
Abstract
An efficient way to alleviate the pollution imposed by petroleum-based supplies like synthetic fibres and plastics is to prepare biocomposites from recyclable forestry waste with a continuous supply. Despite this, it remains a significant challenge in the field of wood-based panel manufacturing to produce high-performance yet environmentally friendly wood-based materials without the addition of chemical adhesives. Lignin can be used as a “natural adhesive” due to its superior bonding properties, but the dispersion of hemicellulose affects cross-linking at the interfacial interface negatively. This study used lignin/cellulose as a matrix and pretreated it with hydrogen peroxide, sodium hydroxide, sodium silicate solution and in situ bonding of wood fibres to create a high-performance biocomposite material for potential mass production. The findings revealed the tensile (106.63 MPa) and bending strengths (148.78 MPa) of the optimised samples were 125.37% and 91.40% higher than the performance before optimisation. Moreover, the biocomposite demonstrated remarkable hydrophobicity, as evidenced by a water contact angle of 99.96°, and exhibited high thermal stability, without any disintegration observed even when subjected to combustion at 1300 °C. These exceptional properties and advantages render it a highly desirable material for eco-friendly homes and construction applications.
Graphical Abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Technological advancements and rapid economic development have resulted in the depletion of global forest resources [1]. As a means of reducing the consumption of these natural resources, efforts have been made to grow small-diameter wood as a potential material for the preparation of biocomposites [2,3,4]. However, this type of wood is low in density and strength, and is particularly susceptible to cracking and deformation. Hence large amounts of adhesives (e.g., aldehyde-based adhesives) are often used to create biocomposites with desirable physical properties [5, 6]. As a result, the material may have emitted unbound formaldehyde, which has negative effects on the atmosphere and well-being of the consumer [7].
In recent decades, the utilisation of green resources such as small-diameter to develop energy-efficient and environmentally friendly biocomposites has gained increasing recognition, with aldehyde-free or glue-free adhesive technology emerging as a promising approach in this field [8]. Notably, the commercial application of formaldehyde-free adhesives in the wood products industry is unrealistic because of the expensive and complicated manufacturing [9]. Therefore, research in biocomposites is currently focused on adhesive-free technology. It is believed that the emergence of formaldehyde-free binders represents the inevitable step that achieving a better product (i.e., biocomposite that has no formaldehyde emission) [10]. Nevertheless, the production of formaldehyde-free binders primarily relies on petrochemical resources, and its extensive use would result in the rapid depletion of petroleum reserves, thereby contradicting the principles of sustainable development [11,12,13]. For this reason, concerted efforts have been invested in developing bio-based adhesives to avoid environmental pollution and ecological damage [14,15,16]. Nevertheless, several technical constraints make the present methods for producing formaldehyde-free panels uncommercially viable due to their high cost and complexity [17,18,19].
Currently, there is a growing interest among scientists in the development of biocomposites using self-reinforced wood fibres [20]. They have taken advantage of the sustainability and unique porous structure of wood biomaterials and, through microstructure optimisation and multi-component synthesis, have enabled wood composites to show great potential for applications in the fields of electromagnetic interference shielding, superhydrophobicity, high flame retardancy and environmental protection. Wood is mainly composed of lignin, hemicellulose and cellulose. Among these, lignin is one of the most abundant natural polymers in nature. Because of its adhesive properties and aromaticity, it can be used as a “natural adhesive” to replace synthetic polymers derived from petroleum resources. Therefore, in the field of adhesive research, lignin can provide more environmentally friendly and sustainable solutions [21]. Unfortunately, the proportion of cellulose and lignin in contact with each other is reduced due to the dispersion of hemicellulose between them [22, 23]. As hemicellulose contains multiple free and hydroxyl groups linked by 1,4-glycosidic bonds [24, 25], removal of hemicellulose can promote close alignment of fibres and increase the density of the material to further improve its properties [26]. In this way, sustainable wood resources could be converted into high-quality biocomposites once the hemicellulose content has been removed. The main treatment methods include biological, acid and alkaline solution methods. These methods have different degrees of degradation of lignin and hemicellulose, and although they can achieve glue-free bonding, the methods are more complicated and cannot effectively produce cleaner biocomposites, and some products also have low physical properties and poor flame retardancy. Among these, the alkali solution method requires milder experimental conditions and is used more to improve interfibre compatibility than other treatment methods [27]. Sodium silicate is the most common silica compound with the characteristics of safety and environmental protection, high thermal stability and small particle size, which can effectively penetrate into the pore structure of wood and has now been proven to have a good flame-retardant effect. Its flame retardancy is mainly achieved by forming a thermal barrier of inorganic silica slag on the surface of wood, but it cannot fully form a stable network structure when used alone, and the flame-retardant effect is not ideal [28, 29]. Based on this consideration, this study utilises the cross-linking effect of lignin, the skeletal effect of cellulose and the flame-retardant effect of sodium silicate to synergistically achieve green enhancement and efficient flame retardancy of wood.
The study aims to develop a clean method of preparing mass-producible high-performance green biocomposites using lignin/cellulose as a matrix to replace traditional petroleum-based materials. The process involves pretreatment with several chemicals and in situ binding of wood fibres. After this, the biocomposite was subjected to extensive analyses and characterisation to investigate the chemical mechanisms. The method is economical and environmentally sustainable since it does not require adhesives for bonding, thus demonstrating the remarkable potential to substitute traditional biocomposites made using resin binders.
2 Materials and methods
2.1 Source of wood for biocomposite fabrication
The Buxus sinica that was utilised in this study was gathered in Guizhou, China, with an original water percentage of about 20. The wood samples exhibited homogeneous construction and no visible surface flaws, like deterioration, discoloration or cracking.
2.2 Fabrication procedures of biocomposite from Buxus sinica
2.2.1 Preliminary processing of Buxus sinica
A crusher was used for crushing the Buxus sinica, which was then sieved 20–80 to yield sinica wood powder, moisture removing at 105 °C until dry, cooled and placed in a desiccator. Then, the wood powder and hydrogen peroxide (wt%: 30%, Analytical Reagent), sodium hydroxide (Analytical Reagent) and sodium silicate solution (Analytical Reagent) were evenly mixed in the ratio of 1%: 0.5%: 0.2% and stirred under the condition of 150 °C treatment for 4 h and finally rinsed with deionised water to get pretreated powder.
2.2.2 Hot-pressing moulding processing
The pretreated powders with 6% and 8% water content were put into moulds (50 × 50 × 5 mm) and then pressed at temperature of 170 °C plus pressure of 75 MPa for 60 min, followed by a passive cooling to ambient temperature with the moulds to retrieve the test specimens densified wood (DW6% and DW8%). Then, the control samples were prepared in the same experimental conditions with regular wood powder (RWP6% and RWP8%) as raw material (Fig. 1).

Schematic of biocomposite preparation
2.3 Characterisation tests
2.3.1 Multiple physical characterisation of biocomposite
A density measurement was computed based on the final weight and volume through the standard GB/T 17657–2013 set by the government of China. Sample was put in a fixed chamber under relative humidity of 65 ± 5% and temperature of 20 ± 1 °C to achieve a consistent weight, and then, the volume and mass of the samples were quantified. The cross-section and surface of samples were reviewed adopting SU6600 SEM manufactured by the Hitachi High-Technologies Corporation in Tokyo, Japan. The tensile and bending strengths were obtained adopting a robust AGS-X universal material testing machine manufactured by Shimadzu in Japan following the standard GB/T 17657–2013 set by the Chinese Government. During the tests, the crosshead velocity was 5 mm/min, while a 25.13-mm span connected to three-point bending equipment was used for the bending tests. In the tensile test, the interval among the two gauges (upper and lower) was 17 mm. Five tests were conducted on each sample, the measurements were then averaged to guarantee the reliability of the evidence collected. The measurable extent of the samples acquired were 2.5 × 8 × 50 mm3 (thickness × width × length). A Quantachrome Autosorb-IQ surface porosity tester by Anton Paar GmbH, Austria, was implemented to ascertain the pore size variations and surface area. The assessment was performed in an inert environment mediated by nitrogen under the following degassing circumstances: approximately 15 h of period, around 102–105 °C, plus the relative pressure (P/P0) ranging 0.01 and 0.995 for adsorption–desorption mechanism [30].
2.3.2 Characterisation of water resistance
As per the standard GB/T 17657–2013 set by the Chinese Government, the water resistivity was scrutinised by submerging the sample in 20–22 °C distilled water for 10 days. Measurements on thickness and weight variations had been gathered once every 24 h. The contact angle and shape of water droplets laying on the sample surface at various periods (0 and 10 s) had been compiled utilising recording device, and the results were used to ascertain and see if the sample was hydrophobic or hydrophilic in accordance with the ASTMD7334 standard [31]. A DSA100S drop shape analyser manufactured by A.KRÜSS Optronic GmbH in Hamburg, Germany, was deployed to assess the sample.
2.3.3 Characterisation of thermal properties
ATGA/DSC 3 + thermogravimetric analyser manufactured by Mettler Toledo in Columbus, USA, was deployed to assess the thermodynamic durability of samples. Prior for the assessment, approximately 10 mg of sample powder was quantified and placed in the sample holder of the analyser. The assessment was then carried out at an operating temperature ranging from 20 to 600 °C with a steady temperature increase of 10 °C per minute. The sample was exposed to the external fire of a butane pistol for half a minute to verify its flame retardancy, and a recording device was adopted to capture the flame pattern. The burning and final debris of the samples at different durations were studied and contrasted.
2.3.4 Characterisation of the chemical compositions
A Nicolet IS50 ATR-FTIR fabricated by Thermo Fisher Scientific in the USA was deployed to ascertain the alterations of the chemical moieties. The sample was repeatedly scanned for 64 times at a consistent speed of 4 cm−1 within the range of 400–4000 cm−1. Determination of the proportion of lignocellulosic components in all samples using the Van Soest washing method (fibre analyser ANKOM 220). A high-resolution nuclear magnetic resonance (NMR) spectroscopy fabricated by Bruker 400 M in Germany was used to characterise the 13C nuclei of the samples. The chemical constituents were assessed through K-alpha XPS using an Axis Ultradld X-ray photoelectron spectroscope manufactured by Shimadzu Enterprise Management Co., Ltd. in China, whereas Peakfit software was employed to assess C1s finding. The cellulose crystallinity was assessed through the use of an X-ray diffractor (XRD) made by Beijing Purvey General Instrument Co. in China at a 2θ scanning angle within 10 to 50 degrees. The transmission pattern was ascertained with a Perkin Lambda 950 UV–Vis spectrophotometer fabricated by PerkinElmer in the USA.
3 Results and discussion
3.1 Density and physical characteristics of biocomposites
Figure 2a displays the physical characteristics of all biocomposites to determine whether biocomposites are feasible for high-strength structural applications. The density of pretreated specimens DW6% (1.39 g/cm3) and DW8% (1.46 g/cm3) was significantly higher than RWP6% (1.32 g/cm3) and RWP6% (1.36 g/cm3), implying the densities of biocomposites can be enhanced via pretreatment followed by hot pressing [32, 33]. Figure 2b, c show the pore size variation and nitrogen adsorption isotherm of the biocomposites. Both materials demonstrate a type III adsorption isotherm and extremely low porosity, which is a consequence of the cell wall collapse under enclosed environments of escalated pressure and temperature. The adsorption capacity and pore size distribution of DW8% are lower than that of RWP8% owing to the deteriorating of the lignin and hemicellulose within the wood cell wall inside an alkaline environment. As a result, this renders adsorption arduous since the cell wall could possibly be more packed and condensed; the finding also concurs with the density analysis results [34].

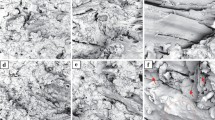
a Density of the biocomposites. b–c Nitrogen isothermal adsorption curves and pore size distribution curves of the biocomposites. d–g Microscopic views of the surface cross-section of the biocomposites
Figure 2d–g show the microscopic images of the cross-section and surface of the biocomposites. What may be seen is the RWP morphology is rough and cracked, while the fibre clusters were firmly wrapped together in a compact configuration on the flattened side of the biocomposites. The dense structure could be formed by lignin melting during the high-temperature thermoforming process (in which the wood fibre cell walls and their depolymerisation also coincide), followed by pressure and cooling [35, 36].
Figure 3a–f show the mechanical property tests of the biocomposites, in which DW8% showed higher performance in the three-point bending and tensile tests, and its static flexural strength and tensile strength reached 141.98 MPa and 106.63 MPa, respectively. Pretreatment samples with higher wood flour moisture content displayed improved mechanical properties. Given that, it is most likely because the higher wood flour water content in hot pressing can soften the wood fibres through heat transfer. As a result, the fibres become more plastic and have greater hydrogen and chemical bonds, resulting in greater mechanical properties. The specific and tensile strengths of DW8% were also evaluated by comparing against those of commonly used composites in this investigation (Fig. 3g). The results revealed that the specific strength of surface DW8% (73 kN∙m/kg significantly greater compared with commonly used composites like wood (6.69 kN∙m/kg), copper alloy (43.7 kN∙m/kg) and mild steel (57.5 kN∙m/kg) [37]. Thus, the biocomposite possesses a remarkable potential for utilisation in furniture and construction.

a–f Mechanical properties of biocomposites. g The specific strength of biocomposites and conventional materials
3.2 Water resistivity of biocomposites
As an effort to broaden the scope of biocomposites in other possibilities, the surface characteristics have been examined systematically. In Fig. 4a, b, it is noted that DW8% has the lowest water absorption (25%) and thickness swelling (14%), which may be due to the formation of a waterproof layer following the hot-pressing treatment on the biocomposites surface. The structure of the closely linked fibre network also prevents water penetration into the composites [38]. Compared to biocomposites made from unprocessed wood flour RWP8% (θ = 80.76°) and RW6% (θ = 77.78°), the preliminary contact angles of biocomposites DW8% (θ = 105.97°) and DW6% (θ = 104.67°) were substantially superior (Fig. 4c–e), proving the pretreatment procedure can improve the hydrophobicity of the biocomposites. DW8% demonstrated the greatest water contact angle and the remarkable hydrophobic efficacy after 10 s (θ = 99.96°). The reason for this is that when the wood cell walls are pretreated, they become more compressible, the fibre components undergo repolymerization during thermoforming reducing the number of surface cracks and tubular pores, and the surface of the biocomposite is flattened and dense, which subsequently reduces the uptake of foreign moisture and improves the water resistance of the biocomposite, which is consistent with previous SEM analysis [39]. The flow diagram of water droplets falling on the sample from high altitude is shown in Fig. 4f. When the biocomposite is placed in a rainy outdoor environment, water droplets in the air can roll on the hydrophobic surface and prevent water penetration.

a–b Thickness expansion rate and intake of water absorption by biocomposites. c–e Contact angle analysis of biocomposites. f Schematic diagram of the flow of water droplets falling from high altitude on a sample
3.3 Heating efficacy and stability of biocomposites
Figure 5 displays the thermal stability variations of biocomposites for comprehension on their thermal degradation [40, 41]. The thermal rate curves (DTG) derived from TGA and of the biocomposites before and after pretreatment are shown in Fig. 5a, b. The released of sample-bound water during heating induced the variations occurred within 30–120 °C, followed by the first rapid decomposition and higher weight loss between 210 and 360 °C, conforming to the dehydration and breaking down of cellulose and hemicellulose. The second large degradation rate in the interval 360–370 °C corresponds to the formation of combustible volatiles and carbons from the decomposition of cellulose and hemicellulose [42, 43]. As may be seen, the sodium silicate treated biocomposites pretreated showed better thermal stability and lower maximum pyrolysis rates than those of the non-pretreated samples.

a Heat loss mass of biocomposites. b The mass loss rate of biocomposites. c The flame resistance of biocomposites. d The mechanism of the flame-retardant process of biocomposites
For the purpose of testing the flame retardancy of the samples, a butane gun burning test was performed in which the specimens were burned for 30 s (Fig. 5c). It can be seen that RWP began to burn at 15 s and intensified at 30 s, whereas DW only began to burn at 30 s. As a result of the combustion, RWP was left with a greater amount of ash, while DW remained unchanged in terms of shape and size [44]. Figure 5d illustrates the flame retardancy progression of the biocomposites. By pretreating boxwood powder with sodium silicate, the carbon content of the material is increased, which results in the enhancement of charring and diminution in wood volatiles. Moreover, the silicate material attached to the interior of the boxwood powder reacts in situ to form a more stable cross-linked carbonised layer, subsequently increasing flame-retardant properties by reducing heat and oxygen diffusion [45, 46].
3.4 Detailed examination of bonding interactions and chemical properties
The cellulose, hemicellulose and lignin contents of all biocomposites are shown in Fig. 6a. Biocomposites pretreated with sodium silicate solution showed increased lignin content while a significant decrease in hemicellulose content. This could be explained by the loss of hemicellulose during solubilisation, followed by exfoliation reactions and alkali hydrolysis in an alkaline environment [47,48,49]. The total nitrocellulose content of the samples before and after pretreatment also differed significantly, with the total nitrocellulose content being significantly higher for DW8% (93.71%) and DW6% (93.15%) compared to RWP8% (87.46%) and RWP6% (84.75%). As a result of the removal of waxes and oils during the pretreatment stage, the fibre contact area was further increased and a stronger bond formed after the hot-pressing process.

a Various lignocellulosic constituents contained in biocomposites. b FTIR spectrum and corresponding chemical moieties presence in the biocomposites. c Variations of XRD spectrum detected for biocomposites. d CP-MAS 13C-NMR spectrum of biocomposites in the range of 20–180 ppm. e The ultraviolet absorption of biocomposites. f–i XPS curves of biocomposites. j Self-enhancement mechanism of the cell wall
The FTIR-ATR absorption peaks at 2900 cm−1, 1956 cm−1, 1425 cm−1, 1268 cm−1 and 1030 cm−1 correspond to symmetric -CH2 stretching, aromatic ring skeleton vibrational C = O stretching, asymmetric bending of C-H in methoxy, C = O stretching of G-lignin and deformation of aromatic C-H planes, respectively (Fig. 6b). The corresponding hemicellulose (1637 cm−1, 1456 cm−1, 1425 cm−1) lignin (1596 cm−1, 1456 cm−1, 1425 cm−1, 1268 cm−1, 1030 cm−1) peaks were all reduced, indicating that alkali disrupts the lignin-carbohydrate polymer and causes degradation of lignin and hemicellulose at high temperature and pressure [50].
A comparison of the crystallinity of biocomposites before and after pretreatment can be seen in Fig. 6c. All samples exhibited diffraction peaks at 2θ = 17.53°, 22.06° and 34.82° that matched to the crystalline types (101), (002) and (004), respectively, where (002) was the main crystalline peak of cellulose I. Despite the hot pressing and pretreatment performed on the Buxus sinica, its native crystalline integrity might still intact. Based on Segal’s empirical equation, the fibre crystallinity (CrI) of the biocomposites is 47.04%, 47.19%, 46.33% and 46.55% for DW8%, DW6%, RWP6% and RWP8%, respectively. Due to the pretreatment with sodium silicate solution, some hemicellulose, lignin and non-fibrous materials in the cell wall have been removed from the amorphous structure. Under high temperatures and pressure, chemical bonds formed between fibres allowed the filaments in the non-crystalline region to be arranged in an orderly fashion, thereby expanding the crystalline region [51]. In addition, the crystalline regions of DW8% and DW6% were larger than those of RWP8% and RWP6%, which indicated that the water content contributed to the formation of crystalline regions and further affected the strength of the biocomposites.
The CP-MAS 13C-NMR spectrum of the biocomposite is depicted in Fig. 6d. Significant hemicellulose degradation can be observed at 172 ppm and 21 ppm due to the signals generated by the carbonyl group in hemicellulose and the methyl group in the acetyl group, respectively [52, 53]. The peaks at 152.6 ppm, 147.6 ppm, 133.2 ppm and 55 ppm represent lignin S3,5 (etherified), S3,5 (non-etherified), S1/4 (non-etherified) and -OCH3, respectively. The intensity of these peaks was enhanced, suggesting the presence of strong vinyl ether bonds in lignin that promote the formation of stronger mechanical substructures [54]. The typical resonance peaks detected at 60 to 110 ppm can be ascribed to cellulose, whereas 105 ppm and 65 ppm can be assigned to cellulose type I C1 and C6. The broad peak at 89–80 ppm is corresponded to C4, while 74–72 ppm is attributed to C2, C3 and C5. Compared with RW8%, the signal peak for the cellulose C4 of pretreated DW8% was enhanced, indicating a decrease in conformational dispersion of cellulose chains. This could probably due to the transformation of cellulose type I into cellulose type II, which agree with the C6 signal results [55, 56].
Figure 6e shows the UV absorption capacity of the biocomposites. It can be seen that the biocomposites treated with sodium silicate exhibit lower UV absorption, which greatly reduces photo-ageing due to UV absorption. This is because the pretreatment process reduces the lignin content, while the photodegradation reaction in wood primarily occurs on lignin. When exposed to UV energy, the alcohol hydroxyl, aromatic and phenolic groups in lignin generate a large number of free radicals. By oxidising these free radicals further with oxygen and water, these free radicals can reduce the mechanical properties of the wood [57, 58].
Figure 6f, j, h and g, display the XPS evaluation performed on the biocomposites surface to ascertain the concentration of components, and four peaks (C1, C2, C3 and C4) were synthesized via the pearl program. Among them, C1 (C–C/C-H) corresponds to the chemical bond among the H and C atoms or only C atoms (C-O), while C2 (C-O), C3 (O-C-O/C = O) and C4 (O = C-O) can be assigned to the chemical bond among O and C (hydroxyl, carbonyl, carboxyl or ester). C1, which is connected to O via double bonds, decreased to a great extent in all the biocomposites prepared after sodium silicate pretreatment, whereas C2, C3 and C4 content increased significantly [59]. Since sodium silicate pretreatment destroyed the lignin–hemicellulose polymer and removed the hemicellulose while wetting and swelling the fibre, more unbound -OH moities became available on the surface of the fibre. As a result of the high temperature and pressure conditions, the -OH moities on the cellulose surface reacted with each other to form more chemical crosslinks. The fibres with lower moisture content cannot form an effective connection, and they are still loose after hot forming. The higher moisture content promotes the formation of stronger chemical bonds inside the biological composites. The higher water content creates more hydroxyl groups during hot pressing, which in turn react with other groups to form more strong chemical bonds between the C and O atoms, resulting in a higher O/C ratio.
As shown from a macroscopic view, Fig. 6j depicts the structural modifications made to the biocomposites throughout processing. First, the pretreatment hydrolysed most of the hemicellulose and increased the proportional amounts of lignin and cellulose. The mechanical strength of the material was significantly increased by closely interacting among the crystalline structures of the fibres through the formation of a tightly bound fibre network with lignin as a fibre binder [60]. Secondly, a dense fibre entangled interfacial framework was created by the homogeneous bonding from each of the shattered fibre cell walls, reducing the inherent porosity and enhancing cellulose materials in biocomposites. During hot pressing, lignin structure fragments self-bond with cellulose fibres, thus strengthening the mechanical attributes of biocomposites [61]. The interunit bonds of β-aryl ether (β-O-4) on lignin were cleaved during pretreatment with sodium silicate solution, leading to a high concentration of phenolic -OH moieties in lignin [6]. As a result, the proportion of hydrogen bonds linking lignin and cellulose was substantially increased and the self-bonding reaction of lignin fragments was also enhanced during hot pressing. Since hot pressing induced the creation of abundant reaction sites on the aromatic ring of lignin, the newly formed higher energy C–C bonds is more favourable than the original aryl ether bonds [62].
4 Conclusion
A clean process was developed using self-adhesive technology to convert waste boxwood powder into biocomposites with excellent properties. The findings revealed the tensile (106.63 MPa) and bending strengths (148.78 MPa) of the optimized samples were 125.37% and 91.40% higher than the performance before optimization. Moreover, the biocomposite demonstrated remarkable hydrophobicity, as evidenced by a water contact angle of 99.96°, and exhibited high thermal stability, without any disintegration observed even when subjected to combustion at 1300 °C. Through the pretreatment process of boxwood powder, hemicellulose and part of the amorphous cell wall structure were removed. This increased the relative content of lignin and cellulose. During further thermoforming, the fibre cell wall collapsed completely. In addition to lignin being used as a fibre binder, lignin was used to strengthen the dimensional stability of the fibre network by chemical bonding between fibre structures. Moreover, the in situ reaction between silicate substances and wood flour produces a more stable cross-linked carbonised layer that is highly resistant to water. In short, biocomposites with excellent properties are produced, which prevent harmful volatiles from being released into the environment from chemical binders, thereby contributing to environmental protection and sustainable development. Therefore, this technology could be a promising approach to utilising green resources for large-scale furniture, decoration and construction applications.
Data availability
The data are available from the corresponding author on reasonable request.
References
Juliebz P (2020) Designing for a green chemistry future. Science 367:397–400. https://doi.org/10.1126/science.aay3060
Chen C, Kuang Y, Zhu S, Burgert I, Keplinger T, Gong A, Li T, Berglund L, Eichhorn SJ, Hu L (2020) Structure-property-function relationships of natural and engineered wood. Nat Rev Mater 5:642–666. https://doi.org/10.1038/s41578-020-0195-z
Li R, Wang Q, Qu G, Zhang Z, Wang H (2023) Green utilization of organic waste resource. Environ Sci Pollut R 1–3. https://doi.org/10.1007/s11356-022-25127-6
Pramreiter M, Nenning T, Malzl L, Konnerth J (2023) A plea for the efficient use of wood in construction. Nat Rev Mater 1–2. https://doi.org/10.1038/s41578-023-00534-4
Xu Y, Zhang X, Liu Z, Zhang X, Luo J, Li J, Shi SQ, Li J, Gao Q (2022) Constructing SiO2 nanohybrid to develop a strong soy protein adhesive with excellent flame-retardant and coating ability. Chem Eng J 446:137065. https://doi.org/10.1016/j.cej.2022.137065
Zeng Y, Yang W, Xu P, Cai X, Dong W, Chen M, Du M, Liu T, Lemstra PJ, Ma P (2022) The bonding strength, water resistance and flame retardancy of soy protein-based adhesive by incorporating tailor-made core-shell nanohybrid compounds. Chem Eng J 428:132390. https://doi.org/10.1016/j.cej.2021.132390
Li T, Chen C, Brozena AH, Zhu JY, Xu L, Driemeier C, Dai J, Rojas OJ, Isogai A, Wagberg L, Hu L (2021) Developing fibrillated cellulose as a sustainable technological material. Nature 590:47–56. https://doi.org/10.1038/s41586-020-03167-7
Chen C, Wu Q, Wan Z, Yang Q, Xu Z, Li D, Jin Y, Rojas OJ (2022) Mildly processed chitin used in one-component drinking straws and single use materials: strength, biodegradability and recyclability. Chem Eng J 442. https://doi.org/10.1016/j.cej.2022.136173
Cottet C, Ramirez-Tapias YA, Delgado JF, de la Osa O, Salvay AG, Peltzer MA (2020) Biobased materials from microbial biomass and its derivatives. Materials 13. https://doi.org/10.3390/ma13061263
Yang Y, Zhang L, Zhang J, Ren Y, Huo H, Zhang X, Huang K, Zhang Z (2023) Reengineering waste boxwood powder into light and high-strength biodegradable composites to replace petroleum-based synthetic materials. ACS Appl Mater Interfaces 15(3):4505–4515. https://doi.org/10.1021/acsami.2c19844
Jiang Z, Ho SH, Wang X, Li Y, Wang C (2021) Application of biodegradable cellulose-based biomass materials in wastewater treatment. Environ Pollut 290:118087. https://doi.org/10.1016/j.envpol.2021.118087
Li C, Wu J, Shi H, Xia Z, Sahoo JK, Yeo J, Kaplan DL (2022) Fiber-based biopolymer processing as a route toward sustainability. Adv Mater 34:e2105196. https://doi.org/10.1002/adma.202105196
Li YE (2019) Sustainable Biomass Materials for Biomedical Applications. ACS Biomater Sci Eng 5:2079–2092. https://doi.org/10.1021/acsbiomaterials.8b01634
Veerasimman A, Shanmugam V, Rajendran S, Johnson DJ, Subbiah A, Koilpichai J, Marimuthu U (2021) Thermal properties of natural fiber sisal based hybrid composites - a brief review. J Nat Fibers 1–11. https://doi.org/10.1080/15440478.2020.1870619
Vigneshwaran S, Sundarakannan R, John KM, Joel Johnson RD, Prasath KA, Ajith S, Arumugaprabu V, Uthayakumar M (2020) Recent advancement in the natural fiber polymer composites: a comprehensive review. J Clean Prod 277. https://doi.org/10.1016/j.jclepro.2020.124109
Xu J, Li X, Liu R, Shang Z, Long L, Qiu H, Ni Y (2020) Dialdehyde modified cellulose nanofibers enhanced the physical properties of decorative paper impregnated by aldehyde-free adhesive. Carbohyd Polym 250:116941. https://doi.org/10.1016/j.carbpol.2020.116941
Koch SM, Pillon M, Keplinger T, Dreimol CH, Weinkötz S, Burgert I (2022) Intercellular matrix infiltration improves the wet strength of delignified wood composites. ACS Appl Mater Interfaces 14:31216–31224. https://doi.org/10.1021/acsami.2c04014
Zhang Y, Xu N, Bai Y, Liu J, Guo Z, Niu Y (2022) Comparison of multidimensional mass transfer models of formaldehyde emissions originating from different surfaces of wood-based panels. Sci Total Environ 848:157367. https://doi.org/10.1016/j.scitotenv.2022.157367
Cai J, Murugadoss V, Jiang J, Gao X, Lin Z, Huang M, Guo J, Alsareii S, Algadi H, Kathiresan M (2022) Waterborne polyurethane and its nanocomposites: a mini-review for anti-corrosion coating, flame retardancy, and biomedical applications. Adv Compos Hybrid Mater 5:641–650. https://doi.org/10.1007/s42114-022-00473-8
Etale A, Onyianta AJ, Turner SR, Eichhorn SJ (2023) Cellulose: a review of water interactions, applications in composites, and water treatment. Chem Rev. https://doi.org/10.1021/acs.chemrev.2c00477
Diblasi C (2008) Modeling chemical and physical processes of wood and biomass pyrolysis. Prog Energ Combust 34:47–90. https://doi.org/10.1016/j.pecs.2006.12.001
Ge S, Ma NL, Jiang S, Ok YS, Lam SS, Li C, Shi SQ, Nie X, Qiu Y, Li D, Wu Q, Tsang DCW, Peng W, Sonne C (2020) Processed bamboo as a novel formaldehyde-free high-performance furniture biocomposite. ACS Appl Mater Interfaces 12:30824–30832. https://doi.org/10.1021/acsami.0c07448
Ge S, Zuo S, Zhang M, Luo Y, Yang R, Wu Y, Zhang Y, Li J, Xia C (2021) Utilization of decayed wood for polyvinyl chloride/wood flour composites. J Mater Res Technol 62–9. https://doi.org/10.1016/j.jmrt.2021.03.026
Ge S, Liang Y, Zhou C, Sheng Y, Zhang M, Cai L, Zhou Y, Huang Z, Manzo M, Wu C, Xia C (2022) The potential of Pinus armandii Franch for high-grade resource utilization. Biomass Bioenergy 158. https://doi.org/10.1016/j.biombioe.2022.106345
Xia Q, Chen C, Yao Y, He S, Wang X, Li J, Gao J, Gan W, Jiang B, Cui M (2021) In situ lignin modification toward photonic wood. Adv Mater 33:2001588. https://doi.org/10.1002/adma.202001588
Chu T, Gao Y, Yi L, Fan C, Yan L, Ding C, Liu C, Huang Q, Wang Z (2022) Highly fire-retardant optical wood enabled by transparent fireproof coatings. Adv Compos Hybrid Mater 5:1821–1829. https://doi.org/10.1007/s42114-022-00440-3
Li X, Tabil LG, Panigrahi S (2007) Chemical treatments of natural fiber for use in natural fiber-reinforced composites: a review. J Polym Environ 15:25–33. https://doi.org/10.1007/s10924-006-0042-3
Li P, Zhang Y, Zuo Y, Lu J, Yuan G, Wu Y (2020) Preparation and characterization of sodium silicate impregnated Chinese fir wood with high strength, water resistance, flame retardant and smoke suppression. J Mater Res Technol 9(1):1043–1053. https://doi.org/10.1016/j.jmrt.2019.10.035
Liu Q, Chai Y, Ni L, Lyu W (2020) Flame retardant properties and thermal decomposition kinetics of wood treated with boric acid modified silica sol. Materials 13(20):4478. https://doi.org/10.3390/ma13204478
Ren Y, Yang Y, Zhang J, Ge S, Ye H, Shi Y, Xia C, Sheng Y, Zhang Z (2022) Innovative conversion of pretreated Buxus sinica into high-performance biocomposites for potential use as furniture material. ACS Appl Mater Interfaces 14:47176–47187. https://doi.org/10.1021/acsami.2c15649
Ji X, Dong Y, Nguyen TT, Chen X, Guo M (2018) Environment-friendly wood fibre composite with high bonding strength and water resistance. R Soc Open Sci 5:172002. https://doi.org/10.1098/rsos.172002
Jiang B, Chen C, Liang Z, He S, Kuang Y, Song J, Mi R, Chen G, Jiao M, Hu L (2019) Lignin as a wood-inspired binder enabled strong, water stable, and biodegradable paper for plastic replacement. Adv Funct Mater 30. https://doi.org/10.1002/adfm.201906307
Li T, Zhai Y, He S, Gan W, Wei Z, Heidarinejad M, Dalgo D, Mi R, Zhao X, Song J (2019) A radiative cooling structural material. Science 364:760–763. https://doi.org/10.1126/science.aau9101
Li K, Wang S, Chen H, Yang X, Berglund LA, Zhou Q (2020) Self-densification of highly mesoporous wood structure into a strong and transparent film. Adv Mater 32:2003653. https://doi.org/10.1002/adma.202003653
Li L, Sun J, Jia G (2012) Properties of natural cotton stalk bark fiber under alkali treating. J Appl Polym Sci 125:E534–E539. https://doi.org/10.1002/app.36987
Yang Y, Ren Y, Ge S, Ye H, Shi Y, Xia C, Sheng Y, Zhang Z (2022) Transformation of Buxus sinica into high-quality biocomposites via an innovative and environmentally-friendly physical approach. Appl Surf Sci 606:154595. https://doi.org/10.1016/j.apsusc.2022.154595
Kaffashsaie E, Yousefi H, Nishino T, Matsumoto T, Mashkour M, Madhoushi M, Kawaguchi H (2021) Direct conversion of raw wood to TEMPO-oxidized cellulose nanofibers. Carbohyd Polym 262. https://doi.org/10.1016/j.carbpol.2021.117938
Khoo SC, Peng WX, Yang Y, Ge SB, Soon CF, Ma NL, Sonne C (2020) Development of formaldehyde-free bio-board produced from mushroom mycelium and substrate waste. J Hazard Mater 400:123296. https://doi.org/10.1016/j.jhazmat.2020.123296
Mi R, Chen C, Keplinger T, Pei Y, He S, Liu D, Li J, Dai J, Hitz E, Yang B (2020) Scalable aesthetic transparent wood for energy efficient buildings. Nat commun 11:3836. https://doi.org/10.1038/s41467-020-17513-w
Kang X, Lu Z, Feng W, Wang J, Fang X, Xu Y, Wang Y, Liu B, Ding T, Ma Y (2021) A novel phosphorous and silicon-containing benzoxazine: highly efficient multifunctional flame-retardant synergist for polyoxymethylene. Adv Compos Hybrid Mater 4:127–137. https://doi.org/10.1007/s42114-020-00198-6
Xu D, Huang Q, Shi Z, Chen Y, Guo L, Wang C, Liu C (2023) Polypyrrole nanotube derived flame-retardant substrate of cellulose nanofiber composites with thermal conductive and electromagnetic interference shielding effect. Composites Communications 38:101507. https://doi.org/10.1016/j.coco.2023.101507
Meng X, Fan W, Wan Mahari WA, Ge S, Xia C, Wu F, Han L, Wang S, Zhang M, Hu Z, Ma NL, Van Le Q, Lam SS (2021) Production of three-dimensional fiber needle-punching composites from denim waste for utilization as furniture materials. J Clean Prod 281. https://doi.org/10.1016/j.jclepro.2020.125321
Migneault S, Koubaa A, Perré P, Riedl B (2015) Effects of wood fiber surface chemistry on strength of wood-plastic composites. Appl Surf Sci 343:11–18. https://doi.org/10.1016/j.apsusc.2015.03.010
Moon RJ, Martini A, Nairn J, Simonsen J, Youngblood J (2011) Cellulose nanomaterials review: structure, properties and nanocomposites. Chem Soc Rev 40:3941–3994. https://doi.org/10.1039/c0cs00108b
NagarajaGanesh B, Ganeshan P, Ramshankar P, Raja K (2019) Assessment of natural cellulosic fibers derived from Senna auriculata for making light weight industrial biocomposites. Ind Crop Prod 139. https://doi.org/10.1016/j.indcrop.2019.111546
Fan Q, Ou R, Hao X, Deng Q, Liu Z, Sun L, Zhang C, Guo C, Bai X, Wang Q (2022) Water-induced self-assembly and in situ mineralization within plant phenolic glycol-gel toward ultrastrong and multifunctional thermal insulating aerogels. ACS Nano 16:9062–9076. https://doi.org/10.1021/acsnano.2c00755
Nasution H, Olaiya NG, Haafiz MKM, Abdullah CK, Bakar SA, Olaiya FG, Mohamed A, HPS AK (2021) The role of amphiphilic chitosan in hybrid nanocellulose-reinforced polylactic acid biocomposite. Polym Advan Technol 32:3446–3457. https://doi.org/10.1002/pat.5355
Nishiyama Y, Wada M, Hanson BL, Langan P (2010) Time-resolved X-ray diffraction microprobe studies of the conversion of cellulose I to ethylenediamine-cellulose I. Cellulose 17:735–745. https://doi.org/10.1007/s10570-010-9415-9
Xia Q, Chen C, Li T, He S, Gao J, Wang X, Hu L (2021) Solar-assisted fabrication of large-scale, patternable transparent wood. Sci adv 7(5):7342–7369. https://doi.org/10.1126/sciadv.abd7342
Panaitescu DM, Frone AN, Chiulan I, Casarica A, Nicolae CA, Ghiurea M, Trusca R, Damian CM (2016) Structural and morphological characterization of bacterial cellulose nano-reinforcements prepared by mechanical route. Mater Design 110:790–801. https://doi.org/10.1016/j.matdes.2016.08.052
Song J, Chen C, Zhu S, Zhu M, Dai J, Ray U, Li Y, Kuang Y, Li Y, Quispe N, Yao Y, Gong A, Leiste UH, Bruck HA, Zhu JY, Vellore A, Li H, Minus ML, Jia Z, Martini A, Li T, Hu L (2018) Processing bulk natural wood into a high-performance structural material. Nature 554:224–228. https://doi.org/10.1038/nature25476
Holtman KM, Hm C, Jameel H, Kadla JF (2006) Quantitative 13C NMR characterization of milled wood lignins isolated by different milling techniques. J Wood Chem Technol 26:21–34. https://doi.org/10.1080/02773810600582152
Mao JD, Holtman KM, Scott JT, Kadla JF (2006) Schmidt R K (2006) Differences between lignin in unprocessed wood, milled wood, mutant wood, and extracted lignin detected by 13C solid-state NMR. J Agric Food Chem 54(26):9677–9686. https://doi.org/10.1021/jf062199q
Santoni I, Callone E, Sandak A, Sandak J, Dirè S (2015) Solid state NMR and IR characterization of wood polymer structure in relation to tree provenance. Carbohyd Polym 117:710–721. https://doi.org/10.1016/j.carbpol.2014.10.057
Focher B, Palma M, Canetti M, Torri G, Cosentino C, Gastaldi G (2001) Structural differences between non-wood plant celluloses: evidence from solid state NMR, Vib Spectrosc and X-ray diffractometry. Ind Crop Prod 13:193–208. https://doi.org/10.1016/s0926-6690(00)00077-7
Garemark J, Perea-Buceta JE, Felhofer M, Chen B, Cortes Ruiz MF, Sapouna I, Gierlinger N, Kilpeläinen IA, Berglund LA, Li Y (2023) Strong, shape-memory lignocellulosic aerogel via wood cell wall nanoscale reassembly. ACS Nano. https://doi.org/10.1021/acsnano.2c11220
Kaschuk JJ, Al Haj Y, Rojas OJ, Miettunen K, Abitbol T, Vapaavuori J (2022) Plant-based structures as an opportunity to engineer optical functions in next-generation light management. Adv Mater 34:2104473. https://doi.org/10.1002/adma.202104473
Li T, Song J, Zhao X, Yang Z, Pastel G, Xu S, Jia C, Dai J, Chen C, Gong A (2018) Anisotropic, lightweight, strong, and super thermally insulating nanowood with naturally aligned nanocellulose. Sci Adv 4(3):3724. https://doi.org/10.1126/sciadv.aar3724
Zhang H, Liu P, Musa SM, Mai C, Zhang K (2019) Dialdehyde cellulose as a bio-based robust adhesive for wood bonding. ACS Sustainable Chem Eng 7:10452–10459. https://doi.org/10.1021/acssuschemeng.9b00801
Ye H, Wang Y, Yu Q, Ge S, Fan W, Zhang M, Huang Z, Manzo M, Cai L, Wang L, Xia C (2022) Bio-based composites fabricated from wood fibers through self-bonding technology. Chemosphere 287:132436. https://doi.org/10.1016/j.chemosphere.2021.132436
Xiao S, Chen C, Xia Q, Liu Y, Yao Y, Chen Q, Hartsfield M, Brozena A, Tu K, Eichhorn SJ (2021) Lightweight, strong, moldable wood via cell wall engineering as a sustainable structural material. Science 374:465–471. https://doi.org/10.1126/science.abg9556
Duan H, Zhuang C, Mei F, Zeng C, Pashameah RA, Huang M, Alzahrani E, Gao J, Han Y, Yu Q (2022) Benzyl (4-fluorophenyl) phenylphosphine oxide-modified epoxy resin with improved flame retardancy and dielectric properties. Adv Compos Hybrid Mater 5:776–787. https://doi.org/10.1007/s42114-022-00491-6
Acknowledgements
Hebei Provincial Scientific Research Project for Introducing of National High-level Innovative Talents (2021HBQZYCXY011), Hunan Province Key R&D (2022NK2043), The Hunan Provincial Science and Technology Innovation Leaders (2021RC4033) and The Science and Technology Talent Support Project of Hunan Province (CN) (2020TJ-Q18).
Funding
Hunan Province Key R&D (2022NK2043), The Hunan Provincial Science and Technology Innovation Leaders (2021RC4033) and The Science and Technology Talent Support Project of Hunan Province (CN) (2020TJ-Q18).
Author information
Authors and Affiliations
Contributions
Yang Yang, Lei Zhang and JiJuan Zhang wrote and revised the main manuscript text. Yi Ren, HongFei Huo and Mashallah Rezakazemi prepared all the figures. Xu Zhang and Kai Huang edited the main manuscript text. Zhongfeng Zhang revised and supported funding. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yang, Y., Zhang, L., Zhang, J. et al. Fabrication of environmentally, high-strength, fire-retardant biocomposites from small-diameter wood lignin in situ reinforced cellulose matrix. Adv Compos Hybrid Mater 6, 140 (2023). https://doi.org/10.1007/s42114-023-00721-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42114-023-00721-5