
Abstract
In this study, we established a QSAR model for studying the antiviral activity of substituted thienopyrimidines derivatives as HCV NS3/4A protease inhibitors. We engaged in random analysis to split the datasets. Statistically, a robust model was generated with R2, Q2, and R2pred values of 0.738, 0.637, and 0.692 respectively. The dependability of these models was verified by appropriate testing limits, and this model also met the Golbraikh and Tropsha standard model conditions. The data derived from the established model was employed in suggesting some promising inhibitors of HCV NS3/4A protease and the designed ligand were found to be excellently fixed when anchored with the target and it has the least binding energy of − 197.8 kcal/mol compared to the binding energy of reference ligand (Voxilaprevir) which is − 159.4 kcal/mol. Our analysis indicates that the designed molecules possess the required drug-likeness, bioavailability, synthetic accessibility, and ADMET features.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Hepatitis C virus (HCV) pathogen remains a serious health issue with a projected 71 million people chronically infected worldwide [1]. HCV is a typical reason for cirrhosis of the liver and liver transplantation in many countries, which is a growing public health concern [2, 3]. In 2015, the World Health Organization (WHO) reported that 71 million people were infected with HCV, accounting for 1% of the global population [4, 5].
HCV is a member of the Flaviviridae family of viruses, comprised of a positive single-strand ribonucleic acid (RNA) that codes for a polyprotein. This polyprotein is cleaved into structural and non-structural (NS) proteins by both host and viral proteolytic proteins. There are 7 HCV genotypes (GTs) and 67 subtypes that have been reported [6, 7]. The geographical distribution of the HCV genotype varies depending on where you live. HCV GT1 is perhaps the most familiar form of HCV in the entire globe, with a geographically diverse distribution and a higher percentage (46%) of HCV cases worldwide. HCV GT3 is the second most common genotype, accounting for 30% of all tract infections (GT). Infections with HCV GT2 and GT4 accounted for 9–13% for most infections, with a narrow distribution pattern [8, 9].
While vaccines exist for some hepatitis viruses, none exist for HCV [2]. Standard interferon (IFN)-free therapeutic regimens also with ribavirin have become widely accepted as a template for antiviral therapy in recent years [10]. However, thyroid deficiency, neurological disorders, stomach problems, and other unpleasant reactions are some of the potential side effects. In recent years, direct-acting antiviral agents have become the mainstay of treatment, with HCV NS protease being the primary focus for designing anti-viral inhibitors [2]. As per Liu et al., ‘‘In 2011, telaprevir and boceprevir were recognized as first-generation direct-acting antiviral agents (DAAs) used as HCV NS3/4A protease inhibitors, initiating progress in the treatment of HCV,” [10,11,12]. When compared to the conventional HCV infection treatment regimen, DAAs have greatly improved tolerability and efficacy [12]. Like antibiotics, the development of resistance mechanisms promotes the quest for new compounds or the modification of current ones [2].
Pyrimidines are 6-membered heterocyclic aromatic benzene and pyridine-like molecules with nitrogens at positions 1 and 3. Heterocycles with pyrimidine substituents are of particular importance since they encompass a diverse range of natural and synthetic products including many that have beneficial bioactivities and medicinal applications. The presence of pyrimidine components in thymine, cytosine, and uracil, the essential components of DNA and RNA nucleic acids, may explain their therapeutic utility [13].
In drug discovery, virtual screening (VS) has proven as an efficient computational technique for testing various databases of organic molecules for specific hits with improved characteristics that could be scientifically verified. VS is commonly used to accelerate the discovery of new organic molecules by reducing the number of options to verify scientifically before justifying their choice. Typically, such methods are used to find targets that are much more essential to obtain successful clinical prospects [14, 15].
The initial phases of the drug development process, which is costly in terms of resources, time, and energy, are preceded by guesswork. Nonetheless, by using computer-aided techniques, the process can be achieved quickly and cost-effectively [16]. The ligand-based strategy has outperformed the random screening of established chemical libraries [17]. It provides a theoretical method for predicting the behavior of known and hypothetical drug molecules. Therese et al. looked at ligand-based and 3D-QSAR methods for discovering special and efficient NS5B inhibitors [18]. In this paper, statistical measures were employed to form a dependable QSAR model, which was then used to design substituted thienopyrimidines derivatives with improved efficacy as NS3/4A protease inhibitors, as well as to examine the binding energy of modeled molecules in reference to Voxilaprevir (a third-generation DAA) via docking studies. Also, the new compounds were screened for drug-likeness, Pharmacokinetic and toxicity properties.
2 Materials and Methods
2.1 Dataset
The dataset used was substituted thienopyrimidines derivatives, which were collected from a dataset repository (https://pubchem.ncbi.nlm.nih.gov/). The datasets are presented in Supplementary material Table S1.
2.1.1 Descriptors Computed
PaDEL-Descriptors software were used to compute the molecular descriptors (structural properties) for each molecule [19].
2.1.2 Dataset Division
The entire dataset was divided into 80% model building set and 20% validation set [20].
2.2 Model Generation
To construct the QSAR model, Genetic Function Approximation (GFA) was used. GFA is a technique that uses the evolution method to produce models. GFA provides different templates for the end-user, as with most extrapolation approaches [21].
2.3 Assessment of the generated model
The following mathematical equations were used to test the defined model;
\(q_{CV}^{2}\) Is the cross-validated correlation coefficient, \(y_{{{\text{obs}}}}\),\( y_{{{\text{pred}}}}\) and \(\overline{y}\) denotes the observed, predicted, and mean data point of observed activity respectively [20].
\(r_{{{\text{pred}}}}^{2}\) is the external explained variance, the observed and predicted responses for the validation set molecules are represented by \(y_{{{\text{obs}}\left( {{\rm{Test}}} \right)}}\) and the mean observed biological response of the model building set is represented by \(\overline{y}_{{{\text{Training}}}}\) [20].
\(cR_{p}^{2}\) is the random R2 value, R, \(R^{2}\), and \(\overline{R}_{r}^{2}\) denote correlation coefficient, coefficient of determination, and the average of the randomized coefficient of determination respectively [20, 22, 23].
2.4 Applicability Domain (AD)
The following mathematical equations were employed to establish the QSAR model's AD.
\(h_{i}\) is the actual reference space of the molecule, \(h^{*}\) the threshold value, X represents the row-matrix descriptor of the query item, n is the total number of query objects, q is the total number of descriptors in the model, Y is the observed response value (model building or testing datasets), \(\overline{Y}\) is the estimated value and SDR is the standardized residual [24, 25].
2.5 Screening of the New Chemical Entities
To ascertain the dependability of these new chemical entities, they were screened by estimation of the drug-likeness and pharmacokinetics properties using online tools SwissADME (www.swissadme.ch/) and PreADMET (https://preadmet.bmdrc.kr/).
2.6 Docking Studies
2.6.1 Preparation of Ligands
ChemBio Ultra 12.0 was used to draw 2D ligand structures [26, 27]. The energy of the query compounds was reduced using Spartan 14's density functional theory (DFT), which was then inputted into Molegro Virtual Docker software [28].
2.6.2 Preparation of the Target Receptor
The model of HCV NS3/4a protease was accessed from the Protein Data Bank, with the structural PDB ID 4A92, the resolution 2.73 Å, the R-value free 0.231, the residue count 1278, and unique protein chains 1. The co-crystallized molecule was removed, hydrogen atoms were added, minor residue structures were removed, and incomplete side chains were substituted with the aid of discovery studio. The structure is saved in PDB format once more for use in the Molegro Virtual Docker tools.
2.6.3 Docking Procedure and Evaluation
The HCV NS3/4a protease enzyme's possible ligand-binding cavity was estimated, and the binding chamber was located within a confined domain of X: 0.13, Y: 11.21, Z: − 35.34 with a radius of 15 and a grid resolution of 0.30 Å. The template, designed and reference molecules prepared according to Sect. 2.6.1 were then introduced into the Molegro Virtual Docker version 6.0 and its bond versatility was positioned along with the amino acid residue, which was also positioned within the confined domain. With a power of 0.90 and an acceptance of 1.10, the versatility was positioned. For the many clusters poses with 100.00 energy consequence measurements, the RMSD cutoff was set to 2.00. The docking algorithm was set up with a 1500 iteration threshold and a simplex conversion magnitude of 50. For the 10 poses, the docking method was evaluated at least 50 times, and the optimal poses were selected depending on defined scoring tasks [28].
3 Results and Discussion
A QSAR technique for studying the structure–activity relationship of 71 substituted thienopyrimidines compounds as active hepatitis-C virus NS3/4a protease blockers was applied in the current study and the QSAR model is described as:
\(N_{{{\text{train}}}} = 56\), \(R_{{{\text{train}}}}^{2} = 0.738\), K = 7, \(Q_{{LOO\left( {{\text{train}}} \right)}}^{2} = 0.637\), \(cR_{p}^{2} = 0.679,\)
\(N_{{{\text{test}}}} = 15\), \(r_{{{\text{test}}}}^{2} = 0.692\), Outliers > \(\pm\) 3.0 = 1, Influential molecules > h* = 4.
Where \(N_{{{\text{train}}}}\) and \(N_{{{\text{test}}}}\) t are the data quantities in the model building and testing datasets, \(R_{{{\text{train}}}}^{2}\) and \(r_{{{\text{test}}}}^{2}\) are the internal and external measurement parameters, \(Q_{LOO}^{2}\) is the squared cross-measurement parameters, and K represents predictor parameters (descriptors) present in the models. The pubcem CID, chemical structure, experimental pEC50, predicted pEC50, and the residual of the dataset used in this research are reported in supplementary file Table S1.
The established model explicates 74% and estimates 69% of the variances of the substituted thienopyrimidines derivatives with antiviral activity against the target receptor (see Table 1). The model parameters defined in Table 1 follow the OECD's requirements for confirming a QSAR model [17, 24]. The internal evaluation of the model yielded R2 and Q2 values of 0.738 and 0.637, respectively. The R2 and Q2 for the model's internal evaluation have been determined to be 0.738 and 0.637, respectively. It shows that the models accurately inferred the data when regressed and that the model can predict the fitted building set for the model, as the model correctly predicted roughly 69% of the dataset, exceeding the necessary limit of 60%. [24]. The y-randomization check shows that the model's random R2 (cR2p = 0.679) is significantly higher than the fundamental expectation of 0.50, indicating that it is not the product of pure chance [16, 22, 23]. Table 2 lists the descriptors used in the model and their descriptions.
The model AD was defined by the square area in Fig. 1. The model cautioning leverage is defined as h∗ (0.43) boundary, and the standardized residual of the models is defined as SDR. The results show that 93% of the molecules studied were within the AD of the model, while 1.4% represented the Outliers, which is compound 10 in Table S1 as shown in Fig. 1 with an SDR of 4.4, and 5.6% of the studied molecules made the influential molecule which is compound 1, 3, 15 and 28 in Table S1 as shown in Fig. 1 with leverage values of 0.75, 0.53, 0.44, and 0.45 respectively, which are greater than cautioning leverage (i.e. 0.75, 0.53, 0.44, and 0.45 > h∗ = 0.43). In conclusion, the proposed models have a lot of potentials and are very effective. As a result, it can be used to improve the activities of any of the compounds under consideration.

The model applicable domain
Figure 2 presented a graph of predicted against observed response for the entire dataset and it shows that there is a strong correlation between the models observed and expected activity figures as well as distribution of the models residual along the line standardized residual equal zero in Fig. 3. As a result of these findings, the models were found to have high internal and external statistical capacity, as well as being structurally bias-free. As a result, they can be used to predict known molecules that aren't active if the molecule is within the AD of the models.

Plot of the model predicted against experimental anti-hepatitis C activity values

Plot of standardized residual against predicted pEC50 values for the entire dataset
3.1 New Molecule Proposed and Activity Estimated
Molecule 67 in Table S1 (see Fig. 4) was used as a standard to define the molecule based on the established and evaluated QSAR model. Since it was carefully selected from Fig. 1, identifying the dataset as more active, low SDR, and discovered within the current model's AD, Molecule 67 was used as a basis for creating new molecules. Using the initially proven QSAR model, the response of the precursor compound, newly developed molecules, and Voxilaprevir was estimated. According to the findings, all modified versions and Voxilaprevir have a higher pEC50 value than the prototype (see Table 3). D3 also has the highest activity across them, much more than Voxilaprevir, as shown in Table 3. Table 3 displays the template structure, newly modified molecules, and licensed molecule (Voxilaprevir), along with their predicted response and Leverages. The leveraging results were rational and lower than the leveraging cut of point (h* = 0.43), meaning the modified molecules, and Voxilaprevir, passed the test and were within the model's application limits.

2-methyl-N-[(E)-1-pyrazin-2-ylethylideneamino]-5,6,7,8-tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidin-4-amine (Compound 67, see Table 1)
3.2 Docking Results and Breakdown
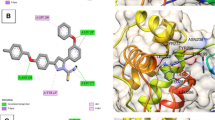
The highest predicted pEC50 value of 8.553 was found in molecule D3, which was verified by a molecular docking analysis using Molegro Virtual Docker version 6.0. The results of the molecular docking study were stated in Table 4. The findings suggest that the designed molecule D3 which is bold in Table 4 has better scores compared to the reference molecule as well as the template molecule. Discovery Studio software was used for virtual evaluation which shed more light on the interaction of molecule 67, D3, and R with the target receptor (HCV NS3/4a protease with PDB ID: 4A92). The virtual image of molecules 67, D3, and R is presented in Figs. 5, 6, and 7 respectively, while the detailed analysis of the interaction is reported in Table 5. Figure 5 shows that ASP454, GLN460, THR287, THR295, CYS431, VAL456, and MET415 residues of the selected macromolecule are implicated in the bonding with the template molecule. Figure 6 shows that HIS293, SER294, GLN460, GLU291, ASP296, THR295, CYS431, and VAL456 are the residues of the selected macromolecule are implicated in the bonding with a designed molecule, while Fig. 7 shows that GLN434, THR433, THR295, ARG393, ASP296, CYS431, and PRO230 are the residues of the selected macromolecule are implicated in the bonding with reference molecule (Voxilaprevir). The main residues ASP, GLN, THR, and CYS are involved in the interaction with all of the docked molecules, according to the docking results shown in Table 5. This indicates that these residues are crucial in inhibiting HCV NS3/4a protease. When compared to the reference molecule, molecule D3 has a higher degree of interaction with the target receptor.

A is the 3D virtual image of template molecule in the binding pocket of HCV NS3/4a protease; B is the 2D view of interaction of the template molecule with HCV NS3/4a protease

A is the 3D virtual image of D3 molecule in the binding pocket of HCV NS3/4a protease; B is the 2D view of interaction of the D3 molecule with HCV NS3/4a protease

A is the 3D virtual image of reference molecule (Voxilaprevir) in the binding pocket of HCV NS3/4a protease; B is the 2D view of interaction of the reference molecule (Voxilaprevir) with HCV NS3/4a protease
3.3 Prediction of Drug-Likeness and Pharmacokinetic Parameters
To ascertain the dependability of these new chemical entities are feasible drugs, the drug-like and pharmacokinetic features (ADMET) were assessed with Voxilaprevir as the standard. The online tool SwissADME was first used to estimate the drug-likeness features as reported in Table 6, and the PreADMET package was implemented to estimate the ADMET features mentioned in Table 7. The key criterion utilized during drug candidates at the preliminary stages of the design process is drug-likeness features [29]. This technique is also defined as the process of correlating physicochemical characteristics of a molecule with the biopharmaceutical feature of the molecule in a human body, especially its effect on oral bioavailability [28]. The concept of five by Lipinski is one of the most common and useful directions at the early clinical phase of drug production, which guess that the molecule is presumed to be poorly absorbed if it violates more than two of these conditions (MW.˂500, HBD ≤ 5, HBA ≤ 10, Log p ≤ 5 and TPSA˂140 Å2) [29]. The targeted molecules are termed to pass on the basis of Lipinski’s conditions since none breaches more than two, and so can be categorized as drug-like molecules. Furthermore, an assessment using the ABS requirements was also carried out, in which the template and all the designed molecules had 0.55 as the value obtained, while the reference molecule had a lower value [28]. This condition was established on the molecule’s probability value to have an optimal permeability and bioavailability outline, where 0.55 shows the total compliance of the Lipinski concept of five and bioavailability value of the rat is 55%, which is a parameter value greater than 10%. The designated molecules were also evaluated on a level between 1 (very easy to synthesize) to 10 (very tough and complicated to synthesize) for their synthetic accessibility [28]. The outcome of this research revealed that the entire molecules considered have synthetic accessibility in the range of 3.54 – 3.80, and hence, easy to synthesize except the reference molecule with synthetic accessibility of 8.83 (see Table 6).
Utilizing web-based SwissADME and preADET tools, the drug-likeness and ADMET features of identified molecules and reference molecule were analyzed and presented in Tables 6 and 7 respectively. The values of BBB penetration offer an understanding of whether or not a substance will move through the blood–brain barrier (BBB). A molecule with a BBB score greater than 2.0 is believed to be extremely permeable to the CNS (Central Nervous System), a molecule with a BBB score within the range 2.0–0.1 is assumed to be moderate permeability to CNS, and a score < 0.1 is considered as poor permeability to CNS [30]. The outcome of this research revealed that molecules D3 and R have high CNS permeability and that molecule 67, D1, D2, D4, D5, and D6 have moderate CNS permeability and none has poor CNS permeability.
As a reliable in vitro model to estimate oral drug permeability, Caco-2 cell absorption is recommended. If the Caco-2 score is < 4, the molecule is poorly permeable, for moderate permeability the score is within the range 4–70, and 70 and above reflect high permeability [30]. The outcome of this research revealed that the entire molecules considered have mild cell absorption toward Caco-2 cells.
The HIA statistics is the measure of the permeability assessed through the total excretion and bioavailability proportion. For poorly absorbed substances, the HIA score is around 0 to 20%, moderately absorbed substances are in the range of 20–70%, and for the well-absorbed substances is 70% above [31]. The outcome of this research revealed that the entire molecules considered have very good HIA scores.
The maximum tolerated dose (MRDT) provides an estimate of the toxic dose threshold of chemicals in humans. For a given compound, a MRTD of less than or equal to 0.477 log(mg/kg/day) is consider low, and high if significantly larger than 0.477 log(mg/kg/day) [28, 30]. In this study, we observed 67, D1, D2, D5, and D6 are highly toxic, while D3, D4, and R have low toxicity.
4 Conclusion
The obtained statistically validated QSAR models offered rationales to describe the antiviral activities of the molecules studied. The models are statistically robust, with R2 values of 0.738 and 0.692 for internal and external validation, respectively, and met the conditions for an effective QSAR model recommended by numerous groups. The binding affinity (− 197.846) of this newly discovered molecule docked into the binding pocket of the crystal structure of Hepatitis C Virus NS3/4A protease (PDB ID: 4A92) were observed to be more significant than that of molecule 67 (− 118.299) in the datasets as well as Voxilaprevir which is -159.365. Our analysis indicates that the designed molecules possess the required drug-likeness and ADMET properties, and bind Hepatitis C Virus NS3/4A protease protein at the main active site effectively. Besides, the pharmacokinetic estimation of ADMET properties has shown that the identified molecules are possible biologically active molecules with specific scaffolds, and could have the potential to inhibit Hepatitis C Virus NS3/4A protease.
References
Jia S, Zhou W, Wu J, Liu X, Guo S, Zhang J, Zhang X (2020) A biomolecular network-based strategy deciphers the underlying molecular mechanisms of Bupleuri Radix/Curcumae Radix medicine pair in the treatment of hepatitis C. Eur J Integ Med 33:101043
El-Kassem LA, Hawas UW, El-Souda S, Ahmed EF, El-Khateeb W, Fayad W (2019) Anti-HCV protease potential of endophytic fungi and cytotoxic activity. Biocatal Agric Biotechnol 19:101170
González-Grande R, Jiménez-Pérez M, Arjona CG, Torres JM (2016) New approaches in the treatment of hepatitis C. World J Gastroenterol 22(4):1421
Kucherenko A, Pampukha V, Romanchuk KY, Chernushyn SY, Bobrova I, Moroz L, Livshits L (2016) IFNL4 polymorphism as a predictor of chronic hepatitis C treatment efficiency in Ukrainian patients. Cytol Genet 50(5):330–333
World Health Organization (2018) Guidelines for the care and treatment of persons diagnosed with chronic hepatitis C virus infection. Geneva. Licence: CC BY-NC-SA 3.0 IGO. WHO, Geneva
Chahine EB, Sucher AJ, Hemstreet BA (2017) Sofosbuvir/velpatasvir: the first pangenotypic direct-acting antiviral combination for hepatitis C. Ann Pharmacother 51(1):44–53
Petruzziello A, Marigliano S, Loquercio G, Cozzolino A, Cacciapuoti C (2016) Global epidemiology of hepatitis C virus infection: an up-date of the distribution and circulation of hepatitis C virus genotypes. World J Gastroenterol 22(34):7824
Coppola N, Alessio L, Onorato L, Sagnelli C, Macera M, Sagnelli E, Pisaturo M (2019) Epidemiology and management of hepatitis C virus infections in immigrant populations. Infect Dis Poverty 8(1):17
Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT, Simmonds P (2014) Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology 59(1):318–327
Liu M, Xu Q, Guo S, Zuo R, Hong Y, Luo Y, Liu Y (2018) Design, synthesis, and structure-activity relationships of novel imidazo [4, 5-c] pyridine derivatives as potent non-nucleoside inhibitors of hepatitis C virus NS5B. Bioorg Med Chem 26(9):2621–2631
Poordad F, McCone J Jr, Bacon BR, Bruno S, Manns MP, Sulkowski MS, Boparai N (2011) Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med 364(13):1195–1206
Bidell MR, McLaughlin M, Faragon J, Morse C, Patel N (2016) Desirable characteristics of hepatitis C treatment regimens: a review of what we have and what we need. Infect Dis Therapy 5(3):299–312
Brown DJ (1984). In: Katritzky AR, Rees CW (eds) Comprehensive Heterocyclic chemistry. Pergamon Press, Oxford
Arthur DE, Ejeh S, Uzairu A (2020) Quantitative structure-activity relationship (QSAR) and design of novel ligands that demonstrate high potency and target selectivity as protein tyrosine phosphatase 1B (PTP 1B) inhibitors as an effective strategy used to model anti-diabetic agents. J Recept Signal Transduct 40:501–520
Neves BJ, Braga RC, Melo-Filho CC, Moreira Filho JT, Muratov EN, Andrade CH (2018) QSAR-based virtual screening: advances and applications in drug discovery. Front Pharmacol 9:1275
Arthur DE, Uzairu A, Mamza P, Abechi S (2016) Quantitative structure–activity relationship study on potent anticancer compounds against MOLT-4 and P388 leukemia cell lines. J Adv Res 7(5):823–837
Roy K, Mitra I, Kar S, Ojha PK, Das RN, Kabir H (2012) Comparative studies on some metrics for external validation of QSPR models. J Chem Inf Model 52(2):396–408
Therese PJ, Manvar D, Kondepudi S, Battu MB, Sriram D, Basu A, Kaushik-Basu N (2014) Multiple e-pharmacophore modeling, 3D-QSAR, and high-throughput virtual screening of hepatitis C virus NS5B polymerase inhibitors. J Chem Inf Model 54(2):539–552
Yap CW (2011) PaDEL-descriptor: an open source software to calculate molecular descriptors and fingerprints. J Comput Chem 32(7):1466–1474
Tropsha A (2010) Best practices for QSAR model development, validation, and exploitation. Mol Inf 29(6–7):476–488
Rogers D (1997) Evolutionary statistics: using a genetic algorithm and model reduction to isolate alternate statistical hypotheses of experimental data. In: Paper presented at the ICGA
Yan F, Liu T, Jia Q, Wang Q (2019) Multiple toxicity endpoint–structure relationships for substituted phenols and anilines. Sci Total Environ 663:560–567
Liu T, Yan F, Jia Q, Wang Q (2020) Norm index-based QSAR models for acute toxicity of organic compounds toward zebrafish embryo. Ecotoxicol Environ Saf 203:110946
Eriksson L, Jaworska J, Worth AP, Cronin MT, McDowell RM, Gramatica P (2003) Methods for reliability and uncertainty assessment and for applicability evaluations of classification-and regression-based QSARs. Environ Health Perspect 111(10):1361–1375
Li F, Li X, Liu X, Zhang L, You L, Zhao J, Wu H (2011) Docking and 3D-QSAR studies on the Ah receptor binding affinities of polychlorinated biphenyls (PCBs), dibenzo-p-dioxins (PCDDs) and dibenzofurans (PCDFs). Environ Toxicol Pharmacol 32(3):478–485
Evans DA (2014) History of the Harvard ChemDraw project. Angew Chem Int Ed 53(42):11140–11145
Li Z, Wan H, Shi Y, Ouyang P (2004) Personal experience with four kinds of chemical structure drawing software: review on ChemDraw, ChemWindow, ISIS/Draw, and ChemSketch. J Chem Inf Comput Sci 44(5):1886–1890
Umar AB, Uzairu A, Shallangwa GA, Uba S (2020) Docking-based strategy to design novel flavone-based arylamides as potent V600E-BRAF inhibitors with prediction of their drug-likeness and ADMET properties. Bull Natl Res Centre 44(1):1–11
Veerasamy R, Rajak H, Jain A, Sivadasan S, Varghese CP, Agrawal RK (2011) Validation of QSAR models-strategies and importance. Int J Drug Des Discov 3:511–519
Danishuddin M, Khan SN, Khan AU (2010) Molecular interactions between mitochondrial membrane proteins and the C-terminal domain of PB1-F2: an in silico approach. J Mol Model 16(3):535–541
Pires DE, Blundell TL, Ascher DB (2015) pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem 58(9):4066–4072
Funding
No fund received.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ejeh, S., Uzairu, A., Shallangwa, G.A. et al. In Silico Design, Drug-Likeness and ADMET Properties Estimation of Some Substituted Thienopyrimidines as HCV NS3/4A Protease Inhibitors. Chemistry Africa 4, 563–574 (2021). https://doi.org/10.1007/s42250-021-00250-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42250-021-00250-y