
Abstract
A series of poly(ionic liquid)s (PILs) incorporating 1,2,3-triazolium and imide units are synthesized in three steps by AA + BB polyaddition between α,ω-diazido tetraethylene glycol and three aromatic bis-imide dipropargyl monomers using copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC) followed by N-alkylation of the resulting poly(1,2,3-triazole imide)s with iodomethane and subsequent anion exchange with lithium bis(trifluoromethylsulfonyl)imide. Structure/properties correlations of poly(1,2,3-triazole imide)s and corresponding poly(1,2,3-triazolium imide)s are discussed based on NMR spectroscopy, differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), size exclusion chromatography (SEC) and solubility measurements.
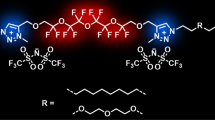
Graphic Abstract

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Poly(ionic liquid)s (PILs) have attracted significant attention in recent years as they ideally merge the properties of ionic liquids (e.g. high thermal, chemical, electrochemical stabilities, tunable viscosity and solubility, enhanced ionic conductivity…) and those of polymer materials (e.g. viscoelasticity, processability, film forming capacities and broad structural design…) [1,2,3,4,5,6]. They have shown promising performances in a broad range of applications including thermoresponsive polyelectrolytes [7], self-assembled colloids [8,9,10], electrolyte gated transistors [11], dye sensitized solar cells [12], anion exchange membranes for fuel cells [13, 14], batteries and supercapacitors [15,16,17,18], sensors and actuators [19,20,21], electrochromic devices [22], gas separation membranes [23, 24], antimicrobial surfaces [25], and catalysis [26, 27]. Their widespread interest rely on the broad array of cations (e.g. ammonium, pyridinium, imidazolium, pyrrolidinium, phosphonium, piperidinium, thiazolium, triazolium…), anions (e.g. halides, carboxylates, sulfonates, phosphates, inorganic fluorides, perfluorinated sulfonimides…) and synthetic approaches (e.g. different chain growth and step growth polymerizations, or post-polymerization chemical modification techniques…) involved in their synthesis.
1,2,3-Triazolium based poly(ionic liquid)s (TPILs) are a recent addition to the PILs family that merge the best attributes of state-of-the-art polymerization and post-polymerization chemical modification techniques with copper(I)-catalyzed azide-alkyne 1,3-dipolar cycloaddition (CuAAC) [28], quantitative N-alkylation of 1,2,3-triazole groups and ion metathesis reactions [2]. So far, a broad variety of side-chain TPILs (e.g. poly(meth)acrylates [29,30,31], poly(N-vinyl triazolium) [32], poly(vinyl ester) [33], poly(aryl ether) [14], polysiloxane [34]) and main-chain TPILs (e.g. ionenes [35,36,37], dendritic structures [38, 39]) have been developed using high precision macromolecular designs. However, up to now there are still important polymer classes that have not been reported as TPIL derivatives. For instance, polyimides are high performance polymer materials that exhibit improved mechanical properties, as well as high chemical stability and heat resistance relevant for many technical applications [40, 41]. Scarce examples of polyimide-based PILs have already been reported so far but none of them included 1,2,3-triazolium cations. Kim and coworkers developed ionic polyimides with side-chain imidazolium groups as well as polyimide networks cross-linked through piperazinium linkages both having enhanced gas separation properties compared to classical polyimides [42,43,44]. Besides, Shaplov and co-workers reported the transformation of polybenzimidazoles into ionic polyimides carrying main-chain benzimidazolium or side-chain quinuclidinium groups and studied their gas separation properties [45].
Herein, we report the synthesis of a series of poly(1,2,3-triazolium imide)s using a three-step synthetic strategy involving: (i) AA + BB CuAAC polyaddition, (ii) N-alkylation of 1,2,3-triazole groups, and (iii) ion metathesis to introduce bis(trifluoromethylsulfonyl)imide (TFSI) counter anions. Their structural and physical properties are discussed based on NMR spectroscopy, size exclusion chromatography (SEC), differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), and solubility measurements.
2 Experimental Section
2.1 Materials
3,3′,4,4′-Biphenyltetracarboxylic dianhydride (97%), propargylamine (98%), sodium acetate (99%), acetic anhydride (99%), diisopropylethylamine (DIPEA, 99%), iodomethane (CH3I, 99%), copper(I) iodide triethylphosphite (CuP(OEt)3I, 97%), lithium bis(trifluoromethylsulfonyl)imide (LiTFSI, 99.95%), as well as all other reagents and solvents were purchased from Merck and used as received. Dipropargyl monomers 2 and 3 [46], and α,ω-diazido tetraethylene glycol 4 [47] were synthesized as described previously.
2.2 Characterization Methods
1H NMR (300 or 400 MHz) and 13C NMR (100 MHz) spectra were recorded on Bruker Avance 300 or 400 spectrometers in deuterated dimethylsulfoxide (DMSO-d6) using as references residual hydrogenated DMSO-d6 for 1H NMR, DMSO-d6 signal for 13C NMR, and the signal of trifluoroethanol for 19F NMR. DSC measurements were carried out under helium using a DSC Q200 apparatus (TA instruments) by applying two cycles in the temperatures range of − 50 to 120 °C at a heating rate of 20 °C/min. Tg values were measured during the second heating cycle. TGA measurements were performed under helium using a TGA Q500 apparatus (TA instruments) at a heating rate of 20 °C/min. SEC measurements were carried out at 50 °C on a setup comprising a Viscotek VE 5111 injector valve bracket, a Viscotek T60 A dual detector, a Viscotek VE 3580 RI detector, a Viscotek VE 1122 solvent delivery system, and a PL gel 5 µm Mixed C (300 × 7.5 mm, polystyrene/divinylbenzene) column using a 0.01 M LiTFSI solution in N,N-dimethylformamide (DMF) as eluent. 2 mg/mL solutions of poly(1,2,3-triazolium imide)s 8–10 in DMF with 0.01 M LiTFSI were prepared and filtered through a 0.45 μm pore size PTFE filter prior to injection. Number average molar masses (Mn), weight average molar masses (Mw) and chain dispersities (Ð) were calculated using a refractive index calibration curve obtained from polystyrene (PS) standards.
2.3 Synthesis of dipropargyl monomer 1
3,3′,4,4′-(Biphenyl-tetracarboxylic) dianhydride (6.00 g, 18.6 mmol) was added portion wise to a solution of propargylamine (2.47 g, 41.0 mmol) in N-methyl-2-pyrrolidone (NMP, 50 mL) and the mixture was stirred for 7 h at 50 °C. A solution of sodium acetate (0.08 g, 0.97 mmol) in a 2:1 vol/vol mixture of acetic anhydride and pyridine (6.7 mL) was added and the mixture was further stirred for 10 h at 70 °C. After cooling down to room temperature, the reaction mixture was precipitated in water and filtered. The crude product was recrystallized from DMF/H2O, filtered and dried under reduced pressure to yield dipropargyl monomer 1 as a white powder (4.20 g, 61.3%). 1H NMR (300 MHz, DMSO-d6): δ 8.37 (s, 2H, H-f), 8.33 (dd, J = 7.8 Hz, J = 1.5 Hz, 2H, H-i), 8.05 (d, J = 7.8 Hz, 2H, H-h), 4.41 (d, J = 2.5 Hz, 4H, NCH2C≡CH), 3.29 (t, J = 2.5 Hz, 2H, NCH2C≡CH) ppm. 13C NMR (400 MHz, DMSO-d6): δ 166.20 (4C, NCO), 144.25 (2C, C-g), 133.67 (2C, C-i), 132.39 (2C, C-e), 131.11 (2C, C-j), 123.97 (2C, C-h), 122.28 (2C, C-f), 78.06 (2C, NCH2C≡CH), 73.87 (2C, NCH2C≡CH), 26.77 (2C, NCH2C≡CH) ppm. HRMS (ESI) m/z: [M + Na]+ calculated for C22H12N2O2Na 391.0689; found, 391.0672.
2.4 Synthesis of Poly(1,2,3-triazole imide)s 5–7
2.4.1 General Procedure for CuAAC Polyaddition. Synthesis of 5
A solution of dipropargyl 1 (866 mg, 2.35 mmol), diazide 4 (580 mg, 2.35 mmol), CuP(OEt)3I (17 mg, 0.047 mmol) and DIPEA (300 mg, 2.35 mmol) in NMP (24 mL) was stirred for 48 h at 60 °C. The crude reaction mixture was precipitated twice in diethyl ether (Et2O), centrifuged, and dried under vacuum to obtain poly(1,2,3-triazole imide) 5 as an orange solid (1.20 g, 79.4%). 1HNMR (300 MHz, DMSO-d6): δ 8.27–8.14 (m, 4H, H-f, H-i), 8.04 (s, 2H, NCH2C=CH), 7.90 (s, 2H, H-h), 4.83 (s, 4H, NCH2C=CH), 4.45 (s, 4H, NCH2CH2OCH2CH2O), 3.75 (s, 4H, NCH2CH2OCH2CH2O), 3.43 (s, 4H, NCH2CH2OCH2CH2O), 3.38 (s, 4H, NCH2CH2OCH2CH2O) ppm.
2.4.2 Synthesis of 6
The general procedure for CuAAC polyaddition was applied to dipropargyl 2 (550 mg, 1.39 mmol), diazide 4 (340 mg, 1.39 mmol), CuP(OEt)3I (10 mg, 0.028 mmol) and DIPEA (180 mg, 1.39 mmol) in NMP (10 mL) to obtain poly(1,2,3-triazole imide) 6 as a dark brown solid (600 mg, 64.4%). 1H NMR (300 MHz, DMSO-d6): δ 8.16 (d, J = 6.3 Hz, 2H, H-i), 8.07–8.04 (m, 4H, H–f, H-h, NCH2C=CH), 4.84 (s, 4H, NCH2C=CH), 4.46 (s, 4H, NCH2CH2OCH2CH2O), 3.76 (s, 4H, NCH2CH2OCH2CH2O), 3.45 (s, 4H, NCH2CH2OCH2CH2O), 3.40 (s, 4H, NCH2CH2OCH2CH2O) ppm.
2.4.3 Synthesis of 7
The general procedure for CuAAC polyaddition was applied to dipropargyl 3 (2.67 g, 5.16 mmol), diazide 4 (1.26 g, 5.16 mmol), CuP(OEt)3I (40 mg, 0.11 mmol) and DIPEA (0.66 g, 5.16 mmol) in NMP (10 mL) to obtain poly(1,2,3-triazole imide) 7 as a dark brown solid (3.10 g, 75.8%). 1H NMR (300 MHz, DMSO-d6): δ 8.08–8.04 (m, 4H, H-i, NCH2C=CH), 7.87 (s, 2H, H-h), 7.70 (s, 2H, H-f), 4.83 (s, 4H, NCH2C=CH), 4.45 (s, 4H, NCH2CH2OCH2CH2O), 3.76 (s, 4H, NCH2CH2OCH2CH2O), 3.46 (s, 4H, NCH2CH2OCH2CH2O), 3.38 (s, 4H, NCH2CH2OCH2CH2O) ppm.
2.5 Synthesis of Poly(1,2,3-triazolium imide)s 8–10
2.5.1 General Procedure for N-alkylation and Anion Exchange. Synthesis of 8
A solution of poly(1,2,3-triazole imide) 5 (1.10 g, 1.74 mmol of 1,2,3-triazole groups) and iodomethane (1.00 mL, 17.4 mmol) in DMF (30 mL) was stirred for 72 h at 60 °C. The reaction mixture was precipitated once in ethyl acetate and centrifuged to afford, after drying under reduced pressure the resulting poly(3-methyl-1,2,3-triazolium iodide) as an orange solid (1.30 g, 80.6%). A solution of this iodide-containing poly(1,2,3-triazolium imide) intermediate (1.00 g, 1.07 mmol of 1,2,3-triazolium groups) and LiTFSI (460 mg, 1.62 mmol) was dissolved in DMF (3 mL) and heated at 45 °C for 20 h. The mixture was precipitated in water, dissolved in CH3CN and precipitated twice in methanol (MeOH) to afford, after centrifugation and drying under reduced pressure, 8 as an orange solid (900 mg, 68.4%). 1H NMR (300 MHz, DMSO-d6): δ 9.02 (s, 2H, NCH2C=CH), 8.39-8.32 (m, 4H, H-f, H-i), 8.06 (s, 2H, H-h), 5.18 (s, 4H, NCH2C=CH), 4.79 (s, 4H, NCH2CH2OCH2CH2O), 4.36 (s, 6H, NCH3), 3.91 (s, 4H, NCH2CH2OCH2CH2O), 3.56 (s, 4H, NCH2CH2OCH2CH2O), 3.48 (s, 4H, NCH2CH2OCH2CH2O) ppm. 13C NMR (100 MHz, DMSO-d6): δ 166.69,166.65 (C-d), 144.17 (C-g), 138.48 (C-b), 133.58 (C-i), 132.80 (C-e), 131.50 (C-j), 130.74 (C-a), 124.05 (C-h), 122.12 (C-f), 119.38 (q, J = 320 Hz, C-TFSI), 69.40 (C-n, C-o), 67.21 (C-m), 53.04 (C-l), 38.24 (C-k), 29.95 (C-c) ppm. 19F NMR (282 MHz, DMSO-d6): –79.40 (s, 12F, CF3SO2N) ppm.
2.5.2 Synthesis of 9
The general procedure for N-alkylation was applied to a mixture of 6 (380 mg, 0.56 mmol of 1,2,3-triazole groups) and iodomethane (0.35 mL, 5.66 mmol) in DMF (7 mL). The general procedure for anion exchange was applied to the poly(3-methyl-1,2,3-triazolium iodide) intermediate (130 mg, 0.136 mmol of 1,2,3-triazolium groups) and LiTFSI (60 mg, 0.21 mmol) in DMF (2 mL) to yield 9 as a dark brown solid (110 mg, 64.1%). 1H NMR (300 MHz, DMSO-d6): δ 9.01 (s, 2H, NCH2C=CH), 8.16-8.13 (m, 6H, H-f, H-h, H-i), 5.19 (s, 4H, NCH2C=CH), 4.80 (s, 4H, NCH2CH2OCH2CH2O), 4.35 (s, 6H, NCH3), 3.92 (s, 4H, NCH2CH2OCH2CH2O), 3.55 (s, 4H, NCH2CH2OCH2CH2O), 3.49 (s, 4H, NCH2CH2OCH2CH2O) ppm. 13C NMR (100 MHz, DMSO-d6): δ 193.16 (C-p), 166.36 (C-d), 141.49 (C-g), 138.27 (C-b), 135.75 (C-i), 134.77 (C-j), 132.01 (C-e), 130.89 (C-a), 123.75,123.62 (C-h, C-f), 119.40 (q, J = 320 Hz, C-TFSI), 69.43 (C-n, C-o), 67.25 (C-m), 53.07 (C-l), 38.30 (C-k), 30.00 (C-c) ppm. 19F NMR (282 MHz, DMSO-d6): –79.40 (s, 12F, CF3SO2N) ppm.
2.5.3 Synthesis of 10
The general procedure for N-alkylation was applied to a mixture of 7 (2.60 g, 3.20 mmol of 1,2,3-triazole groups) and iodomethane (2.00 mL, 32.0 mmol) in DMF (3 mL). The general procedure for anion exchange was applied to the poly(3-methyl-1,2,3-triazolium iodide) intermediate (2.50 g, 2.32 mmol of 1,2,3-triazolium groups) and LiTFSI (1.00 g, 3.48 mmol) in DMF (4 mL) to yield 10 as a yellow solid (2.00 g, 62.5%). 1H NMR (300 MHz, DMSO-d6): δ 8.99 (s, 2H, NCH2C=CH), 8,14 (d, J = 7.6 Hz, 2H, H-i), 7.89 (d, J = 6.8 Hz, 2H, H-h), 7.69 (s, 2H, H-f), 5.17 (s, 4H, NCH2C=CH), 4.80 (s,4H, NCH2CH2OCH2CH2O), 4.35 (s, 6H, NCH3), 3.91 (s, 4H, NCH2CH2OCH2CH2O), 3.55 (s, 4H, NCH2CH2OCH2CH2O), 3.47 (s, 4H, NCH2CH2OCH2CH2O) ppm. 13C NMR (100 MHz, DMSO-d6): δ 166.12,166.02 (C-d), 138.20 (C-b), 137.22 (C-g), 135.84 (C-h), 133.03 (C-j), 132.62 (C-e), 130.91 (C-a), 124.31 (C-i), 123.34 (C-f), 121.79 (C-q), 119.40 (q, J = 320 Hz, C-TFSI), 69.63,69.47,69.16 (C-n, C-o), 67.27 (C-m), 64.49 (t, J = 26 Hz, C-p), 53.13 (C-l), 38.36 (C-k), 30.08 (C-c) ppm. 19F NMR (282 MHz, DMSO-d6): –63.56 (s, 6F, CF3C), –79.40 (s, 12F, CF3SO2N) ppm.
2.6 Membrane Preparation
Poly(1,2,3-triazolium imide) membranes were hot pressed at ca. Tg + 20 °C using first a 250 µm thick hollow spacer and then without any spacer directly between two Teflon sheets using 20 bars of pressure. The resulting membranes were ca. 25 µm thick.
3 Results and Discussions
3.1 Synthesis of Dipropargyl Monomers 1–3
While dipropargyl monomers 2 and 3 have been previously reported, dipropargyl monomer 1 was synthesized from 3,3′,4,4′-biphenyltetracarboxylic dianhydride and propargylamine adapting the same experimental conditions [46]. After reaction of these two compounds in NMP for 7 h at 50 °C, the cyclodehydration of the amide-acid intermediate was promoted by the addition of a sodium acetate solution in a 2:1 mixture of acetic anhydride/pyridine and further heating at 70 °C for 10 h (Scheme 1). Dipropargyl monomer 1 was then obtained as a white powder in 61.3% yield after purification by recrystallization. Its structure and purity were confirmed by 1H and 13C NMR spectroscopy (Figs. S1 and S2) as well as by high-resolution electrospray ionization mass spectrometry.

Synthesis of dipropargyl monomer 1
3.2 Synthesis of Poly(1,2,3-triazole imide)s 5–7
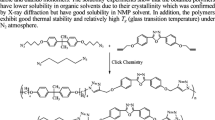
PTIs 5–7 were synthesized by AA + BB CuAAC polyaddition between stoichiometric amounts of dipropargyl monomers 1–3 and α,ω-diazido tetraethylene glycol 4 (Scheme 2). Polyaddition reactions were carried out during 48 h at 60 °C in NMP (6 wt% of AA + BB monomers) using CuIP(OEt)3 and DIPEA as catalytic system (respectively 0.02 and 1.0 equiv. according to alkyne and azide functionalities) [48]. Polymerization media remained homogeneous throughout the reaction and poly(1,2,3-triazole imide)s (PTIs) 5–7 were obtained in yields ranging from 64 to 79% after two precipitations in Et2O and drying under reduced pressure. At room temperature PTIs 5–7 are soluble at 1 mg/mL in polar aprotic solvents such as DMF, DMSO and NMP, while insoluble in heptane (nC7H16), MeOH, Et2O and ethyl acetate (EtOAc) (Table 1). However, while PTI 7 is soluble at 1 mg/mL in acetone, tetrahydrofuran (THF) and dichloromethane (CH2Cl2), and soluble at 10 mg/mL in chloroform (CHCl3) and acetonitrile (CH3CN), PTIs 5 and 6 are insoluble in these solvents. The broader solubility of 7 most probably results from the presence of the hexafluoroisopropylidene groups.

Synthesis of poly(1,2,3-triazolium imide)s 8–10
1H NMR spectroscopy of PTIs 5–7 was thus carried out in DMSO-d6 (Fig. 1). Occurrence of the CuAAC polyaddition reaction was confirmed by the appearance of 1,2,3-triazole signals at ca. 8.05 ppm. This was also corroborated by the significant downfield shifts of the methylene proton signals neighboring the 1,2,3-triazole groups after polyaddition (see as example Fig. S3 for the stack of 1H NMR spectra of monomers 1, 4 and PTI 5). While 1H NMR spectra of PTIs 6 and 7 did not show any chain-end signals, propargyl chain-end signals could be observed at 4.39 and 3.27 in the 1H NMR spectrum of PTI 5. The number average polymerization degree (Xn) of PTI 5 could thus be calculated by comparing the signal of the propargyl chain-ends at 4.39 ppm and the signals of e.g. the N-1 methylene groups at 4.82 ppm. The obtained value (Xn = 31) corresponds to a number average molar mass (Mn) of ca. 9.8 kg/mol. Due to the absence of chain-end signals, the Mn values of PTIs 6 and 7 should thus be significantly higher than the value calculated for 5.

1H NMR spectra (25 °C, DMSO-d6) of PTIs 5–7 (*peaks assigned to propargyl chain-ends)
3.3 Synthesis of Poly(1,2,3-triazolium imide)s 8–10
PTIs 5–7 were then submitted to N-alkylation reaction using iodomethane and subsequent anion exchange reaction using LiTFSI to afford the corresponding TFSI-containing poly(1,2,3-triazolium imide)s (TPTIs) 8–10 (Scheme 2). As generally observed [2], TPTIs 8–10 are soluble in a broader range of solvents compared to their neutral PTI precursors 5–7 due to the presence of highly delocalized 1,2,3-triazolium/TFSI ion pairs. Indeed, while still insoluble in heptane and EtOAc, and still soluble at 1 mg/mL in DMF, DMSO and NMP such as PTIs 5–7, TPTIs 8–10 are also soluble at 1 mg/mL in CH3CN (Table 1). Solubility in other tested solvents is highly structure-dependent and TPTI 10, most probably due to the presence of hexafluoroisopropylidene groups, exhibit the broadest solubility as it is soluble at 1 mg/mL in acetone, THF and CH2Cl2 while soluble at 10 mg/mL in MeOH and CHCl3.
Solubility of TPTIs 8–10 in DMF allowed their characterization by SEC using 0.01 M LiTFSI in DMF as eluent and calibration against PS standards (Fig. 2). TPTIs 8–10 exhibit monomodal traces with Mn values ranging from 15.4 to 46.9 kg/mol and chain dispersities (Ð) ranging from 1.47 to 1.95 (Table 2). However, Mn values are biased by the use of PS calibration as the distinct chemical structures of dipropargyl monomers most probably induce different solubility, chain stiffness and radius of giration of the chains in the SEC eluent. For instance, TPTI 8 exhibit an intermediate Mn value of 22.6 kg/mol although it is the only sample of the series where propargyl chain-end signals could be detected by 1H NMR. Its comparably low Ð value most probably stems from the fractionation of polymer chains during precipitations but in this case higher Mn values should be obtained. There is however a fair agreement between the Mn values obtained from 1H NMR of 5 and SEC of 8 (Mn = 18.6 and 22.6 kg/mol, respectively). TPTI 9 exhibit the highest Mn value (Mn = 46.9 kg/mol) and a Ð value which is the closest to the theoretical value of 2.0 (Ð = 1.95) expected for a step growth polymerization process. However, whereas no chain-ends could be observed by 1H NMR, TPTI 10 exhibit the smallest Mn value (Mn = 15.4 g/mol). Therefore, whereas SEC provides reliable chain dispersity values, the Mn values should be addressed carefully and only the Mn value calculated by 1H NMR of TPTI 8 should be considered. It can thus be concluded that TPTIs 9 and 10 should have comparable chain lengths much higher than for TPTI 8 due to the absence of noticeable chain-end signals in their 1H NMR spectra.

SEC traces (25 °C, DMF 0.01 M LiTFSI) of TPTIs 8 (solid line), 9 (dashed line) and 10 (dotted line)
1H NMR spectroscopy of TPTIs 8–10 was carried out in DMSO-d6 (Fig. 3). The quantitative downfield shifts of the chemical displacement of the 1,2,3-triazolium protons (from ca. 8.05 ppm for PTIs 5–7 to ca. 9.01 ppm for TPTIs 8–10) and the appearance of the N-3 methyl signal at ca. 4.36 ppm with a 1:3 ratio attested the completion of the N-alkylation reaction. Additionally, the chemical displacements of most proton signals of the polymer backbones were shifted downfield as a result of the different electron delocalization of the 1,2,3-triazolium groups compared to the parent 1,2,3-triazole ones (see as example Fig. S4 for the stack of 1H NMR spectra of PTI 5 and TPTI 8). The structures of TPTIs 8–10 were further corroborated by 13C and 19F NMR spectroscopy (Figs. S5 and S6).

1H NMR spectra (25 °C, DMSO-d6) of TPTIs 8–10 (*peak assigned to propargyl chain-ends)
3.4 Physical Properties of Poly(1,2,3-triazole imide)s 5–7 and Poly(1,2,3-triazolium imide)s 8–10
Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were used to investigate the thermal properties of PTIs 5–7 and TPTIs 8–10 (Table 2). All samples are amorphous materials as their DSC traces exhibit a single transition corresponding to their glass transition temperature (Tg). Due to the presence of flexible tetraethylene glycol segments Tg values are rather low for polyimide materials as they range from – 19 to 97 °C for PTIs 5–7 and from 27 to 44 °C for TPTIs 8–10. The differences in Tg values for each series are mainly related to the flexibility of the dipropargyl monomer. As generally observed [2], thermal stability of TPTIs 8–10 is lower than their PTIs 5–7 neutral precursors (Fig. 4). After a weight loss at 200 °C that could not be precisely attributed, all samples exhibit a steep weight loss at ca. 400 °C for PTIs 5–7 and ca. 350 °C for TPTIs 8–10. Temperatures for 20% weight loss values (Td20) show that the main difference in thermal stability for both series is due to the lower stability of 1,2,3-triazolium groups compared to 1,2,3-triazole ones. For both series Td20 values decreases according to the following order Td20(5 and 8) > Td20(6 and 9) > Td20(7 and 10). The generally observed enhanced thermal stability characteristic of polyimide materials is thus significantly altered by the non-aromatic structure of the triethylene glycol segments as well as the weaker thermal stability of 1,2,3-triazole/1,2,3-triazolium units compared to imide ones.

TGA traces of (top) PTIs 5–7 and (bottom) TPTIs 8–10
3.5 Elaboration of TPTI 8–10 Self-Standing Membranes by Hot Press
TPTIs 8–10 were pressed between two Teflon sheets at temperatures ca. 20 °C above their Tgs. Although ca. 25 µm thick self-standing membranes could be obtained from TPTIs 8 and 9, TPTI 10 yielded brittle membranes that broke into pieces after trying to separate the membrane from the Teflon sheets (Fig. 5). This is probably due to the lower Tg value of 10 although the differences in Mn, chain stiffness and entanglement might also impact the processability and mechanical properties of the different membranes. Nevertheless, even if self-standing membranes could be obtained from TPTIs 8 and 9, their mechanical properties were insufficient to mount them on a permeation cell in order to perform the characterization of their gas separation properties which was the initial motivation of this study.

Optical images of ca. 25 µm thick membranes of TPTIs 8 (left), 9 (middle) and 10 (right)
4 Conclusion
We have developed a series of TPTIs by a three-step synthetic approach combining CuAAC polyaddition, N-alkylation of the resulting PTIs and anion exchange to introduce TFSI counter anions. The structures of neutral PTIs 5–7 were confirmed by 1H NMR spectroscopy while those of TPTIs 8–10 were characterized by SEC as well as 1H, 13C and 19F NMR spectroscopy. However, although having bisphtalimide segments, the presence of triethylene glycol segments and 1,2,3-triazole/1,2,3-triazolium units afforded materials with Tg values and thermal stabilities significantly lower than other class of neutral and ionic polyimides. These moderate properties impeded the preparation of robust polymer membranes required for the study of their gas permeation properties. Further studies regarding the synthesis and gas permeation properties of cross-linked TPTI membranes are currently undergoing.
References
Yuan J, Mecerreyes D, Antonietti M (2013) Poly(ionic Liquid)s: an Update. Prog Polym Sci 38:1009–1036
Obadia MM, Drockenmuller E (2016) Poly(1,2,3-triazolium)s: a new class of functional polymer electrolytes. Chem Commun 52:2433–2450
Shaplov AS, Marcilla R, Mecerreyes D (2015) Recent advances in innovative polymer electrolytes based on poly (ionic liquid)s. Electrochim Acta 175:18–34
Shaplov AS, Ponkratov DO, Vygodskii YS (2016) Poly(ionic liquid)s: synthesis, properties, and application. Polym Sci Ser B 58:73–142
Qian W, Texter J, Yan F (2017) Frontiers in poly(ionic liquid)s: syntheses and applications. Chem Soc Rev 46:1124–1159
Chen M, White BT, Kasprzak CR, Long TE (2018) Advances in phosphonium-based ionic liquids and poly(ionic liquid)s as conductive materials. Eur Polym J 108:28–37
Kohno Y, Saita S, Men Y, Yuan J, Ohno H (2015) Thermoresponsive polyelectrolytes derived from ionic liquids. Polym Chem 6:2163–2178
Zhang W, Kochovski Z, Lu Y, Schmidt BVKJ, Antonietti M, Yuan J (2016) Internal morphology-controllable self-assembly in poly(ionic liquid) nanoparticles. ACS Nano 10:7731–7737
Cordella D, Ouhib F, Aqil A, Defize T, Jerome C, Serghei A, Drockenmuller E, Aissou K, Taton D, Detrembleur C (2017) Fluorinated poly(ionic liquid) diblock copolymers obtained by cobalt-mediated radical polymerization-induced self-assembly. ACS Macro Lett 6:121–126
He H, Rahimi K, Zhong M, Mourran A, Luebke DR, Nulwala HB, Moeller M, Matyjaszewski K (2017) Cubosomes from hierarchical self-assembly of poly(ionic liquid) block copolymers. Nat Commun 8:14057
Choi J-H, Xie W, Gu Y, Frisbie CD, Lodge TP (2015) Single ion conducting, polymerized ionic liquid triblock copolymer films: high capacitance electrolyte gates for n-type transistors. ACS Appl Mater Interfaces 7:7294–7302
Lee C-P, Ho K-C (2018) Poly(ionic liquid)s for dye-sensitized solar cells: a mini-review. Eur Polym J 108:420–428
Ran J, Wu L, Varcoe JR, Ong AL, Poynton SD, Xu T (2012) Development of imidazolium-type alkaline anion exchange membranes for fuel cell application. J Membr Sci 415–416:242–249
Liu L, He S, Zhang S, Zhang M, Guiver MD, Li N (2016) 1,2,3-triazolium-based poly(2,6-dimethyl phenylene oxide) copolymers as anion exchange membranes. ACS Appl Mater Interfaces 8:4651–4660
Yin K, Zhang Z, Yang L, Hirano S-I (2014) An imidazolium based polymerized ionic liquid via novel synthetic strategy as polymer electrolytes for lithium ion batteries. J Power Sour 258:150–154
Zhang P, Li M, Yang B, Fang Y, Jiang X, Veith GM, Sun X-G, Dai S (2015) Polymerized ionic networks with high charge density: quasi-solid electrolytes in lithium-metal batteries. Adv Mater 27:8088–8094
Tiruye GA, Muñoz-Torrero D, Palma J, Anderson M, Marcilla R (2015) All-solid state supercapacitors operating at 3.5 V by using ionic liquid based polymer electrolytes. J Power Sour 279:472–480
Hernandez G, Isik M, Mantione D, Pendashteh A, Navalpotro P, Dejarav S, Marcilla R, Mecerreyes D (2017) Redox active poly(ionic liquid)s as active materials for energy storage applications. J Mater Chem A 5:16231
Margaretta E, Fahs GB, Inglefield DL, Jangu C, Wang D, Heflin JR, Moore RB, Long TE (2016) Imidazolium-containing ABA triblock copolymers as electroactive devices. ACS App Mater Interfaces 8:1280–1288
Lin H, Gong J, Eder M, Schuetz R, Peng H, Dunlop JWC, Yuan J (2017) Programmable actuation of porous poly(ionic liquid) membranes by aligned carbon nanotubes. Adv Mater Interfaces 4:1600768
Guterman R, Ambrogi M, Yuan J (2016) Harnessing poly(ionic liquid)s for sensing applications. Macromol Rapid Commun 37:1106–1115
Shaplov AS, Ponkratov DO, Aubert P-H, Lozinskaya EI, Plesse C, Maziz A, Vlasov PS, Vidal F, Vygodskii YS (2014) Truly solid state electrochromic devices constructed from polymeric ionic liquids as solid electrolytes and electrodes formulated by vapor phase polymerization of 3,4-ethylenedioxythiophene. Polymer 55:3385–3396
Zulfiqar S, Sarwar MI, Mecerreyes D (2015) Polymeric ionic liquids for CO2 capture and separation: potential, progress and challenges. Polym Chem 6:6435–6451
Dai Z, Noble RD, Gin DL, Zhang X, Deng L (2016) combination of ionic liquids with membrane technology: a new approach for CO2 separation. J Membr Sci 497:1–20
Munoz-Bonilla A, Fernandez-Garcia M (2018) Poly(ionic liquid)s antimicrobial materials. Eur Polym J 105:135–149
Lambert R, Coupillaud P, Wirotius A-L, Vignolle J, Taton D (2016) Imidazolium-based poly(ionic liquid)s featuring acetate counter anions: thermally latent and recyclable precursors of polymersupported N-heterocyclic carbenes for organocatalysis. Macromol Rapid Commun 37:1143–1149
Liu Y, Cheng W, Zhang Y, Sun J, Zhang S (2017) Controllable preparation of phosphonium-based polymeric ionic liquids as highly selective nanocatalysts for the chemical conversion of CO2 with epoxides. Green Chem 19:2184–2193
Arslan M, Tasdelen MA (2019) Click chemistry in macromolecular design: complex architectures from functional polymers. Chem Afr 2:195–214
Sood R, Zhang B, Serghei A, Bernard J, Drockenmuller E (2015) Triethylene glycol-based poly(1,2,3-triazolium acrylate)s with enhanced ionic conductivity. Polym Chem 6:3521–3525
Tejero R, Arbe A, Fernandez-García M, Lopez D (2015) Nanostructuration by self-assembly in N-Alkyl thiazolium and triazolium side-chain polymethacrylates. Macromolecules 48:7180–7193
Kallel Elloumi A, Abdelhedi Miladi I, Serghei A, Taton D, Aissou K, Ben Romdhane H, Drockenmuller E (2018) Partially biosourced Poly(1,2,3-triazolium)-based diblock copolymers derived from levulinic acid. Macromolecules 51:5820–5830
Adzima BJ, Taylor SC, He H, Luecbke DR, Martyjaszewski K, Nulwala HB (2014) Vinyl-triazolium monomers: versatile and new class of radically polymerizable ionic monomers. J Polym Sci, Part A: Polym Chem 52:417–423
Obadia MM, Colliat-Dangus G, Debuigne A, Serghei A, Detrembleur C, Drockenmuller E (2015) Poly(vinyl ester 1,2,3-triazolium)s: a new member of the poly(ionic liquid)s family. Chem Commun 51:3332–3335
Jourdain A, Serghei A, Drockenmuller E (2016) Enhanced ionic conductivity of a 1,2,3-triazolium-based poly(siloxane ionic liquid) homopolymer. ACS Macro Lett 5:1283–1286
Abdelhedi-Miladi I, Obadia MM, Allaoua I, Serghei A, Ben Romdhane H, Drockenmuller E (2014) 1,2,3-triazolium-based poly(ionic liquid)s obtained through click chemistry polyaddition. Macromol Chem Phys 215:2229–2236
Colliat-Dangus G, Obadia MM, Vygodskii YS, Serghei A, Shaplov AS, Drockenmuller E (2015) Unconventional poly(ionic liquid)s combining motionless main chain 1,2,3-triazolium cations and high ionic conductivity. Polym Chem 6:4299–4308
Flachard D, Serghei A, Fumagalli M, Drockenmuller E (2019) Main-chain poly(1,2,3-triazolium hydroxide)s obtained through AA + BB click polyaddition as anion exchange membranes. Polym Int 68:1591–1598. https://doi.org/10.1002/pi.5865
Wu J, Chen J, Wang J, Liao X, Xie M, Sun R (2015) Synthesis and conductivity of hyperbranched poly(triazolium)s with various end-capping groups. Polym Chem 7:633–642
Wang C, Li H, Zhang H, Sun R, Song W, Xie M (2018) Enhanced ionic and electronic conductivity of polyacetylene with dendritic 1,2,3-triazolium-oligo(ethylene glycol) pendants. Macromol Chem Phys 219:1800025
Liaw D-J, Wang K-L, Huang Y-C, Lee K-R, Lai J-Y, Ha C-S (2012) Advanced polyimide materials: syntheses, physical properties and applications. Prog Polym Sci 37:907–974
Favvas EP, Katsaros FK, Papageorgiou SK, Sapalidis AA, Mitropoulos AC (2017) A review of the latest development of polyimide based membranes for CO2 separations. React Funct Polym 120:104–130
Kammakakam I, Yoon HW, Nam S, Park HB, Kim T-H (2015) Novel piperazinium-mediated crosslinked polyimide membranes for high performance CO2 separation. J Membr Sci 487:90–98
Kammakakam I, Nam S, Kim T-H (2016) PEG-imidazolium-functionalized 6FDA-durene polyimide as a novel polymeric membrane for enhanced CO2 separation. RSC Adv 6:31083
Gye B, Kammakakam I, You H, Nam S, Kim T-H (2017) PEG-imidazolium-incorporated polyimides as high-performance CO2-selective polymer membranes: the effects of PEG-imidazolium content. Sep Purif Technol 179:283–290
Shaplov AS, Morozova SM, Lozinskaya EI, Valsov PS, Gouveia ASL, Tomé LC, Marrucho IM, Vygodskii YS (2016) Turning into poly(ionic liquid)s as a tool for polyimide modification: synthesis, characterization and CO2 separation properties. Polym Chem 7:580–591
Hajipour AR, Abrishami F (2012) Synthesis and characterization of novel polyimides containing triazoles units in the main chain by click chemistry. J Appl Polym Sci 124:1757–1763
Goswami LN, Houston ZH, Sarma SJ, Jalistgi SS, Hawthorne MF (2013) Efficient synthesis of diverse heterobifunctionalized clickable oligo(ethylene glycol) linkers: potential applications in bioconjugation and targeted drug delivery. Org Biomol Chem 11:1116–1126
Binauld S, Fleury E, Drockenmuller E (2010) Solving the loss of orthogonality during the polyaddition of α-azide-ω-alkyne monomers catalyzed by Cu(PPh3)3Br: application to the synthesis of high-molar mass polytriazoles. J Polym Sci Part A 48:2470–2476
Acknowledgements
The authors gratefully acknowledge the financial support from the Tunisian Ministry of High Education and Scientific Research and IDEXLYON.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Anaya, O., Haddane, A., Drockenmuller, E. et al. Poly(1,2,3-triazolium imide)s Obtained Through AA + BB Click Polyaddition. Chemistry Africa 2, 713–721 (2019). https://doi.org/10.1007/s42250-019-00090-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42250-019-00090-x