
Abstract
Finger millet blast caused by Pyricularia oryzae is the most globally important disease of finger millet. The disease ravages cultivated finger millet, posing a threat to one of the world’s most important, climate-resilient, and nutrient-rich cereal crops, which serves as an important source of food security. Both pathogenic and mutualistic fungi colonize plant tissues and compete for the same niche, subsequently influencing the disease pathogenesis. We analyzed the natural dynamics of the fungal community associated with finger millet blast. Fungal culture was performed on blast-diseased finger millet tissues collected from different agro-ecological regions in Kenya. Fifty-five isolates were obtained from finger, leaf and neck tissues with blast symptoms and characterized based on morphology and DNA sequencing of the internal transcribed spacer (ITS) and 28S rDNA regions. The identified fungal genera corresponded to Exserohilum, Sarocladium, Fusarium, Epicoccum, Cochliobolus, Curvularia, Bipolaris, Penicillium, Setosphaeria, Phoma, and Alternaria, in addition to two unnamed fungal species. Many fungal pathogens of other crops that are not inherently expected to be present on finger millet were encountered, including Penicillium citrinum. Multi-gene phylogenetic studies using concatenated sequences for the ITS and 28SrDNA regions were analyzed using IQ-tree on maximum likelihood basis and aligned using MAFFT to generate a tree with two distinct groups, which were further segregated into several subgroups. The outcome of this study provides useful information on the type of fungal community associated with finger millet blast, which may open avenues for understanding and protecting against finger millet blast disease in Kenya.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Finger millet blast disease, caused by the ascomycetous fungus Pyricularia oryzae (syn. Magnaporthe oryzae), is the most devastating disease of finger millet and threatens global finger millet production. The disease typically causes grain yield losses of 30–50% but can cause 100% yield loss during endemic seasons (Mbinda and Masaki 2020). Apart from finger millet, P. oryzae also infects other crops of the Poaceae family such as rice, wheat and oats and their wild relatives. However, pathogenicity experiments have demonstrated that P. oryzae strains are host-specific, although cross-specificity has been established on a limited range of hosts (Klaubauf et al. 2014; Longya et al. 2020). The fungal infection occurs in the fingers, leaves and neck, with the first symptoms of the disease being small gray or brownish dots on the leaves that transition into diamond-shaped lesions with a white or grayish center after 2–3 days of infection. Leaf infection results in severely blasted leaves that eventually die off, while neck and panicle infections significantly reduce finger length, seed weight, number of seeds per finger, and total grain yield (Manyasa et al. 2019).
Finger millet (Eleusine coracana) is a staple food for many resource-poor farming households in the tropical semi-arid regions of southern Asia and sub-Saharan Africa because of its ability to grow in poor soils with minimal farm inputs. In addition, its superb nutritional quality, excellent storage characteristics and climate resilience make finger millet an excellent crop for use as a staple food and famine reservoir (Gupta et al. 2017). Sadly, the incidence of blast disease has been on the rise due to the ability of the fungus to cross-infect other graminaceous species, climate change and an open free-trade market (Langner et al. 2018). Blast disease management has incorporated the use of approaches such as planting of resistant cultivars, chemical control, crop residue destruction and even biological control, but with limited success (Imam et al. 2015). This is due to the breakdown of pathogen resistance after some years as a result of the rapid and frequent genetic mutation of the pathogen to escape plant immunity, combined with the high cost of fungicides for already resource-poor farmers (Yadav et al. 2019). Understanding the structure and genetic diversity of finger millet blast fungus populations is vital for the development of durable, resistant cultivars that are regionally acclimatized, which would contribute significantly to the control and management of the disease (Gladieux et al. 2018).
Plant tissues are colonized by highly diverse microorganisms whose pathogenesis may be suppressed or aided by the presence of other microbes. Such microbial populations seem to be ubiquitous among plant species and are thought to coevolve within the plant in a mutually dependent manner (Card et al. 2016). Their interactions are not clearly understood owing to the complex web of relationships found within plant tissues, including differences between types of microbial communities and plant genotypes. It is therefore essential to understand their relationships and behavior during disease outbreaks, as they share the same biological niche as the pathogenic microbe in question. Discerning which microbial species are present, how and when they occur, and the crucial roles that they play during these interactions is key (Busby et al. 2015). If this approach is to be considered, it would be necessary to thoroughly understand their behavior when the pathogen appears and when the disease occurs (Latz et al. 2018). This novel microbiome perspective to plant–microbe interactions offers a more integrated strategy to plant disease control and would open up new avenues for combating plant diseases. Moreover, a comprehensive understanding of the roles of these fungi would provide significant progress towards the deployment of improved management strategies for plant diseases, including finger millet blast.
The use of morphological characteristics is a significant component of identifying fungal species. The discovery of molecular tools has, however, transformed fungal taxonomy. These tools provide highly discriminating information with good reproducibility and are relatively abundant. Most fungal species have a high degree of similarity based on their morphological characteristics, often causing misidentification. Accurate identification of fungal species is extremely critical for crop disease management (Kusai et al. 2016). The internal transcribed spacer (ITS) and 28S rDNA regions are frequently used as phylogenetic markers for taxonomic analysis of fungi (Chen et al. 2021). Considering the lack of information on the identities of fungal species associated with finger millet blast pathogen in Kenya, this study sought to use both morphological and molecular approaches to assess the overall fungal composition coexisting with P. oryzae in plants with finger millet blast disease.
Materials and methods
Isolation of fungi from finger millet plants
Sampling was conducted during the planting season from May–June 2018 and in February and June 2019 in five finger-millet-growing counties of Kenya (Machakos, Makueni, Kisii, Busia and Bungoma). A purposive sampling technique was employed to select finger millet farms, and at least six plant tissues infected with finger millet blast were randomly sampled from each farm surveyed per county. A total of 290 finger millet plant tissues with typical blast symptoms (diamond-shaped lesions on the neck and leaves and reddish brown to rusty brown color on the inflorescence, Fig. 1) were collected and placed in clean sample collection bags. Approximately 4-mm segments of each plant tissue were aseptically excised, rinsed with tap water to reduce surface contaminants and sterilized with 3% sodium hypochlorite followed by three consecutive rinses with sterile distilled water to remove any other contaminants and excess sodium hypochlorite. The samples were dried on sterile blotting paper and cultured on oatmeal agar (OMA) media supplemented with 50 mg/L of chloramphenicol at 27 °C for 7–10 days. Emerging colonies were streaked on 4% water agar (WA) with a sterile 10-μl loop. After 48 h, the streaks were observed under a stereomicroscope until the most extended and germinated single conidium was observed. A single germinated conidium was then removed using a scalpel blade by cutting the agar closely around the conidium. The agar piece containing the spore was lifted and transferred onto OMA supplemented with chloramphenicol and incubated with a 12/12 h light/dark photoperiod at 27 °C to induce growth and sporulation. All of the resultant pure cultures were maintained in 30% glycerol containing half strength potato dextrose broth at -20 °C.

Typical blast disease symptoms on finger millet plant in Kenya a Rusty brown necrosis on finger and stem while b Brownish spots on severely infected leaf. c Healthy finger millet crop that is yet to mature
Morphological identification
The fungal isolates’ macro- and micro-morphological characteristics were observed. Each pure fungal isolate was cultured on OMA amended with 50 mg/L chloramphenicol and incubated at 27 °C for five days. The mycelial characteristics on the plates, such as colony color, margination and elevation, were then recorded. Conidia production was induced by scraping the surface of the fungal mycelium with a sterile spreader, and the isolates were incubated in the dark for a further 5 days. Conidia characteristics such as conidia shape and hyphae were observed under a Leica ICC 50E compound microscope at 100 × magnification.
Molecular characterization based on ITS and 28S sequencing
Fungal genomic DNA was extracted using a modified CTAB method (Panda et al. 2017). PCR amplification was carried out using two primer sets, ITS1/ITS4 (ITS1: 5´-TCCGTAGGTGAACCTGCGG-3´ and ITS4: 5´- TCCTCCGCTTATTGATATGC-3´) and NL1/4 (NL1: 5’GCATATCAATAAGCGGAGGAAAAG-3’ and NL4: 5’GGTCCGTGTTTCAAGACGG 3’), for the fungal ITS and 28S rDNA regions, respectively (White et al. 1990). PCR conditions were set as follows: initial denaturation at 95 °C for 5 min followed by 35 cycles of denaturation, annealing and extension at 95 °C for 30 s; 55 °C for 15 s (for ITS1) or 53 °C for 40 s (for 28S rDNA); 72 °C for 30 s (for ITS1) or 72 °C for 1 min (for 28S rDNA); and a final extension at 72 °C for 5 min (for ITS1) or 7 min (for 28S rDNA). The PCR products were resolved by 1% agarose gel electrophoresis. ITS amplicons were sequenced by Macrogen Europe B.V. using an Applied Biosystems 3730xl DNA Analyzer and 28SrDNA amplicons by Inqaba Biotec using an ABI 3500XL Genetic Analyzer platform. Both the forward and reverse ITS and 28S rDNA sequence data were assembled and manually edited using BioEdit sequence alignment editor software. The taxonomic relationships between the isolates were ascertained with standard nucleotide BLAST searches against the nucleotide database in the UNITE fungal database (http://www.unite.ee), NCBI RefSeq (http://www.ncbi.nlm.gov/refseq/targetedloci/) and GenBank (http://www.ncbi.nih.gov/blast). The fungal ITS and 28S rDNA gene sequences obtained in this study were deposited in GenBank under accession numbers MW151763-MW151817 and MW644907-MW644961, respectively.
Community structure analysis
The sequences obtained from the ITS and 28S rDNA primers were analyzed both separately and in concatenated form to confirm their phylogenetic placement, using Methanoculleus thermophillus as the outgroup. Two phylogenetic trees were constructed using a neighbor-joining method with Molecular Evolutionary Genetic Analysis (MEGA X) software, while another tree was constructed using concatenated sequences for both ITS and 28SrDNA with MAFFT alignment software version 7.407. To evaluate the best DNA model substitution pattern, a Bayesian information criterion score (BIC) was considered, which revealed a K2 + G + 1 model as the best fit (Kumar et al. 2018). All ambiguous positions were removed for each sequence pair (pairwise deletion option). The evolutionary history was inferred using the Maximum Likelihood method. The tree was drawn to scale with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Maximum Composite Likelihood method and are in the units of the number of base substitutions per site (Tamura et al. 2004). The differences in composition bias among sequences were considered in evolutionary comparisons (Tamura and Kumar 2002). The concatenated sequences were analyzed using MAFFT alignment v.7.407 (Katoh and Standley 2013); alignment curation was performed via the Block Mapping and Gathering with Entropy (BMGE) package v.1.12 (Criscuolo and Gribaldo 2010); the IQ-Tree v2.03 (Nguyen et al. 2015) algorithm was used to infer the tree by maximum likelihood and visualized in Figtree v.1.4.4.
Data analysis
SPSS version 25 (SPSS Inc., Chicago, IL, USA) was used to determine the percentages for the variables of interest based on the descriptive data. The chi-squared test (α = 0.05) was used to compare the proportion of fungal compositions from different plant tissues, namely leaf, finger and neck. Phylogenetic analysis was performed using MEGA X. and MAFFT alignment v.7.407.
Results
Morphological characteristics
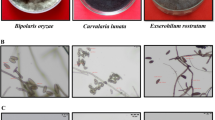
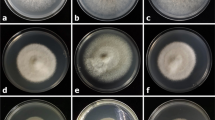
A total of 55 pure fungal isolates were recovered from 290 plant tissues sampled from Machakos (5), Makueni (11), Kisii (13), Busia (16) and Bungoma (10) counties, consisting of different blast-diseased tissues (finger, leaf and neck) of finger millet (Table 1). The percentages of fungal isolates obtained from the different host tissues were 45.4%, 38.2% and 14.4% for the finger, leaf and neck tissues, respectively, showing typical blast symptoms. The macroscopic characteristics (colony color, margin and elevation) and microscopic characteristics (conidia shape) of the 55 fungal isolates associated with finger millet blast disease were examined (Table 1). These strains were purified based on their distinct morphology on OMA and PDA media and differentiated based on their macroscopic and microscopic characteristics. At the macroscopic scale, the isolates cultured on OMA media showed mycelia with varied features in terms of appearance and color. Five major mycelial colors were observed, gray (27), white (6), pinkish/purplish (8), grayish white (7) and black/grayish black (5), while 2 were brownish (Table 1, Fig. 2). The number of mycelial growth patterns, characterized by colony color, elevation and margin, also varied across the blast-infected plant tissues. The highest number of mycelial growth pattern combinations was observed in the finger tissues (46%), followed by the leaf (38%) and neck tissues (16%), as shown in Fig. 3. Hyphal characteristics were differentiated as either septate or non-septate. Forty fungal isolates had septate hyphae while 15 were aseptate. None of the fungal isolates showed a pseudo-hyphae type of septation in their hyphal structures. The conidial shapes observed under the microscope were differentiated into four main patterns: globose, elliptical, irregular and cylindrical (Fig. 4). Most of the conidia shapes were either cylindrical (25.5%) or globose (23.5%), while 18.2% had an irregular pattern, 16.4% had an elliptical shape and 16.4% had a round shape (Table 1, Fig. 5). All of the conidia patterns lacked observable appendages and had smooth surfaces. The conidia color ranged from pale yellow to deep pigmentation, but none were hyaline (Fig. 4). Some of the conidia were either simple (lacking septa; Fig. 4a, e) or septate (having cross walls; Fig. 4b, c, f). These cross walls were either transverse (Fig. 4b), pseudo septate (Fig. 4 c, d) or longitudinal (Fig. 4f). Isolates having similar conidial features and morphology were grouped in the same genus. However, due to the similarity of fungal morphological characteristics and the risk of misidentification, molecular identification was pursued for confirmation of fungal identities.

Morphological variation in selected fungal mycelia on OMA medium, showing differences in colors and mycelial growth patterns. a: grey, entire, raised; b: white, rhizoid, raised; c: pinkish, undulate, raised; d: greyish white, undulate, flat; e: black, entire, raised (a: is isolate BU229-G, b: BU2-42, c: BU29-46, d: F1-M5, e: BG17-58)

The mycelial growth patterns on different plant parts. The proportion of mycelial growth pattern combinations exhibited by different fungal genera isolated from different plant parts sampled from five finger millet growing counties in Kenya

Variation in conidia shape under a light microscope. a, globose; b, elliptical; c, irregular; d, cylindrical; e, round and f, cylindrical with longitudinal septate. (a: is isolate F17-2 b: isolate MK49-20; c: isolate KIS47-32; d: isolate BU85-47; e: isolate KIS53-27; f: isolate KIS2-M24)

Phylogenetic tree for the concatenated sequences of ITS and 28SrDNA regions
Molecular analysis determined by the ITS and 28S rDNA regions
Genomic DNA was extracted from 55 samples of fungal strains cultured from blast-infected finger millet tissues for molecular analysis. The DNA was successfully amplified and sequenced. The sizes of the 28S rDNA and ITS regions ranged from 557–616 and 484–601 bp, respectively, after sequence editing (Supplementary material 1). After a nucleotide BLAST search, the ITS and 28S rDNA sequences exhibited high percentage similarities of between 99 and 100% with the UNITE and GenBank databases. The identities of the isolates based on the ITS and 28S rDNA sequences are as shown in Table 1. The isolates were assigned to three classes of Phylum Ascomycota, namely Eurotiomycetes, Dothiodeomycetes and Sordariomycetes, and further separated into 10 genera and two unnamed fungi. For the ITS sequences, the isolates are related to Exserohilum rostratum (3), Sarocladium sp. (2), Fusarium equiseti (1), Fusarium sp. (1), Fusarium annulatum (1), Fusarium oxysporum (1), Fusarium incarnatum (1), Fusarium chlamydosporum (1), Epicoccum sp. (3), Epicoccum sorghinum (4), Cochliobolus sp. (1), Curvularia lunata (12), Curvularia mebaldsii (2), Curvularia panici (1), Curvularia trifolii (4), Curvularia clavata (2), Curvularia petersonii (1), Curvularia hominis (1), Curvularia akaiiensiis (1), Bipolaris bicolor (2), Bipolaris cynodontis (2), Bipolaris simmondsii (1), Bipolaris sp. (1), Penicillium citrinum (1), Setosphaeria rostrata (1), Alternaria alternata (1), and Alternaria sp. (1). The 28S rRNA sequences are related to Exserohilum rostratum (3), Sarocladium kiliense (2), Sarocladium sp. (1), Fusarium equiseti (1), Fusarium pseudocircinatum (1), Fusarium annulatum (1), Fusarium oxysporum (1), Fusarium chlamydosporum (1), Epicoccum nigrum (2), Epicoccum proteae (1), Epicoccum sorghinum (4), Cochliobolus kusanoi (1), Cochliobolus hawaiiensis (1), Curvularia lunata (16), Curvularia akaiiensis (1), Curvularia hawaiiensis (1), Curvularia trifolii (2), Curvularia geniculata (2), Curvularia coatesiae (2), Bipolaris woodii (4), Bipolaris cynodontis (2), Phoma (1), Penicillium citrinum (1), Setosphaeria rostrata (1), Alternaria alternata (1), and Alternaria alstroemeriae (1).
Three phylogenetic trees were constructed and the relationships between all isolates evaluated for ITS, 28SrDNA, and their concatenated sequences, as shown in Supplementary material 2 and Fig. 5. Each dataset consisted of the 55 fungal isolates recovered after culture. A further 33 and 19 sequences for ITS and 28S rDNA, respectively, including the outgroup species, were retrieved from GenBank. Several clustering methods including neighbor-joining, maximum-evolution, and unweighted pair group method with arithmetic mean (UPGMA) generated nearly identical topologies in MEGA. The optimal trees generated by the individual ITS and 28S rDNA sequences as well as their concatenated sequences together with their relatives yielded similar topologies. The trees were divided into two major groups based on the three classes of Phylum Ascomycota. The first category consisted of Fusarium species, Sarocladium and Penicillium of classes Sordariomycetes and Eurotiomycetes (Fig. 5, Supplementary material 2). Notably, isolate BU188-37 was the only isolate in the three trees that clustered with Penicilium citrinum, a member of the Aspergillaceae family, which are known for secondary metabolite production. The second category was assigned to members belonging to class Dothiomycetes including Curvularia species, Exserohilum, Bipolaris, Cochliobolus, Alternaria, Epicoccum and Dothidiomycete sp. (Supplementary material 2). Setosphaeria (synonym for Exserohilum) and Phoma formed an ambiguous branch and were therefore removed from the tree. All trees displayed a high species richness of genus Curvularia, since a majority of the isolates clustered with a number of its species. Methanoculleus thermophilus was used as an outgroup to root the tree. BLASTN and phylogenetic analyses both indicated that a total of 11 genera for 28SrDNA and 10 together with two unnamed fungi for ITS were generated from this study. Although the total numbers of genera found were the same for both genes (Fig. 6a, b), the total species count was slightly higher for 28S rDNA than for ITS (Table 1).

Proportional distribution of different fungal species obtained using a ITS and b 28S rDNA primers from finger millet samples
Distribution of different fungal species on different blast-infected finger millet tissues
The fungal composition varied among the different blast-infected finger millet tissues, as shown in Fig. 6. The genus Curvularia made up the highest proportion of the species obtained from all of the sampled plant tissues compared to the other genera, representing 44% of the composition of fungal isolates followed by Epicoccum (13%) and Fusarium (13%). Notably, genera Curvularia and Epicoccum were represented by species in all of the sampled blast-infected finger millet tissues (finger, leaf and neck). The genera Sarocladium, Bipolaris, unassigned fungal strain and Fusarium each had at least one species obtained from either finger and neck or finger and leaf but not from all three blast infected plant tissues (Table 1). The rest of the genera, namely Setosphaeria, Cochliobolus, Exserohilum, Penicillium and Alternaria each had only one species from the sampled blast infected plant tissues (Table 1). The results of the chi-square test on fungal compositions in the finger, leaf and neck tissues indicated that they were not statistically significant at p = 0.05, χ2 = 25.5, df = 24. The fungal communities recovered from the five sampling sites did not show a distinct distribution, and the fungal taxa were randomly distributed across the different sites. Fungal richness was mainly observed in the western agro-ecological zone made up of Busia, Bungoma and Kisii, where a total of 39 fungal species were recovered, compared to the eastern agro-ecological zone (Machakos and Makueni) with only 16 fungal species.
Discussion
This is the first report of fungal species associated with the finger millet blast disease pathogen P. oryzae that combines multigene sequence phylogenetic analysis with a morphological approach. Our study identified twenty-six species based on the 28SrRNA gene and twenty-eight species plus two unnamed fungal species with the ITS gene as fungi coexisting with P. oryzae during finger millet blast infection. The genera identified included Cochliobolus (C. kusanoi, C. hawaiiensis, Cochliobolus sp.); Setosphaeria (S. rostrata); Sarocladium (S. kiliensis, Sarocladium sp.); Curvularia (C. hawaiiensis, C. geniculatus, C. lunata, C. coatesiae, C. hominis, C. petersonii, C. panici, C. clavata, C. trifolii, C. mebaldsii); Bipolaris (B. woodii, B. cynodontis, B. bicolor, B. simmondsii, Bipolaris sp.); Fusarium (F. annulatum, F. pseudocircinatum, F. chlamydosporum, F. oxysporum, F. equiseti, F. incarnatum, Fusarium sp.); Alternaria (A. alstroemeriae, A. alternate, Alternaria sp.); Epicoccum (E. nigrum, E. proteae, E. sorghinum); Exserohilum (E. rostratum); Penicillium (P. citrinum) and Phoma. Overall, these fungal species were grouped into three classes of Phylum Ascomycota—Eurotiomycetes, Dothiodeomycetes and Sordariomycetes—for both genes.
The results from this work show that Curvularia species were the most frequently isolated in all the blast-infected tissues. P. oryzae is known to be a weak saprophyte (Rajashekara et al. 2016). It is therefore challenging to detach the blast fungus from these two genera using conventional host tissue transplant isolation techniques. Growth of the blast fungus on media was likely eclipsed by the genus, which is why most of the fungal isolates were from the genus Curvularia at the expense of P. oryzae. Accurate identification of P. oryzae causing finger millet blast can therefore easily be clouded by the necrotic spots exhibited by Curvularia species on blast-infected finger millet samples. However, in culture, the visual characteristics of the two can be easily differentiated. The mycelium for Curvularia tends to have a greyish black appearance on oatmeal agar media with a black underside (Kusai et al. 2016), while that of blast fungus displays features ranging from white to greyish/black (Gowrisri et al. 2019; Longya et al. 2020). Saprophytic fungi such as Alternaria and Curvularia species are of great interest because they primarily infect necrotic lesion centers from behind the advancing front of parasitic mycelium (Rajashekara et al. 2016). Our findings further identified Curvularia lunata as the most common compared to other Curvularia species. This genus is made up of more than 40 species, which include saprophytes, endophytes and pathogens. This pathogenic group has been implicated in heavy losses among cereals and vegetables because they form necrotic spots on the leaves of these plants (Kusai et al. 2016; Kee et al. 2020; Khemmuk et al. 2016; Schoch et al. 2020). From these findings, we questioned the role played by the identified fungal isolates in P. oryzae pathogenesis. Perhaps some of the microbes could be enhancing the aggressiveness of P. oryzae during colonization. For example, Epicoccum nigrum has been identified as a beneficial co-inhabiting fungus in sugarcane due to its secretion of cell-wall-degrading enzymes that facilitate the successful invasion of plant cell walls and ultimately cause disease, especially during the saprophytic phase of the fungal cycle (Kubicek et al. 2014). A comprehensive study is therefore recommended to understand the impacts of some of the identified species on the severity of the blast disease and the likely endophytic co-inhabitants in finger millet.
A. alternata has a wide host range and is known to be prevalent on the surface of finger millet seeds as well as a causative agent for leaf spot and other diseases in several plants (Jain 2020). This fungus has been identified as seed-borne mycoflora in sorghum and foxtail millet together with Curvularia lunata, Aspergillus flavus, and Phoma sp. in Korea (Yago et al. 2011). A. alstroemeriae has also been implicated as the causative agent for black spot on Alstroemeria, a perennial ornamental plant (Yamagishi et al. 2009). This is the first report of its presence in the finger millet plant, a finding that requires further investigation.
Fusarium is a widely distributed genus of pathogens that affects several plants. Finger millet and other millets including proso, pearl and foxtail millets have been identified as hosts to various species of this pathogen. F. equiseti is known to cause ear rot in foxtail millet, while F. oxysporum is very destructive to most millet varieties (Chala et al. 2019). Other than causing diseases, Fusarium species are also known to produce mycotoxins that contaminate the millet, making it unfit for human consumption (Akanmu et al. 2013). F. chlamydosporum has been implicated in mycotoxin contamination of millet, although it tends to be host-selective and region-specific. F. solani has previously been obtained from African millet (Choi et al. 2021). Several other Fusarium species not isolated in this study have also been implicated as pathogens associated with millets. Some of these include F. nygamai, F. compactum, F. fujikuroi, and F. semitectum (Choi et al. 2021).
Bipolaris species have also been recorded as causative agents for a number of diseases in cereals and grasses. B. australiensis, for example, is pathogenic in Chloris and Pennisetum species in Australia, India and Kenya. This species has also been implicated as the causative agent of brown leaf spot in Cynodon spp. and pearl millet as well as leaf spot in betel vine (Piper betle) and date palm from China, India, Pakistan and Iraq (Fang et al. 2007). More recently, it was also established as a pathogen in foxtail millet in Iran (Mirzaee et al. 2020). Other species such as B. oryzae, B. cynodontis, B. sorghicola and B. victoriae have been involved as causal agents of rice brown spot disease in Iran (Nazari et al. 2015). In finger millet, B. setariae has been reported as a pathogen (Jain 2020) but not B. bicolor, B. cynodontis, B. woodii or B. simmondsii. In Kodo millet, Sarocladium oryzae has been implicated as the causal agent for sheath rot, which causes grain discoloration, thus lowering grain quality (Nagaraja et al. 2016). This fungus has also been detected in rice residues and several weed species present in rice fields in India (Yadav and Thrimurty 2006). Cochliobolus lunatus has been detected as the causal agent of rice sheath rot, which causes black kernel disease in India and Bangladesh (Bigirimana et al. 2015). In this study, C. kusanoi, C. hawaiiensis, and Cochliobolus sp. were identified in finger millet.
Exserohilum rostratum is reported in finger millet for the first time in this study. Some Exserohilum species have been reported as pathogenic among the small millets, including E. oryzicola, which has been identified as the causal agent of leaf spot in barnyardgrass in Japan. This species has also been pathogenic in rice, barley, bread wheat and durum wheat, with milder but nonpathogenic foliar symptoms in soybean (Tomioka et al. 2021). E. turcicum is the causal agent for maize northern leaf blight (NLB) and has been reported as a serious pathogen in maize in East Africa and elsewhere (Nwanosike et al. 2015). Isolate BU188-37 clustered with Penicillium citrinum. Although this fungus causes mold in maize and rice (Gowrisri et al. 2019), the genus Penicillium is largely known for producing commercially valuable secondary metabolites such as alkaloids, antibiotics, hormones and mycotoxins (Shahid et al. 2020). The results therefore suggest that isolate BU188-37 could possess valuable secondary metabolites whose potential needs to be exploited.
Although the distribution of isolated fungi varied across the agro-ecological zones, Curvularia, Epicoccum, Bipolaris and Fusarium were the most dominant genera in all four of the agro-ecological zones under study. Moreover, the samples collected from Kisii County showed higher fungal community richness than those from Machakos. The observed variation in fungal composition could be associated with the environmental characteristics of the different agro-ecological zones where the samples were collected. For example, certain areas in eastern Kenya experience a dryer environment as opposed to the wet and humid conditions in western Kenya. In our previous study on the occurrence, distribution and severity of finger millet blast disease in Kenya, we reported that blast disease varied according to ecological characteristics (Odeph et al. 2020). The high humidity in some agro-ecological zones could facilitate the relatively high percentage of finger, leaf and neck infections. A similar observation was reported for Colletotrichum species associated with anthracnose in avocado (Sharma et al. 2017). Our findings further revealed that of the three sampled tissues infected with finger millet blast, the percentage of fungi isolated from fingers was significantly higher (46%) compared to leaves (38%) and neck tissues (16%). P. oryzae is a hemibiotrophic pathogen, and it combines both biotrophic and necrotrophic characteristics. The pathogen attacks all plant tissues by sustaining both lifestyles concurrently (Marcel et al. 2010). The mechanism of pathogenesis is hypothesized to be the same in all plant tissues. However, it is now clear that the fungus has tissue invasion preferences. The ability of P. oryzae to use either hyphopodia or appressoria allows the pathogen to modify its invasion mechanisms to the physiological and nutritional characteristics of the target organ (Tucker et al. 2010). This raises questions as to the degree of similarity between finger, leaf and neck infection approaches. The fungus therefore combines the common and organ-specific components described in the P. oryzae genetic toolbox to achieve its initial infiltration. From our findings, we could not specifically deduce why more fungi species were isolated from finger tissues compared to the leaf and neck.
Conclusions
In summary, findings of this work sheds light on the existence of diverse fungal plant species associated with finger millet blast pathogen in Kenya, which may lay the foundation for future studies related to management of the pathogen. A total of 10 fungal genera and 2 unnamed fungal species are reported. All the isolates recovered belong to the same group as species that are known to be serious plant pathogens. The diverse genetic structure of fungal communities associated with blast disease attests to the probability of their association with P. oryzae during pathogenesis. Future work may include exploiting the potential of isolate BU188-37 which clustered with P. citrinum: a member of one of the highly prolific secondary metabolites producing fungal genera.
Availability of data and material
The datasets used and/or analyzed during the current study are available from the corresponding author on request.
References
Akanmu AO, Abiala MA, Akanmu AM, Adedeji AD, Mudiaga PM, Odebode AC (2013) Plant extracts abated pathogenic Fusarium species of millet seedlings. Arch Phytopathol Plant Prot 46(10):1189–1205. https://doi.org/10.1080/03235408.2013.763613
Bigirimana VDP, Hua GK, Nyamangyoku OI, Höfte M (2015) Rice sheath rot: an emerging ubiquitous destructive disease complex. Front Plant Sci 6:1066. https://doi.org/10.3389/fpls.2015.01066
Busby PE, Ridout M, Newcombe G (2015) Fungal endophytes: modifiers of plant disease. Plant Mol Biol 90(6):645–655. https://doi.org/10.1007/s11103-015-0412-0
Card S, Johnson L, Teasdale S, Caradus J (2016) Deciphering endophyte behaviour: the link between endophyte biology and efficacious biological control agents. FEMS Microbiol Eco 92(8):1–19. https://doi.org/10.1093/femsec/fiw114
Chala A, Degefu T, Brurberg MB (2019) Phylogenetically diverse Fusarium species associated with sorghum (Sorghum bicolor L. Moench) and finger millet (Eleusine coracana L. Garten) grains from Ethiopia. Diversity 11(6):93. https://doi.org/10.3390/d11060093
Chen LH, Yan W, Wang T, Wang Y, Liu J, Yu Z (2021) Analysis of small and large subunit rDNA introns from several ectomycorrhizal fungi species. PloS One 16(3):e0245714. https://doi.org/10.1371/journal.pone.0245714
Choi JH, Nah JY, Lee MJ, Jang JY, Lee T, Kim J (2021) Fusarium diversity and mycotoxin occurrence in proso millet in Korea. LWT, 141, 110964. https://doi.org/10.1016/j.lwt.2021.110964
Criscuolo A, Gribaldo S (2010) BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol Biol 10(210):1–21. https://doi.org/10.1186/1471-2148-10-210
Fang KF, Huang JB, Hsiang T (2007) First report of brown leaf spot caused by Bipolaris australiensis on Cynodon spp. in China. Plant Pathology 56:349. https://doi.org/10.1111/j.1365-3059.2007.01538.x
Gladieux P, Ravel S, Rieux A, Cros-Arteil S, Adreit H, Milazzo J, Thierry M, Fournier E, Terauchi R, Tharreau D (2018) Coexistence of multiple endemic and pandemic lineages of the rice blast pathogen. MBio 9(2): 1–18. https://doi.org/10.1128/mBio.01806-17.
Gowrisri N, Kamalakannan A, Malathi VG, Rajendran L, Rajesh S (2019) Morphological and molecular characterization of Magnaporthe oryzae B. Couch, inciting agent of rice blast disease. Madras Agric J 106:255–260. https://doi.org/10.29321/MAJ2019.000256
Imam J, Alam S, Mandal NP, Maiti D, Variar M, Shukla P (2015) Molecular diversity and mating type distribution of the rice blast pathogen Magnaporthe oryzae in North-east and Eastern India. Indian J Microbiol 55(1):108–113. https://doi.org/10.1007/s12088-014-0504-6
Jain AK (2020) Seed borne mycoflora of finger millet and their management: A review
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30(4):772–780. https://doi.org/10.1093/molbev/mst010
Klaubauf S, Tharreau D, Fournier E, Groenewald JZ, Crous PW, De Vries RP, Lebrun MH (2014) Resolving the polyphyletic nature of Pyricularia (Pyriculariaceae). Stud Mycol 79:85–120. https://doi.org/10.1016/j.simyco.2014.09.004
Kubicek CP, Starr TL, Glass NL (2014) Plant cell wall–degrading enzymes and their secretion in plant-pathogenic fungi. Annu Rev Phytopathol 52:427–451. https://doi.org/10.1146/annurev-phyto-102313-045831
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol 35(6): 1547–1549. https://doi.org/10.1093/molbev/msy096.
Kusai NA, Azmi MM, Zulkifly S, Yusof MT, Zainudin NA (2016) Morphological and molecular characterization of Curvularia and related species associated with leaf spot disease of rice in Peninsular Malaysia. Rend Fis Acc Lincei 27(2):205–214. https://doi.org/10.1007/s12210-015-0458-6
Langner T, Białas A, Kamoun S (2018) The blast fungus decoded: Genomes in flux. MBio9:e00571–18. https://doi.org/10.1128/mbio.00571-18
Latz MA, Jensen B, Collinge DB, Jørgensen HJ (2018) Endophytic fungi as biocontrol agents: elucidating mechanisms in disease suppression. Plant Ecol Divers 11(5–6):555–567. https://doi.org/10.1080/17550874.2018.1534146
Longya A, Talumphai S, Jantasuriyarat C (2020) Morphological characterization and genetic diversity of rice blast fungus, Pyricularia oryzae, from Thailand using ISSR and SRAP markers. J Fungi 6(38):1–13. https://doi.org/10.3390/jof6010038
Manyasa EO, Tongoona P, Shanahan P, Githiri S, Ojulong H, Njoroge SM (2019) Exploiting Genetic Diversity for Blast Disease Resistance Sources in Finger Millet (Eleusine coracana). Plant Heal Prog 20(3):180–186. https://doi.org/10.1094/PHP-11-18-0068-RS
Marcel S, Sawers R, Oakeley E, Angliker H, Paszkowski U (2010) Tissue-adapted invasion strategies of the rice blast fungus Magnaporthe oryzae. Plant Cell 22(9):3177–3187. https://doi.org/10.1105/tpc.110.078048
Mbinda W, Masaki H (2020) Breeding strategies and challenges in the improvement of blast disease resistance in finger millet. A current review. Front Plant Sci 11. 602882. https://doi.org/10.3389/fpls.2020.602882
Mirzaee MR, Zare R, Nasrabad AA (2020) A new leaf and sheath brown spot of foxtail millet caused by Bipolaris australiensis. Australas Plant Dis Notes 5:19–20. https://doi.org/10.1071/DN10008
Nagaraja A, Kumar B, Jain AK, Sabalpara AN (2016) Emerging diseases: Need for focused research in small millets. J Mycopathol Res 54(1):1–9
Nazari S, Javan-Nikkhah M, Fotouhifar KB., Khosravi V, Alizadeh A (2015) Bipolaris species associated with rice plant: pathogenicity and genetic diversity of Bipolaris oryzae using rep-PCR in Mazandaran province of Iran. J Crop Prot 4(4):497–508. https://jcp.modares.ac.ir/article-3-4588-en.html
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32(1):268–274. https://doi.org/10.1093/molbev/msu300
Nwanosike MRO, Mabagala RB, Kusolwa PM (2015) Effect of northern leaf blight (Exserohilum turcicum) severity on yield of maize (Zea mays L.) in Morogoro, Tanzania. Int J Sci Res 4(9):466–475
Odeph M, Luasi WW, Kavoo A, Mweu C, Ngugi M, Maina F, Nzilani N, Mbinda WM (2020) Occurrence, distribution and severity of finger millet blast caused by Magnaporthe oryzae in Kenya. Afr J Plant Sci 14(4):139–149. https://doi.org/10.5897/AJPS2020.1970
Panda G, Sahu C, Yadav MK, Aravindan S, Umakanta N, Raghu S, Prabhukarthikeyan SR, Lenka S, Tiwari JK, Kar S, Jena M (2017) Morphological and molecular characterization of Magnaporthe oryzae from Chhattisgarh. ORYZA 54(3):330–336. https://doi.org/10.5958/2249-5266.2017.00045.5
Schoch CL, Ciufo S, Domrachev M, Hotton CL, Kannan S, Khovanskaya R, Leipe D, Mcveigh R, O’Neil K, Robbertse B, Sharma S, Soussov V, Sullivan JP, Sun L, Tumer S, Karsch-Mizrachi I (2020) NCBI Taxonomy: A comprehensive update on curation, resources and tools. Database 2020 (2):1-21. https://doi.org/10.1093/database/baaa062
Shahid MG, Nadeem M, Gulzar A, Saleem M, Ghafoor GZ, Hayyat MU, Shahzad L, Arif R, Nelofer R (2020) Novel ergot alkaloids production from Penicillium citrinum employing response surface methodology technique. Toxins 12(7):1–19. https://doi.org/10.3390/toxins12070427
Sharma G, Maymon M, Freeman S (2017) Epidemiology, pathology and identification of Colletotrichum including a novel species associated with avocado (Persea americana) anthracnose in Israel. Sci Rep 7(1):1–16. https://doi.org/10.1038/s41598-017-15946-w
Tamura K, Kumar S (2002) Evolutionary distance estimation under heterogeneous substitution pattern among lineages. Mol Biol Evol 19(10):1727–1736. https://doi.org/10.1093/oxfordjournals.molbev.a003995
Tamura K, Nei M, Kumar S (2004) Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci 101(30):11030–11035. https://doi.org/10.1073/pnas.0404206101
Tomioka K, Asami H, Chiba M, Kobayashi H, Nagata K, Mori S, Yamazaki R, Kawasaki Y, Yasumoto S, Masunaka A, Sekiguchi H (2021). Leaf spot of barnyardgrass caused by Exserohilum oryzicola in Japan and the fungal influence on rice, barley, bread wheat, durum wheat, and soybean. J Gen Plant Pathol 1–6. https://doi.org/10.1007/s10327-021-01009-6
Tucker SL, Besi M, Galhano R, Franceschetti M, Goetz S, Lenhert S, Osbourn A, Sesma A (2010) Common genetic pathways regulate organ-specific infection-related development in the rice blast fungus. Plant Cell 22(3):953–972. https://doi.org/10.1105/tpc.109.066340
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: MA Innis, DH Gelfaud, JJ Sninsky, TJ White (eds) PCR protocols. A guide to methods and applications. Academic Press, San Diego, CA, USA, 1:315–322
Yadav MK, Aravindan S, Raghu S, Prabhukarthikeyan SR, Keerthana U, Ngangkham U, Pramesh D, Banerjee A, Adak T, Kar MK, Parameswaran C (2019) Assessment of genetic diversity and population structure of Magnaporthe oryzae causing rice blast disease using SSR markers. Physiol Mol Plant P 2019:157–165. https://doi.org/10.1016/j.pmpp.2019.02.004
Gupta SM, Arora S, Mirza N, Pande A, Lata C, Puranik S et al (2017) Finger millet: a “Certain” crop for an “uncertain” future and a solution to food insecurity and hidden hunger under stressful environments. Front Plant Sci 8:643. https://doi.org/10.3389/fpls.2017.00643
Prakash G, Srinivasa N, Sankar SM, Singh SP, Satyavathi CT (2016) Standardization of pearl millet blast (Magnaporthe grisea) phenotyping under artificial conditions. Ann Agric Res Series 37:1–6
Khemmuk W, Shivas RG, Henry RJ, Geering AD (2016) Fungi associated with foliar diseases of wild and cultivated rice (Oryza spp.) in northern Queensland. Australas Plant Pathol 45(3):297–308. https://doi.org/10.1007/s13313-016-0418-3
Yadav VK, Thrimurty VS (2006) Weeds as alternate host for Sarocladium oryzae. Ann Plant Prot Sci 14(2):514–515
Yago JI, Roh JH, Bae SD, Yoon YN, Kim HJ, Nam MH (2011) The effect of seed-borne mycoflora from sorghum and foxtail millet seeds on germination and disease transmission. Mycobiology 39(3):206–218. https://doi.org/10.5941/MYCO.2011.39.3.206
Yamagishi N, Nishikawa J, Oshima Y, Eguchi N (2009) Black spot disease of alstroemeria caused by Alternaria alstroemeriae in Japan. J Gen Plant Pathol 75(5):401–403. https://doi.org/10.1007/s10327-009-0182-0
Acknowledgements
This research work was financed by the National Research Fund, Kenya. Special thanks to Steven Runo of Department of Biochemistry, Microbiology and Biotechnology, Kenyatta University for assisting in microscopy work. Authors also thank the Institute of Biotechnology Research, Jomo Kenyatta University of Agriculture and Technology for providing facilities for the research.
Funding
This work was supported financially by a grant from the National Research Fund, Kenya, Grant No. NRF/1/MMC/278 and International Centre for Genetic Engineering and Biotechnology (ICGEB) through grant No. CRP/KEN18-01.
Author information
Authors and Affiliations
Contributions
MO performed all the experiments, AK, CM and WM supervised the study, MO wrote the draft manuscript, AK and WM revised the manuscript, conceptualized idea. AK, and WM obtained of funding, contributed with experimental design, coordination and manuscript writing. All authors agreed on the final appearance of the manuscript after careful review.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Odeph, M., Kavoo, A., Mweu, C. et al. Identification and characterization of fungal species associated with the finger millet pathogen Pyricularia oryzae in Kenya. J Plant Pathol 103, 1153–1167 (2021). https://doi.org/10.1007/s42161-021-00900-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42161-021-00900-7