
Abstract
Single crystals of a new organic–inorganic compound (C10H10N2) CdCl4 were grown by the slow evaporation technique and characterized by X-ray diffraction, Hirshfeld surface, Infrared absorption, Solid state 13C NMR, photoluminescence (PL) properties, differential scanning calorimetry (DSC) and dielectric measurements. The title compound belongs to the monoclinic space group P21/c with the following unit cell parameters: a = 7.29420(1), b = 13.9206(3), c = 14.3880(3) Å, β = 100.247(2)° and Z = 4. The structure can be described by the alternation of two different cationic-anionic layers. It consists of isolated [CdCl4]2− tetrahedral anions and 2.2′ bipyridinium (C10H10N2)2+ cations, which are connected via N–H⋯Cl hydrogen bonds. The Hirshfeld surface analysis was conducted to investigate intermolecular interactions and associated 2D fingerprint plots, revealing the relative contribution of these interactions in the crystal structure quantitatively. Furthermore, the room temperature infrared (IR) spectrum of the title compound was recorded and analyzed on the basis of detailed vibrational studies found in the literature; the detailed assignment confirms the presence of the organic groups. Solid state 13C NMR spectrum shows four signals, confirming the solid state structure determined by X-ray diffraction. Besides, photoluminescence measurements showed a strong emission line at 2.95 eV associated with radiative recombinations of excitons confined within the [CdCl4]2− which were investigated at room temperature. Finally, the thermal analysis studies were performed, and phase transition was found in the temperature range between 300 and 550 K, while the electrical measurements were performed to discuss the phase-transition mechanism.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The field of crystal engineering of new organic–inorganic architectures has gradually outlined the promising perspective to attain the purposive design and synthesis of a novel quality of non-linear optical (NLO) hybrid materials with interesting electronic properties [1,2,3]. Organic–inorganic hybrid compound based on metal halide units have received increasing attention in recent research, particularly because of their ability to combine the specific properties of inorganic frameworks (such as band gap tunability, good electrical mobility, mechanical and thermal stability, and dielectric or magnetic transitions) and the features of organic molecules (such as plastic mechanical property, structural diversity, ease of processing and efficient luminescence) including the formation of weak interactions [4,5,6,7,8,9,10,11,12,13,14,15,16,17,18]. Indeed, they present interesting properties due to their unusual topological properties, excellent magnetic, luminescence, catalytic, electrical and ferroelectric properties [19,20,21,22,23]. In the case of cadmate halide systems, some interest has been directed towards halocadmate (II) compound in combination with organic cations, due to their specific physical and chemical properties, [24,25,26,27]. In fact, they can occur as simple tetrahedral anions [CdCl4]2− or form the backbone of chain polymers. This is due to the fact that the Cd2+ configuration, exhibits a variety of coordination numbers and geometries, depending on crystal packing and ligands.
The 2.2′-bipyridine has got good non-linear optical (NLO) properties in recent years with respect to their future potential applications in the field of optoelectronic such as optical communication, optical computing, optical switching and dynamic image processing [28]. However, in conjunction with some current researches on these metal halide compounds, the crystal structure and physical properties of many organic–inorganic materials have been investigated [29,30,31].
Within this context, we report here the synthesis of a new organic–inorganic compound, (C10H10N2) CdCl4 as well as the chemical preparation and structural study. Here, we performed X-ray diffraction measurements to determine the complete crystal structure. This structural study was accompanied with an insight into Hirshfeld surface analysis, X-ray powder, Scanning electron microscopy (SEM), Transmission electron microscopy (TEM), Infrared (IR) spectroscopy, Solid-state Cross-Polarization Magic Angle Spinning Carbon-13 Nuclear Magnetic Resonance CP/MAS-NMR measurements as well as photoluminescence and dielectric analyses.
2 Experimental
2.1 Chemical preparation
The (C10H10N2) CdCl4 compound was synthesized by dissolving 0.156 g of 2.2′-bipyridine in a Hydrochloric Acid (HCl) solution (37%) and 0.183 of Cadmium Chloride (CdCl2) in water with a molar ratio of 1:1 then stirred for 5 min. The resulting aqueous solution was evaporated at room temperature, which finally led to the growth of needle crystals within 2 weeks.
2.2 Crystal chemistry
The chemical analysis of cadmium and chloride atoms was performed in order to confirm the formula determined by the structural refinement [32]. Density was measured at room temperature by flotation in CCl4. The value of the density measured (Dm = 1.892 (5) g cm−3) was very close to that calculated (Dx = 1.905 g cm−3).
2.3 Investigation techniques
The characterization of the investigated compound was performed using X-ray diffraction (XRD), Scanning electron microscopy (SEM), Transmission electron microscopy (TEM), Energy-dispersive X-ray spectroscopy (EDX), Spectroscopic measurements as well as photoluminescence and dielectric properties.
2.3.1 Single-crystal X-ray diffraction
The diffraction data from a selected single crystal was collected at room temperature on Oxford Diffraction Xcalibur Gemini S diffractometer equipped with Mo Kα radiation (0.71073 Å) at 298(2) K. The data were processed with CrysAlis software and lattice parameters were refined from the setting angles of 158 reflections in the 2.8 < θ < 31.5 range. The empirical absorption corrections were based on a multi-scan. A total of 21,567 reflections was collected using the scanning technique of which 3605 had I > 2σ(I) and were used for the structure determination. The structure analyses were carried out with the monoclinic symmetry, space group P 21/c, according to the automated search for space group available in WinGX [33]. Transition metal [Cd (II)] and chlorine atoms were located using the Patterson methods with SHELXS-86 program [34]. The oxygen atoms and the organic moieties were determined from successive Fourier calculations using SHELXL-97. All the hydrogen atoms were placed geometrically and refined isotropically. The pertinent experimental details of the structure determination for the new compound are presented in Table 1. The refinement was carried out by full-matrix least squares method (SHELXL-97 program) and converged to an acceptable final agreement factor. The last cycle of refinement included the atomic coordinates of all the atoms, isotropic thermal and anisotropic thermal parameters whose values are listed in Tables 2 and 3, respectively. The structural graphics were created with ORTEP [35] and with the DIAMOND program [36].
2.3.2 Hirshfeld surface
The Hirshfeld surface defined as the volume of space where the molecule electron density exceeds that from all neighboring molecules [37, 38] and quantified by means of the Crystal Explorer package using the CIF file format [39].
For each point on the iso-surface two different types of distances are defined: ‘de’ which is the distance to the nearest atoms outside and ‘di’ which is the distance to the nearest nucleus inside the surface, are readily defined. The normalized contact distance (dnorm) based on both ‘de’ and ‘di’ was given by the following equation:
where \(r{i^{vdw}}\) and \(r{e^{vdw}}\) are the van der Waals radii of the atoms.
2.3.3 Powder X-ray diffraction
The powder X-ray diffraction (PXRD) was recorded on a Siemens D5000 powder diffractometer, using Cu Kα radiation (1.542 Å) with a 2θ range of 5–60. The simulation of the PXRD spectra was carried out by the single-crystal data and Oscail (4.6.1) program.
2.3.4 TEM, micrographs and X-ray microanalysis
The TEM micrographs were obtained on a JEOL 2011 transmission electron microscope. Micrographs and X-ray microanalysis (SEM/EDX) were recorded with a JEOL- 6610LV electron microscope operating at 30 kV coupled with an Oxford X-Max microanalysis system (EDX).
2.3.5 Solid-state cross-polarization magic angle spinning carbon-13 nuclear magnetic resonance (CP/MAS-RMN) spectroscopy
The 13C NMR spectra were recorded at room temperature by means of a Brucker DSX-300 spectrometer. The spectra were acquired with the use of cross-polarization for protons with 12 ms contact time. A powdered sample was packed in a 4 mm diameter rotor and set to rotate at a speed of up to 8 kHz in a Doty MAS probe head. The chemical shifts were referenced with respect to dimethyl sulfoxide (DMSO).
2.3.6 Spectroscopic measurements
The Infrared spectrum (IR) were recorded in the range of 4000–400 cm−1 using PerkinElmer FTIR spectrophotometer 1000 for samples dispersed in spectroscopically pure KBr pressed into a pellet. Moreover, Solid photoluminescence spectra were taken using a time-resolved Edimbugh Instrument FLSP920 spectrofluorimeter with a Red-PMT detector and a Xenon bulb as an excitation source.
2.3.7 Differential scanning calorimetry and dielectric measurements
The DSC measurements were recorded on a NETZSCH apparatus (Model 204 Phoenix) with the heating/cooling rates at 5 K min−1. Electrical impedances were measured in the range 50 Hz–40 KHz using HP 4192A impedance analyzer. Our measurements were done in the 298–440 K temperature range.
3 Results and discussion
3.1 Description of the structure
At ambient temperature, the title compound crystallizes in the monoclinic space group P21/c with lattice parameters a = 7.29420 (1), b = 13.9206 (3), c = 14.3880 (3) Å, β = 100.247 (2)°. The unit cell volume is 1437.65 (5) Å3 with Z = 4 formula units.
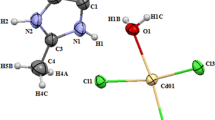
Figure 1 illustrates the asymmetric unit of the structure drawn with 50% probability thermal ellipsoids, for non-hydrogen atoms, was found to contain own cations and one type of chlorocadmate tetrahedra.

Asymmetric unit of (C10H10N2) CdCl4
The packing of (C10H10N2) CdCl4 viewed along (a, c) plane created with DIAMOND (Fig. 2) demonstrates that an alternation of organic–inorganic corrugated layers, is made up of (C10H10N2)2+ groups and isolated [CdCl4]2− tetrahedron.

View of an inorganic–organic sheet in (C10H10N2) CdCl4. (Color figure online)
The organic group was located in the (a, c) plane at approximately y = 0 and y = 2/4. Besides, the [CdCl4]2− anions were arranged in parallel layers separated by the (C10H10N2)2+ cations. As each Cd atom is surrounded by four Cl atoms, forming a weakly distorted tetrahedral configuration, with Cd–Cl distances varying between 2.4201 (8) and 2.4779 (7) Å with an average of 2.4507 Å (Table 4). The Cl–Cd–Cl angle values were in the 104.69 (2)°–112.10 (3)° range. Taking into account the geometrical characteristics of the entities, the calculated average of the Baur distortion indices (DI) are [40]:
where “d” is the (Cd–Cl) distance, “a” is the (Cl–Cd–Cl) angle, m is the average value, n1 = 4, and n2 = 6. The values of the distortion indices were DI (Cd–Cl) = 0.0016 and DI (Cl–Cd–Cl) = 0.0044. Lower values of the distortion indices indicate that the coordination geometry of the metal is slightly distorted octahedral; this can be explained by the stereochemical inactivity of the 5s2 lone pair of Cd.
The organic part of the (C10H10N2) CdCl4 compound was formed by one type of cation, i.e. (C10H10N2)2+. The selected, measured bond lengths and bond angles are grouped in Table 4. The organic molecule exhibited a regular spatial configuration with normal C–C and C–N distances in the range between 1.3348 (3) and 1.4783 (3) Å respectively, and C–C–C and C–C–N angles in the range between 116.88 (2)° and 124.30 (2)°, respectively. These values are in good agreement with those observed in similar compounds [41, 42].
The weak intermolecular hydrogen bonding contacts N–H⋯Cl reported in Table 5 and Fig. 3, provide a linkage between the cationic (C10H10N2)2+ entities, and [CdCl4]2− anions. The N⋯Cl distances varied between 3.061 Å and 3.088 Å and the N–H–Cl angle values varied between 150.66° and 157.27°.

Perspective view of the (C10H10N2) CdCl4 compound (the red lines represent hydrogen bonds). (Color figure online)
3.2 Hirshfeld surface analysis
Hirshfeld surfaces comprising d norm surface and finger print plots were generated and analyzed for the title compound in order to explore the packing modes and intermolecular interactions. The ‘dnorm’, ‘di’, ‘de’, shape index and curvedness curves are mapped on Hirshfeld surface for visualizing the intermolecular interactions are shown in the Fig. 4.

Hirshfeld surface analysis of (C10H10N2) CdCl4 a dnorm, b de, c di, d shape-index and e curvedness
The 2D fingerprint plots of (C10H10N2) CdCl4 (Fig. 5), which analyses the strong indication for all intermolecular contacts, revealed that the main intermolecular interactions were contacts.

Fingerprint plots of major contacts in (C10H10N2) CdCl4
The fingerprint plots for different atom–atom contacts and the percentage contribution of each contact to the Hirshfeld surfaces in the title compound showed that the contribution to the Hirshfeld surfaces from different contact id different (Fig. 6). Globally, H⋯Cl and H⋯H intermolecular interactions were the most abundant in the crystal packing (54.8 and 13.5%, respectively). This is evidence that van der Waals forces exert an important influence on the stabilization of the packing in the crystal structure, other interconnects, such as C⋯H/H⋯C (11%), Cd⋯H/H⋯Cd (2.1%), C⋯C (0.9%) and N⋯H/H⋯N (0.4%), contribute less to the Hirshfeld surfaces.

The relative contributions to the Hirshfeld surface area for (C10H10N2) CdCl4
3.3 Molecular electrostatic potential
The molecular electrostatic potential (MEP) is an important property that can be derived from electron density distribution. MEP is generated using the Avogadro software [43], on an asymmetric unit. The MEP is used as reactivity map displaying most probable regions for the electrophilic attack of charge point-like reagents on hybrid molecules [37, 44]. The blue and red colors indicate the positive and negative potentials, respectively. Figure 7 shows that a positive electrostatic potential is localized over the organic cation while the [CdCl4]2− part is more electronegative.

Electrostatic potential (red: negative potential, blue: positive potential). (Color figure online)
Based on this result, we can say that there is a global electrostatic attraction between the [CdCl4]2− anion and the organic cation which adds up to the favorable H⋯Cl hydrogen bonding.
3.4 X-ray powder diffraction patterns
The XRPD pattern of the (C10H10N2) CdCl4 compound is shown in Fig. 8. The sample is of single phase without any detectable impurity and narrow peaks indicate the good crystallinity of the material. The overlay simulated XRD pattern using single crystal XRD data closely resembles the experimental pattern. Most of the peak positions in powder XRD and simulated pattern from single crystal XRD coincide.

Simulated and experimental XRD of (C10H10N2) CdCl4. (Color figure online)
3.5 Particle size and morphology measurements (TEM, SEM and EDXS)
The surface morphology of the grown crystal of (C10H10N2) CdCl4 was taken at 180x and 550x magnifications with an acceleration voltage of 3–4 kV by means of scanning electron microscope (SEM). As can be seen from this figure, the compound consists of an assembly of crystal fragments having uniform distribution and a flat surface which indicates good crystal quality (Fig. 9a, b). Also, the size and morphology of the crystals are determined by analyzing the recorded TEM images. Figure 9c highlights the transmission electron microscope (TEM) image of the sample. The crystals are made up of uniform distribution of morphology with sizes are in good argument with those obtained from the SEM data.

SEM images (a, b), TEM (c) and a typical EDX spectrum (d) of (C10H10N2) CdCl4
However, the EDX spectrum of the title compound revealed the presence of all non-hydrogen atoms: Cadmium, Chloride, Carbon, Nitrogen and Oxygen. Element analyses for the observed atoms were C: 16.2%, N: 8.1%, Cd: 33.2%, Cl: 35.6% and O: 6.9% while, the calculated values were C: 15.34%, N: 8.94%, Cd: 35.92%, Cl: 33.89% and O: 5.11% (Fig. 9d).
3.6 Infrared spectroscopy
In order to give more information on the crystal structure, we have studied the vibrational properties using infrared absorption. The infrared spectrum recorded at room temperature is shown in Fig. 10. Based on some studies carried out for previous works and reported on similar compounds containing the same cations [45,46,47,48,49,50], we propose an attempt of assignment of the observed bands (Table 6).

FT-IR spectra of (C10H10N2) CdCl4
The high-frequency bands at 3041 and 2932 cm−1 assignable to (NH+) stretching modes while that of deformation around 1591 cm−1. The two broadest and most intense bands at 2891 and 2763 cm−1 are attributed respectively to asymmetrical and symmetrical vibrations of the (CH) group, whereas those towards 1537 cm−1 correspond to the vibration δ (C–H). An intense peak around 1441 and 1388 cm−1 favors the ν (C=C) vibration of the aromatic nucleus. The vibrations ν (C–C) and ν (C–N) appear around 1282, 1250 cm−1 and around 1154 and 1036 cm−1 respectively. The fine peaks observed in the region 930–771cm−1 combine the deformation modes δ (C–C–N) and δ (C–N–C). Finally, the two lines corresponding to τ (C–C) and β (C–N) are shown around 620 and 482 cm−1 respectively.
3.7 Solid state NMR spectroscopy
The 13C CP-MAS NMR spectrum of tetrachlorocadmate (II) 2.2′-bipyridinium recorded between 10 and 165 ppm, reported in Fig. 11, is in good agreement with the X-ray structure. Indeed, it exhibited four well-defined resonance signals corresponding to the ten different carbon environments. This shows the existence of only one organic cation in the asymmetric unit of the compound.

13C CP-MAS NMR spectrum of (C10H10N2) CdCl4
The chemical shifts are reported in Table 7, the highest frequency of chemical shift which are observed at 152.62 and 149.82 ppm, can be explained by the fact the four carbon atoms C(5)/C(6) and C(1)/C(7) of the bipyridinium ring is linked to the electronegative atoms N1 and N2. The isotropic shift at about 139.59, 125.81 and 122.01 ppm can be assigned to the carbon atoms away from any interaction.
These results agree with the crystal data, while the peaks at 40 ppm correspond to the presence of dimethyl sulfoxide (DMSO).
3.8 Photoluminescent properties
The luminescent coordination hybrid compound is of great current interest because of their various applications in chemical sensors, photochemistry, and electroluminescent display [51]. The luminescent properties of chloride cadmium complexes have been investigated [52, 53]. The title compound exhibits own emission band at 420 nm upon excitation at 320 nm. This emission band may be assigned to the photoluminescence emission of Cd(II) ion. In the cadmate chloride based hybrid, the lowest exciton state arises from excitation between the Valence Band (VB), which consists of a mixture of Cd (4d) and Cl (3p) states, and the Conduction Band (CB) which derives primarily from Cd (5s) states. Under excitation of 320 nm irradiation, an electron (−) is excited from the VB to the CB, leaving a hole (+) in the VB. The exciton (−) and the hole (+) move freely in the CB and VB, forming an exciton, the recombination of the electron and hole in the exciton yields a blue emission at 420 nm (Fig. 12).

Excitation and Emission fluorescence profile of (C10H10N2) CdCl4. (Color figure online)
3.9 Differential scanning calorimetry
The result of the calorimetric study of this compound (Fig. 13) reveals two distinct endothermic peaks detected at 420 and 512 K. The first peak denotes a phase transition at 420 K whereas the second peak corresponds to the decomposition of the product below the melting point.

Differential scanning calorimetry curve of (C10H10N2) CdCl4
This transition may be interpreted by a dynamical order disorder which involve the structural modification of both anion and cation groups. Then, to approve our point of view and to gain more information on the nature of this transition, we have undertaken a dielectric study at several temperatures between 298 and 440 K in the frequency range of 50 Hz–40 KHz.
3.10 Dielectric study
The complex permittivity formalism may be the useful tool in the determination of the electrical and dielectric properties, and it is expressed as:
where ɛ′ and ɛ″ are the real and imaginary parts of the dielectric constant, respectively.
The temperature dependence of the real part ɛ′ and the imaginary part ɛ″ of the permittivity are given in Fig. 14a, b. These products are plotted at selected frequencies respectively.

Temperature dependence of the real part ɛ′ (a) and ɛ″ (b) of the complex permittivity at several frequencies. (Color figure online)
From the qualitative analysis, we can observe at low temperatures that the permittivity reaches a constant value. This may be explained by the restricted reorientation motions of the cation with respect to the direction of the applied electric field, acquiring a weak contribution to the polarization. By increasing the temperature, at 420 K an anomaly above this transition can be detected. This behavior confirms the results detected by DSC method. As the temperature rises (T > 420 K), the variation of ɛ′ and ɛ″ increases and shows a strong dispersion. This behavior is attributed to the disorder increase in the (C10H10N2) CdCl4 compound. The corresponding induced disorder weakens the Van Der Waals forces and the electrostatic interaction. For this reason, the charge carriers can orient themselves with respect to the direction of applied electric field [54].
4 Conclusion
In summary, we have synthesized a new organic–inorganic hybrid material (C10H10N2) CdCl4. The formula unit of the compound is formed by a tetrahedral [CdCl4]2− entities and an organic diprotonated cation (C10H10N2)2+. The material cohesion of the compound is assured by hydrogen bonds (N–H⋯Cl) established between anions and cations. The Hirschfield surface analysis reveals the percentage of intermolecular contacts of the title compound. The powder XRD is in agreement with the simulated pattern obtained from the crystal structure. The assignment of the vibration modes frequencies of homologous compounds. The 13C CP-MAS NMR spectrum is in agreement with X-ray structure. Moreover, we found a strong blue fluorescence property at room temperature which can be associated with radiative recombination of strongly bond excitons in [CdCl4]2−. Finally, the result of the calorimetric study and dielectric measurements reveals a phase transition at 420 K.
5 Supporting information available
Crystallographic data for the structural analysis have been deposited with the Cambridge Crystallographic data Center, CCDC 1525108 for the complex. Copies of the data can be obtained free of charge at http://www.ccdc.cam.ac.uk/conts/retrieving.html.
References
Y. Baklouti, N. chaari, H. Feki, N. C-Boudjada, F. Zouari, Spectrochim. Acta Part A. 136, 397 (2015)
S. Hajlaoui, I. Chaabane, J. Lhoste, A. Bulou, K. Guidara, J. Alloys Compd. 679, 302 (2016)
S. Karoui, S. Kamoun, A. Jouini, J. Solid State Chem. 197, 60 (2013)
S. Liu, J. Chen, L. Liou, J. Wang, Inorg. Chem. 40, 6499 (2001)
T. Dammak, M. Koubaa, K. Boukheddaden, H. Bougzhala, A. Mlayah, Y. Abid, J. Phys. Chem. C 113, 19305 (2009)
Q. Sun, S. Liao, J. Yao, J. Wang, Q. Fang, Acta Cryst. E68, m1160 (2012)
T. Dammak, S. Elleuch, H. Bougzhala, A. Mlayah, R. Chtourou, Y. Abid, J. Lumin. 129, 893 (2009)
L. Dobrzycki, J. Mol. Struct. 921, 18 (2009)
D.B. Mitzi, K. Chondroudis, C.R. Kagan, IBM J. Res. Dev. 45, 29 (2001)
Y. Li, G. Zheng, C. Lin, J. Lin, Cryst. Growth Des. 8, 1991 (2008)
C.R. Kagan, D.B. Mitzi, C. Dimitrakopoulos, Science. 286, 945 (1999)
W. Cui, K. Wua, X. Liua, L. Wena, B. Ren, Thermochim. Acta. 521, 80 (2011)
K. Pradeesh, M. Agarwal, K.K. Rao, G. Vijaya Prakash, Solid State Sci. 12, 95 (2010)
A. Vishwakarma, P. Ghalsasi, A. Navamoney, Y. Lan, A. Powell, Polyhedron 30, 1565 (2011)
Y. Wang, X. Wu, W. Zhang, C. Luo, J. Li, Y. Wang, Mater. Res. Bull. 98, 59 (2018)
G. Wu, H. Wu, K. Wang, C. Zheng, Y. Wang, A. Feng, RSC Adv. 6(63), 58069 (2016)
H. Wu, G. Wu, Y. Ren, X. Li, L. Wa, Chem. Eur. J. 22, 8864 (2016)
G. Wu, Y. Cheng, Z. Yang, Z. Jia, H. Wu, L. Yang, H. Li, P. Guo, H. Lv, Chem. Eng. J. 333, 519 (2018)
G.C. Papavassiliou, Prog. Solid State Chem. 25, 125 (1997)
M. Era, S. Morimoto, T. Tsutsui, S. Saito, Appl. Phys. Lett. 65, 676 (1994)
M. Wojtas, R. Jakubas, Z. Ciunik, W. Medycki, J. Solid State Chem. 177, 1775 (2004)
G. Bator, Th Zeegers-Huyskens, R. Jakubas, J. Zaleski, J. Mol. Struct. 570, 61 (2001)
B. Kulicka, T. Lis, V. Kinzhybalo, R. Jakubas, A. Piecha, Polyhedron. 29, 2014 (2010)
J. Pons, J.G. Antón, M.F. Bardia, T. Calvet, J. Ros, Inorg. Chim.Acta. 362, 2698 (2009)
I.D. Olekseyuk, L.D. Gulay, I.V. Dydchak, L.V. Piskach, O.V. Parasyuk, O.V. Marchuk, J. Alloys Compd. 340, 141 (2002)
F. Neve, O. Francescangeli, A. Crispini, Inorg. Chim. Acta. 338, 51 (2002)
N. Hannachia, A. Bulou, C. Chassenieux, K. Guidara, F. Hlel, Physica A 390, 2987 (2011)
E. Conterosito, C. Magistris, C. Barolo, G. Croce, M. Milanesio, J. Mol. Struct. 1107, 337 (2016)
B.F. Ali, R. Al-Far, J. Chem. Cryst. 38, 689 (2008)
A. Linden, M.A. James, M.B. Millikan, L.M. Kivilighon, A. Petridis, B.D. James, Inorg. Chim. Acta. 284, 215 (1999)
A. Angeloni, P.C. Crawford, A.G. Orpen, T.J. Podesta, B.J. Shore, Chem. Eur. J. 10, 3783 (2004)
G. Charlot, Chimie Analytique Quantitative, vol. 2 (Masson, Paris, 1974).
L.J. Farrugia, J. Appl. Crystallogr. 32, 837 (1999)
G.M. Sheldrick, SHELXS-97, Programs for Crystal Structure Solution (University of Gottingen, Germany, 1997)
L.J. Farrugia, J. Appl. Cryst. 30, 565 (1997)
K. Brandenburg, Diamond Version 2.0 Impact Gbr, Bonn, Germany (1998)
M.S. Lassoued, M.S.M. Abdelbaky, A. Lassoued, A. Gadri, S. Ammar, A. Ben Salah, S. Garcia Granda, J. Mol. Struct. 1141, 390 (2017)
M.S. Lassoued, W. Ben Soltan, S.M. Mohammed, S. Abdelbaky, A. Ammar, A. Gadri, S. Ben Salah, Garcia Granda, J. Mater. Sci. 28, 12698 (2017)
S.K. Wolff, D.J. Grimwood, J.J. McKinnon, D. Jayatilaka, M.A. Spackman, CrystalExplorer 2.1. (University of Western Australia, Perth, Australia, 2007)
W. Baur, J. Acta Crystallogr. Sect. B 30, 1191 (1974)
I. Chaabane, F. Hlel, K. Guidara, PMC Phys. B 11, 11 (2008)
A. Kessentini, M. Belhouchet, J.J. Suñol, Y. Abid, T. Mhiri, Spectrochim. Acta Part A 134, 28 (2015)
M.D. Hanwell, D.E. Curtis, D.C. Lonie, T. Vandermeersch, E. Zurek, G.R. Hutchison, J. Cheminform. 4, 1 (2012)
M.S. Lassoued, M.S.M. Abdelbaky, R. Mendoza merona, A. Gadri, S. Ammar, A. Ben Salah, S. Garcia Granda, J. Mol. Struct. 1142, 73 (2017)
A. Kessentini, M. Belhouchet, Y. Abid, C. Minot, T. Mhiri, Spectrochim. Acta Part A 122, 476 (2014)
S. Banerjee, A. Ghosh, B. Wu, P.G. Lassahn, C. Janiak, Polyhedron 24, 593 (2005)
I. Mkaouar, B. Hamdi, N. Karâa, R. Zouar, Polyhedron. 87, 424 (2015)
M.O. Sinnokrot, E.F. Valeev, C.D. Sherrill, J. Am. Chem. Soc. 124, 1088 (2002)
H. Feki, Z. Elaoud, T. Mhiri, Y. Abid, A. Mlayah, Spectrochim. Acta Part A 69, 743 (2008)
S. Yurdakul, K. Arıcı, J. Mol. Struct. 691, 45 (2004)
D. Sun, N. Zhang, R.-B. Huang, L.-S. Zheng, Cryst. Growth Des. 10, 3699 (2010)
G. Wei, Y. F. Shen, Y.-R. Li, X.-C. Huang, Inorg. Chem. 49, 9191 (2010)
B. Machura, I. Nawrot, R. Kruszynski, M. Dulsk, Polyhedron 54, 272 (2013)
B. Sundarakannan, K. Kakimoto, H. Ohsato, J. Appl. Phys. 94, 5182 (2003)
Acknowledgements
Financial support from Faculty of Science in University of Gabes, Tunisia, Spanish Ministerio de Economía y Competitividad (MINECO-13-MAT2013-40950-R, and FPI Grant BES-2011-046948 to MSM.A.) and Gobierno del Principado de Asturias (GRUPIN14-060) are acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Lassoued, M.S., Lassoued, A., Abdelbaky, M.S.M. et al. Synthesis, crystal structure, photoluminescence and dielectric properties of a new organic–inorganic compound: tetrachlorocadmate (II) 2.2′-bipyridinium. J Mater Sci: Mater Electron 29, 5413–5426 (2018). https://doi.org/10.1007/s10854-017-8507-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-017-8507-9