lengths ranging [1.324(14); 1.429(14)] Å and [1.308(14); 1.357(15)] Å, respectively. The main values of the C–Cl bond lengths vary from 1.697(9) to 1.712(11) Å. Additionally, the experimental values of C–C–C and N–C–C and N–C–Cl and C–C–Cl angles vary from 116.3(9)° to 120.8(10)°and 121.4(9)° to 123.3(8)° and 115.3(8)° to 116.3(7)° and 122.4(8)° to 123.2(8)°, respectively. It is worth noting that C–N–C angle is of the order 123.3(8)°. These values go in good agreement with those reported in previous literature [7, 10, 34, 35].
The mercuric ion Hg (+ II) adopts the geometry of a regular tetrahedral (Table 2). The Hg–Cl bond distances range from 2.447(3) to 2.476(3) Å whereas the Cl–Hg–Cl angles fall in the range 104.43(8)° to 115.95(10)° [9, 36].
Interestingly enough, a study of the calculated geometric characteristics revealed a good convergence with the experimental parameters. According to Table 2, the majority of the calculated link values are slightly longer than those of the experimental links, which can be explained by the fact that the theoretical calculation considers that the molecule is isolated in the gaseous phase at 0 K while the experimental result examines the molecule in the solid state, presenting therefore intermolecular interactions. Thus, the accuracy of the calculation is satisfactory and the B3LYP/LanL2DZ level is suitable for the compound investigated in this manuscript.
Figure 4 and Table 3 foreground the interactions of hydrogen bonding values present in this structure. The cohesion of the structure is ensured by hydrogen bonds: on the one hand between the organic part and the inorganic part and on the other hand with the water molecule and both organic–inorganic entities. These bonds are interatomic and are of the type O–H…Cl, C–H…Cl and N–H…Cl (chlorine of the anion).

Packing diagram showing the capped sticks of the (CAP)2[HgCl4]•H2O compound with dotted red lines represent hydrogen bonds
In addition, the surface mapped on the shape index (Fig. 5a) discloses the rationalized information from hydrogen bond interactions, such as the deep red area which highlights the hydrogen bond acceptor (concave) and the blue area which corresponds to the hydrogen bond donor (convex); thus indicating the stabilization of the crystal studied here [37]. Again, the presence of red and blue red triangles (Fig. 4b) adjacent with summit in the shape index goes in tandem with the presence of π– π interactions interacting between aromatic rings [8]. within this framework, these intermolecular interactions involved within the structure go in good agreement with the 2-D fingerprint plots (Fig. 6). The fingerprint plots for the new hybrid indicated the H…Cl/Cl…H interactions, with the most important one being 55.9% of the total Hirshfeld surface region. These values proved to be de = 1.6 Å, di = 1.00 Å / de = 1.00 Å, di = 1.6 Å, this yielded the de > di (acting as a donor) and the other de < di (acting as an acceptor). As a matter of fact, Fig. 7 emphasizes the domination of these interactions; allowing to conclude that hydrogen bonds play a very crucial role in the stability of the building. The rates of intermolecular interactions H…H and C…H/H…C amount to 14.1% and 9.1%, respectively of the Hirshfeld surface area. Thus; the Hirshfeld surface area revealed Cl…Cl and C…C interactions in the order of 4.8% and 3.2%. C…C interactions suggested that the value for the de = di = 1.8 Å is indicative of the presence of π–π interaction between organic molecules, which is portrayed in Fig. 8. Basically, the results found by the analyses of intermolecular interactions using the Hirshfeld surface go in good conformity with the previous structural description. The proportion of H…N/N…H interactions comprises 3.1%, whereas 2.2% of contribution comes from the Cl…C/C…Cl intermolecular interactions. We also outline the following interactions with their percentages: C…N/N…C, Cl…O/O…Cl, Cl…N /N…Cl, H…O/O…H, H…Hg/Hg…H and Cl…Hg/Hg…Cl and N…N, 1.6%, 1.5%, 1.3%, 1.1%, 1%, 0.8% and 0.1%, respectively. We equally record the following interactions with their percentages: C…N/N…C, Cl…O/O…Cl, Cl…N /N…Cl, H...O/ O…H, H…Hg/Hg…H and Cl…Hg/Hg…Cl and N…N, 1.6%, 1.5%, 1.3%, 1.1%, 1%, 0.8% and 0.1%, respectively.

The dnorm a and shape index b Hirshfeld surfaces and the full fingerprint plot of (CAP)2[HgCl4]•H2O compound

2–D Fingerprint plots analysis of (CAP)2[HgCl4]•H2O compound

The Hirshfeld surface mapped dnorm and the fingerprint plot of the H…Cl/Cl…H interactions of (CAP)2[HgCl4]•H2O compound

The π–π interactions between the chains of aromatic ring and the Hirshfeld surface mapped shape index the π–π interactions of (CAP)2[HgCl4]•H2O compound
The contributions of the different interactions to the Hirshfeld surfaces were calculated, suggesting the dominance of H…Cl/Cl…H interactions on the total surface of Hirshfeld (Fig. 9).

Relative contribution of various intermolecular interactions to the Hirshfeld surface area of (CAP)2[HgCl4]•H2O compound
The Crystal Explorer allowed the calculation of the porosity of this material, whose visualization of the voids with the isosurfaces of electron density were exposed to a level of 0.002 a.u. As can be inferred from Fig. 10, the resulting volume of the void and its surface area surface, were determined as: Volume (93.91 Å3) (20.24%) and area (358.13 Å2) (78.15%) per unit cell. This result may be assigned to the response of molecular crystals to external pressure.

Crystal void of (CAP)2[HgCl4]•H2O compound
3.2 MAS–NMR
The experimental study using the 13C NMR technique (Fig. 11) of the studied compound revealed the presence of five intense peaks (blue spectra) between 90 and 170 ppm. Owing to the Dmfit software [38], the deconvolution of the experimental spectrum entailed the appearance of ten peaks, which coincided with the total number of carbon atoms in unequal positions in this structure present in the asymmetric unit of this hybrid crystal. This result can confirm the structural results and purity of the crystalline phase of the synthetized product. Table 4 highlights an attempt of displacement of the pyridinic ring attribution. The latter was enacted in the same way as the molecule presented in previous works [5,6,7, 34, 35].

Plots of experimental and fitted curves of the isotropic band of the 13C spectrum of (CAP)2[HgCl4]•H2O compound
3.3 Vibrational Studies
The study of the vibration modes of the structural architecture of this compound, using infrared absorption and Raman diffusion, confirmed the presence of the envisaged chemical elements. This study was supported by the theoretical calculation of vibratory spectra using functional density theory with the B3LYP/LanL2DZ assembly base. The theoretical and experimental studies of each technique are plotted in Figs. 12 and 13. This part was followed by an attempt to assign the majority of the peaks to their corresponding vibratory mode while relying on previous works [5,6,7, 17, 34, 35]. These attributions are listed in Table 5 and discussed further in what follows: the stretching vibrations O–H, N–H and C–H were detected by the presence of peaks in the high frequency [3800–3000] cm−1. The IR spectrum indicated the presence of several bands in the 2800 cm−1–1700 cm−1 region, which are characteristic of the combined harmonics and vibrations. The latter are not present in the Raman spectrum from which they are excluded in the assignments. The NH2 deformation and C = C stretching vibrations were detected by the presence of peaks at 1655 cm−1 and 1631 cm−1, respectively. The region located between 800 and 400 cm−1 was assigned to NH scissoring and CH bending vibrations. The IR and Raman spectrum displayed strong peaks at 786 cm−1 and 717 cm−1, which were attributed probably to the carbonyl scissoring mode, and out of plane bending mode, respectively.

Superposition of the experimental (black) and the DFT computed (red) FT-IR spectra of (CAP)2[HgCl4]•H2O compound

Superposition of the experimental (red) and the DFT computed (black) FT-Raman spectra of (CAP)2[HgCl4]•H2O compound
Using Raman spectrum (Fig. 13), the Tetrahedron [HgCl4]2− vibration modes proved to be in the range of (40–400) cm−1. The attribution was difficult but relied on references [19, 36]. The bands observed at 258 cm−1 and 218 cm−1 can be attributed to asymmetric and symmetric stretchings, respectively. Asymmetric and symmetrical strain vibration peaks of the Hg–Cl bond in the [HgCl4]2− anion appeared at 186 cm−1 and 108 cm−1, respectively.
3.4 Optical Properties
3.4.1 UV–Vis Absorption
The experimental (black spectrum) and theoretical (red spectrum) UV–visible spectra at room temperature are displayed in Fig. 14. These spectra indicated a satisfactory agreement between the experimental and calculated results. It can be inferred that the (CAP)2 HgCl4·H2O exhibited a single intense band around 266 nm (4.66 eV) and 273 nm (4.53 eV) respectively, which refers to the optical energy band gap value equal to 4.66 eV, as illustrated in Fig. 15. The difference among the measured and calculated transition energies is 0.13 eV. This may account for the characteristic feature of mercury (II) complexes with a 5 d10 electronic configuration. According to literature studies [19, 39], this band was allocated to n → π* transitions, appearing in the corresponding free ligand, as a consequence of the presence of the aromatic ring. In addition, the energy gap value allowed the assumption that this hybrid material can be considered as an optically potential material with spectroscopic applications.

Experimental and theoretical optical absorption spectra at room temperature of (CAP)2[HgCl4]•H2O compound

Molecular orbital surfaces for the HOMO and LUMO of (CAP)2[HgCl4]•H2O compound
3.4.2 Molecular Electrostatic Potential Analysis
In order to explore the relative polarity of the molecule, we examined the molecular electrostatic potential (MEP), which is a visual method, unveiling an interesting property that can be derived from the density distribution of electrons. The mapped MEP surface was used for the qualitative interpretation of the electrophilic and nucleophilic sites [51]. The chemical polarity qualifier and the assessment of the charge distribution in the crystal the MEP map, were calculated by B3LYP, as presented in Fig. 16. The MEP figure portraying the visual representation of chemically active sites, presents different colors such as: electrophilic reactivity which is in red (negative regions) as well as nucleophilic reactivity which is in blue (positive regions). The potential increases through moving from red–orange–yellow–green–blue color, where the color code of these cards varies between − 0.053 a.u. and 0.053 a.u. deepest red and deepest blue, respectively.

Molecular electrostatic potential map of (CAP)2[HgCl4]•H2O compound
In this respect, the negative regions are mainly localized on the chlorine atoms, whereas the positive regions are mainly localized on the amino atoms N. This region may constitute a suitable site for the nucleophilic attack. These results provide useful information on the region from which the compound can have an intra- and an intermolecular interaction. This result converges towards the interpretation that this crystal presents a global electrostatic attraction between the anion [HgCl4]2− and the organic cation that is added to the favorable H…Cl hydrogen bond.
3.4.3 Mulliken Atomic Charges
It is well known that the Mulliken population indicates charge distributions on atoms and quantifies how the electronic structure changes under atomic displacement [44].
In addition, the Mulliken charge is directly related to the chemical bonds present in the studied crystalline structure. Figure 17 depicts the charge distribution of the different atoms of this complex (CAP)2[HgCl4]•H2O with all the carbon atoms displaying negative charges, except C atoms between both nitrogen ones. The latter are positively charged due to the negative charges of the nitrogen atoms. The metal Hg has a positive charge due to the fixation of halogens Cl which are negatively charged. Finally, all hydrogen atoms carry positive charges while both chlorine atoms carry negative charges. It is well confirmed that the load transfer by the hydrogen bonds (N–H…Cl) of the hybrid material has been analysed.

Atomic charge distribution of (CAP)2[HgCl4]•H2O compound
3.4.4 Photoluminescence Behavior
We measured solid-state photoluminescence to determine its optical properties, which corresponds to the result of the absorption of photons by the material. We computed the photoluminescence (PL) of our compound at room temperature, with an excitation wavelength of 285 nm, as presented in Fig. 18. A wide fluorescence band from 346 to 381 nm emissions by our hybrid lies approximately in the luminescence range observed for other mercury-containing materials [HgCl4], corresponding described in literature [40, 41]. The corresponding CIE coordinates (0.149; 0.201); are located in the blue region of the CIE chromaticity diagram [42].

Emission spectrum of (CAP)2[HgCl4]•H2O compound
In hybrid compounds based on haberdashery chloride, the luminescence of the excitons comes from the electronic transitions of the inorganic part. Excitation by a laser follows an electron (-) which will be excited from the valence band (VB). The latter is made up of a combination of Hg (6d) and Cl (4p), which is in the direction of the conduction band (CB) for the Hg(5 s) states, leaving a ( +) gap in (VB). The (-) electron and ( +) hole move freely in (CB) and (VB) forming an exciton [6]. Recombination of the electron with the hole in the exciton results in blue emissions. To illustrate the process of formation and recombination of excitons in our compound, we built up a simple model, as revealed in Fig. 19.

Simple model for the formation and recombination of the exciton in the (CAP)2[HgCl4]•H2O compound
3.5 The Thermal Study
3.5.1 ATG and DSC Studies
The thermal behavior of this compound is the central focus of this part. Notably, the combination of gravimetric and calorimetric behavior pave the way for the following discussion.
The TGA curve (Fig. 20a) displays a weight loss of 3.06% between 360 and 423 K, which coincides with the unit formula of the studied compound. The calorimetric study (Fig. 20b) presents a small bump at 340 K, which can probably be ascribed to the loss of surface water molecule. The same curve has an intense peak at 370 K, indicating the loss of the structural water molecule that is detected in the thermogravimetric study. This endothermic peak at an enthalpy ΔH = 70.61 J/g implies that the compound exhibits thermal stability at higher temperatures.

a ATG spectrum and (b) DSC spectrum of (CAP)2[HgCl4]•H2O compound
The calorimetric study is followed by a cooling cycle on the same sample. This demonstrates the total absence of an endothermic peak (Fig. 20b), after which, it undergoes another warming sweep cycle that shows the total absence of an endothermic peak. Thus, this confirms that the observed peaks are due to the loss of surface and structural water molecules.
3.5.2 Electrical Study
Electrical analysis is used to provide useful information about the behaviour of localised electrical charge carriers, leading to a better understanding of conduction processes.
The electrical properties study of a material is denoted by the expression of the complex impedance parameter term Z*with Z’and Z" represent respectively the real and imaginary parts of the impedance the complex impedance.
Rs: series resistance. ω = 2π f (f = resonance frequency). Cs: capacitance in series. j = (− 1)1/2 the imaginary factor. C0: vacuum capacitance of the circuit elements.
Figure 21 presents the Nyquists diagram (plot of Z’ versus Z”) at different temperatures (303–398 K). These spectra reveal that the increase in temperature leads to a decrease in Z". This decrease is well marked at temperatures above T = 363 K.

The impedance plots at T = [303–398] K of (CAP)2[HgCl4]•H2O compound
These spectra demonstrate on the one hand, in the field of scanned frequency and in the lower temperature in 358 K, that the new organic–inorganic compound has a capacitive behavior from which it is impossible to determine the resistance. On the other hand, these spectra follow the cole–cole law for any temperature greater than 366 K. This corroborates that the electrode polarization phenomena have an outstanding contribution [44].
The Arrhenius modelling equation was utilised to understand the conduction mechanism.
Ea is the activation energy, σ0 is the pre-exponential factor, Kb is the Boltzmann constant, and T is the absolute temperature.
The variation of activation energy as a function of 1000/T is depicted in Fig. 22. This variation is separated into three regions as follows, Region (I): ∆Ea1 = 0.004 eV and Region (II): ∆Ea2 = 0.27 eV and Region (III): ∆Ea3 = 0.8 eV. These values are suggestive that the H+ ion transport in the material is due probably to a jump mechanism [45].

Arrhenius plots Ln(σT) = f(1000/T) of (CAP)2[HgCl4]•H2O compound
The thermal study undertaken TGA and DSC depicts a single endothermic peak assigned to the evaporation of water molecules, in the temperature range 298–420 K. From this perspective, it helps us draw the conclusion that the variation in activation energy with temperature cannot be due to a phase transition. The observed change is rather due to a change in the conduction mechanism. Departing from Fig. 22, the activation energy of the region (III) involved in this jump is greater than that calculated in the regions (II) and region (I). This refers to the fact the observed time in this frequency range is too short for all jumps (proton) to be successful, the neighborhood cannot be therefore comfortable with the new position of the proton after it jumps.
4 Conclusions
In the current research work, single crystals of (CAP)2[HgCl4]·H2O were synthetised from an aqueous solution by a slow evaporation at room temperature. The edifice was stabilized by not only hydrogen bonds (N–H…Cl, O–H…Cl and C–H…Cl) but also by the presence of cation–cation π– π interactions. The number of solid state 13C, MAS-NMR components goes in good agreement with that of the crystallographically independent site. The cohesion within this material was also examined by the Hirshfeld surface calculation method. The DFT calculations, optimized geometry and vibration spectrum coincided well with experimental data, which were interpreted by analogy with pervious works. The decrease in the HOMO and LUMO energy gap accounts for intraligand n→π* transitions.
The thermogravimetric and calorimetric measurements with differential scanning revealed the absence of phase transition. The obtained peaks are due to the evaporation of the molecules. The studies of luminescence activities confirmed that this compound exhibited a powerful fluorescence property at room temperature. Thus, the new crystal displays an outstanding electrical behavior, where the activation energy emitted by the impedance spectra can be assigned by the H+ proton jump mechanism.
Data availability
The data set generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Šimon T, Czakó A (2014) Influence of long-term application of organic and inorganic fertilizers on soil properties. J Plant Soil Environ 60(7):314–319. https://doi.org/10.17221/264/2014-PSE
Maughan AE, Kurzman JA, Neilson JR (2015) Hybrid inorganic–organic materials with an optoelectronically active aromatic cation:(C7H7) 2SnI6 and C7H7PbI3. J Inorganic Chem (1): 370–378. https://doi.org/10.1021/ic5025795.
Mahmood Q, Hassan M, Ahmad SHA, Bhamu KC, Mahmood A, Ramay SM (2019) Study of electronic, magnetic and thermoelectric properties of AV2O4 (A= Zn, Cd, Hg) by using DFT approach. J Phys Chem Solids 128:283–290. https://doi.org/10.1016/j.jpcs.2017.08.007
Thomas T, Hartmann B, Lunkenheimer P, Schubert H, Schlueter JA, Müller J (2019) Low-Frequency Charge carrier dynamics in ferroelectric κ-(BEDT-TTF) 2X–A Comparative Study of X= Cu [N (CN)2] Cl and X= Hg (SCN) 2Cl. J Phys Status Solidi (b). 256(9):1800746. https://doi.org/10.1002/pssb.201800746
Aziz AAA, Elantabli FM, Moustafa H, El-Medani SM (2017) Spectroscopic, DNA binding ability, biological activity, DFT calculations and non linear optical properties (NLO) of novel Co (II), Cu (II), Zn (II), Cd (II) and Hg (II) complexes with ONS Schiff base. J Mol Struct 1141:563–576. https://doi.org/10.1016/j.molstruc.2017.03.081
Sakka A, Mhadhbi N, Hamdi B, Ahmed AB, Tozri A, Nasr S, Naïli H (2023) Structure/function relationships of a new stannate (IV) complex based on 5, 7-dichloro-8-hydroxyquinolinium, accomplished with DFT calculations. J Mol Struct 1277:134811. https://doi.org/10.1016/j.molstruc.2022.134811
Jellali A, Elleuch S, Hamdi B, Zouari R (2019) Experimental and theoretical investigations of the molecular structure, the spectroscopic properties and TD-DFT analysis of a new semiconductor hybrid based iron (III). J Saudi Chem Soc 23(5):600–611. https://doi.org/10.1016/j.jscs.2018.10.006
Hamdi B, Zouari R, Ben Salah A (2018) Preparation, molecular structure, thermal properties, electrical conductivity analysis and dielectric relaxation of a new hybrid compound (NH 2C5H3 ClNH)2 ZnBr 4· H 2 O. J Chem Papers. 72:2795–2811. https://doi.org/10.1007/s11696-018-0521-8
Jellali A, Hamdi B, Salah N, Zouari R (2018) Experimental, theoretical, characterization and TD-DFT analysis of a complex [Cu (CAP) 2 Cl2]. J Inorg Organomet Polym Mater 28:1636–1647. https://doi.org/10.1007/s10904-018-0822-y
Egger DA, Kronik L (2014) Role of dispersive interactions in determining structural properties of organic–inorganic halide perovskites: insights from first-principles calculations. J Phys Chem Lett 5(15):2728–2733. https://doi.org/10.1021/jz5012934
Soroceanu M, Barzic AI, Stoica I, Sacarescu L, Ioanid EG, Harabagiu V (2017) Plasma effect on polyhydrosilane/metal interfacial adhesion/cohesion interactions. Int J Adhes Adhes 74:131–136. https://doi.org/10.1016/j.ijadhadh.2017.01.00
Utschig LM, Silver SC, Mulfort KL, Tiede DM (2011) Nature-driven photochemistry for catalytic solar hydrogen production: a photosystem I–transition metal catalyst hybrid. J Am Chem Soc 133(41):16334–16337. https://doi.org/10.1021/ja206012r
Shukla D, Adiga SP, Ahearn WG, Dinnocenzo JP, Farid S (2013) Chain-amplified photochemical fragmentation of N-alkoxypyridinium salts: proposed reaction of alkoxyl radicals with pyridine bases to give pyridinyl radicals. J Org Chem 78(5):1955–1964. https://doi.org/10.1021/jo301975j
Karâa N, Ben Ahmed A, Hamdi B (2023) New semiconductor halocadmate [CdnXm](2n–m) crystal structure, molecular conformation and theoretical investigations. J Solid State Chem 322:123954. https://doi.org/10.1016/j.jssc.2023.123954
Yan B, Wang QM (2007) Molecular fabrication and photoluminescence of novel terbium co-polymer using 4-vinyl pyridine as the efficient second ligand. J Opt Mater 30(4):617–621. https://doi.org/10.1016/j.optmat.2007.01.015
Smith MD, Watson BL, Dauskardt RH, Karunadasa HI (2017) Broadband emission with a massive stokes shift from sulfonium Pb–Br hybrids. J Chem Mater 29(17):7083–7087. https://doi.org/10.1021/acs.chemmater.7b02594
Vidyasagar CC, Muñoz Flores BM, Jiménez Pérez VM (2018) Recent advances in synthesis and properties of hybrid halide perovskites for photovoltaics. J Nano-Micro Lett 10:1–34. https://doi.org/10.1007/s40820-018-0221-5
Aruta C, Licci F, Zappettini A, Bolzoni F, Rastelli F, Ferro P, Besagni TJAP (2005) A. Journal of Growth and optical, magnetic and transport properties of (C 4 H 9 NH 3) 2 MCl 4 organic-inorganic hybrid films (M= Cu, Sn). J Appl Phys A. 81:963–968. https://doi.org/10.1007/s00339-004-3102-3
Karaa N, Hamdi B, Ben Salah A, Zouari R (2012) Synthesis, infra-red, MAS-NMR characterization, structural study and electrical properties of the new compound [C5H6ClN2] 2Cd3Cl8. J Mol Struct 1013:168–176. https://doi.org/10.1016/j.molstruc.2011.12.053
Saleh TA (2015) Isotherm, kinetic, and thermodynamic studies on Hg (II) adsorption from aqueous solution by silica-multiwall carbon nanotubes. J Environ Sci Pollut Res 22:16721–16731. https://doi.org/10.1007/s11356-015-4866-z
Temoçin Z, Yiğitoğlu M (2010) Studies on selective uptake behavior of Hg (II) and Pb (II) by functionalized poly (ethylene terephthalate) fiber with 4-vinyl pyridine/2-hydroxyethylmethacrylate. J Water Air Soil Pollut 210:463–472. https://doi.org/10.1007/s11270-009-0271-x
Argent SP, Adams H, Riis-Johannessen T, Jeffery JC, Harding LP, Clegg W, Ward MD (2006) Complexes of Ag (I), Hg (I) and Hg (II) with multidentate pyrazolyl-pyridine ligands: from mononuclear complexes to coordination polymers via helicates, a mesocate, a cage and a catenate. J Dalton Trans 42:4996–5013. https://doi.org/10.1039/B607541J
McAuliffe, C. A. (Ed.). (2016). The chemistry of mercury. Springer. ISBN 1349024899, 9781349024896
Elwej R, Nasri S, Hlel F (2016) Impedance spectroscopic investigation on phase transition and electrical conduction mechanism of the new inorganic-organic complex: (C6H9N2)2HgCl4 (I), (C6H9N2)2(Hg0.75Cd0.25)Cl4 (II) and (C6H9N2)2(Hg0.12Zn0.88)Cl4 (III). J Alloys Compounds 684:389–396. https://doi.org/10.1016/j.jallcom.2016.05.172
Farrugia LJ (1999) WinGX suite for small-molecule single-crystal crystallography. J Appl Crystallogr 32:837–838. https://doi.org/10.1107/S0021889899006020
Farrugia LJ (1997) It ORTEP-3 for windows—a version of it ORTEP-III with a graphical user interface (GUI). J Appl Crystallogr 30:565–565. https://doi.org/10.1107/S0021889897003117
K. Brandenburg (1998) Diamond Version 2.0. Impact Gbr, Bonn.
Allen FH, Davies JE, Galloy JJ, Johnson O, Kennard O, Macrae CF, Mitchell EM, Mitchell GF, Smith JM, Watson DG (1991) The development of versions 3 and 4 of the Cambridge Structural Database System. J Chem Inform Comput Sci 31:187–204. https://doi.org/10.1021/ci00002a004
McKinnon, J.J, Mitchell. A. S, Spackman. M. A. Hirshfeld Surfaces: A New Tool for Visualising and Exploring Molecular Crystals. Journal of Chemistry—A European. 4 (1998) 2136. https://doi.org/10.1002/(SICI)1521-3765(19981102)4:11%3C2136::AID-CHEM2136%3E3.0.CO;2-G
Seth SK (2014) Discrete cubic water cluster: an unusual building block of 3D supramolecular network. J Inorganic Chem Commun 43:60–63. https://doi.org/10.1016/j.inoche.2014.02.014
Seth SK (2014) Structural elucidation and contributio n of intermolecular interactions in O-hydroxy acyl aromatics: Insights from X-ray and Hirshfeld surface analysis. J Mol Struct 1064:70–75. https://doi.org/10.1016/j.molstruc.2014.01.068
McKinnon JJ, Jayatilaka D, Spackman MA (2007) Towards Quantitative Analysis of Intermolecular Interactions with Hirshfeld Surfaces. J Chem Commun 37:3814–3816. https://doi.org/10.1039/B704980C
Spackman MAS, McKinnon JJ (2002) Fingerprinting intermolecular interactions in molecular crystals. J Cryst Eng Comm 4:378–392. https://doi.org/10.1039/b203191b
Spackman MA, Byrom PG (1997) A novel definition of a molecule in a crystal. J Phys Chem Lett 267:215–220. https://doi.org/10.1016/S0009-2614(97)00100-0
Lee C, Yang WT, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. J Phys Rev B. 37:785–788. https://doi.org/10.1103/PhysRevB.37.785
Leopold N, Szabó L, Pîrnău A, Aluaş M, Leopold LF, Chiş V, Cozar O (2009) Raman spectroscopic and DFT theoretical study of 4-(2-pyridylazo) resorcinol and its complexes with zinc(II) and copper(II). J Mol Struct 919:94–99. https://doi.org/10.1016/j.molstruc.2008.08.022
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) GAUSSIAN 09, Revision A.1, GAUSSIAN, Inc, Wallingford CT
National Institute of Standards and Technology (NIST), Computational chemistry comparison and benchmark database: precomputed vibrational scaling factors. http://cccbdb.nist.gov/vibscalejust.asp.
Dennington R, Keith T, Millam J (2009) GaussView, Version 5, Semichem Inc, Shawnee; Mission, KS
Fleming I (1976) Frontier orbitals and organic chemical reactions. John Wiley and Sons, New York
Runge E, Gross EKU (1984) Journal. Density-functional theory for time-dependent systems. Phys Rev Lett. 52 (1984) 997–1000. https://doi.org/10.1103/PhysRevLett.52.997.
Karâa N, Hamdi B, Oueslati A, Salah AB, Zouari R (2010) Preparation, infra-red, MAS-NMR and structural characterization of a new copper based inorganic organic hybrid compound: [C5H6N2Cl]2CuCl4. J Inorg Organomet Polym Mater 20:746–754. https://doi.org/10.1007/s10904-010-9409y
Karâa N, Hamdi B, Ben Salah A, Zouari R (2013) Synthesis, Infra-red, CP/MAS-NMR characterization, structural study and electrical properties of the bis(4-amino-2-chloropyridinium)tetrachlorozincate(II) monohydrate. J Mol Struct 1049:48–58. https://doi.org/10.1016/j.molstruc.2013.06.003
Ben Debabis R, Amamou W, Chniba-Boudjada N, Zouari F (2019) Synthesis, crystal structure, and vibrational and magnetic properties of Co (II) and Hg(II) complexes with an 8-hydroxyquinoline unit. J Phys Chem Solids 124:296–304. https://doi.org/10.1016/j.jpcs.2018.09.038
Rigane I, Walha S, Ben Salah A (2016) Hydrogen bonding in thiobenzamide synthon and its Cadmium complex: Crystal structure and Hirshfeld analysis. J Chem Sci 9:1395–1404. https://doi.org/10.1007/s12039-016-1133-x
Massiot D, Fayon F, Capron M, King I, Lecalve S, Alonso B, Durand JO, Bujoli B, Gan Z, Hoatson G (2002) Modeling one- and two-dimensional solid-state NMR spectra. J Chem 40:70–76. https://doi.org/10.1002/mrc.984
Estévez-Hernández O, Duque J, Rodríguez-Hernández J, Reguera E (2015) J Polyhedron 97:148–156. https://doi.org/10.1016/j.poly.2015.05.028
Ayari C, Mrad MH, Alotaibi AA, Precisvalle N, Othmani A, Ben Nasr C (2023) Investigation on crystal structure, spectroscopic characterization, thermal analysis, conductivity study and antioxidant activity of a novel compound containing cluster entities (C7H10N)2·(HgCl2)2·(Hg2Cl6). J Cluster Sci. https://doi.org/10.1007/s10876-023-02459-y
Loseva OV, Rodina TA, Antzutkin ON, Ivanov AV (2018) Chemisorption Activity of Mercury (II) Cyclopentamethylenedithiocarbamate: Synthesis, Structure, and Thermal Behavior of the [Hg 2 {S2CN (CH2)5}4] and [Au3 {S2CN(CH2)5}6][Au {S2CN(CH2)5}2][Hg2Cl6]2 Complexes. Russian J General Chem 88:2540–2549. https://doi.org/10.1134/S1070363218120149
Hasabeldaim EHH, Swart HC, Kroon RE (2023) Luminescence and stability of Tb doped CaF2 nanoparticles. J RSC Adv 13:5353–5366. https://doi.org/10.1039/D2RA07897J
Okulik N, Jubert AH (2005) Theoretical study on the structure and reactive sites of non-steroidal anti-inflammatory drugs. J Mol Struct (Thoechem) 682:55–62. https://doi.org/10.1016/j.theochem.2004.04.069
Mancuso JL, Mroz AM, Le KN, Hendon CH (2020) Electronic structure modeling of metal–organic frame works. J Chem Rev 120(16):8641–8715. https://doi.org/10.1021/acs.chemrev.0c00148
Mahamoud H, Louati B, Hlel F, Guidara K (2011) Impedance spectroscopy study of Pb2P2O7 compound. J Ionics 17:223–228. https://doi.org/10.1007/s11581-011-0540-8
Chowdari BVR, Gopalakrishnan R (1987) AC conductivity analysis of glassy silver iodomolybdate system. J Solid State Ionics 23:225–233. https://doi.org/10.1016/0167-2738(87)90055-5
Acknowledgements
The authors thank the members of units of common services, at the University of Sfax for their assistance in terms of the measurements for X-ray diffraction and Raman. The authors are equally grateful to the members of Laboratory of Applied Physic, Department of Physics, Faculty of Sciences of Sfax, University of Sfax—Tunisia for their valuable assistance in the Photoluminescence (PL) spectroscopy measurements.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jellali, A., Karâa, N., Ghalla, H. et al. Physical and Chemical Characterisations, Optical Properties and Dielectric Studies of a New Organic–Inorganic Material: bis(4-amino-2-chloropyridinium) Tetrachloromercurate (II) Monohydrate. Chemistry Africa 7, 1649–1665 (2024). https://doi.org/10.1007/s42250-023-00813-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42250-023-00813-1